

Abstract
5-Amino-N-aryl-3-[(4-methoxyphenyl)amino]-1H-pyrazole-4-carboxamides 4a–c were synthesized by the reaction of N-(aryl)-2-cyano-3-[(4-methoxyphenyl)amino]-3-(methylthio)acrylamides 3a–c with hydrazine hydrate in ethanol. The reaction of 5-amino-N-aryl-1H-pyrazoles 4a–c with acetylacetone 5 or 2-(4-methoxybenzylidene)malononitrile 8 yielded the pyrazolo[1,5-a]pyrimidine derivatives 7a–c and 10a–c, respectively. The structures of the synthesized compounds were established based on elemental analysis and spectral data (IR, MS, 1H-NMR, and 13C-NMR). Representative examples of the new synthesized products were screened for their in vitro cytotoxic activity against Ehrlich Ascites Carcinoma (EAC) cells.
Keywords: N-Substituted cyanoacetamide; Ketene N,S-acetals; Pyrazolo[1,5-a]pyrimidines; 5-Aminopyrazoles; Cytotoxic activity
Introduction
The design and synthesis of novel mono-, di-, and polycyclic fused nitrogen heterocyclic compounds is among the active principles in chemical materials, particularly those displaying strategic roles in the development of different industries, especially from the biological point of view. Pyrazoles and related fused heterocyclic derivatives have great importance in the medicinal field as biological agents such as antimicrobial, anti-inflammatory, and anticancer agents [1–6]. The importance of pyrazole and pyrazolopyrimidine in the pharmacological industry as antitumor agents [7, 8] promoted us to synthesize new derivatives that may serve as new chemotherapeutic drugs.
A literature survey revealed that some drugs bearing pyrazole and pyrazolopyrimidine moieties are considered as the most active in drug manufacture such as Celecoxib [9], Pyrazofurin [10], Ramifenazone [11], Zaleplon [12], and Indiplon [13], respectively [Fig. 1].
Fig. 1.
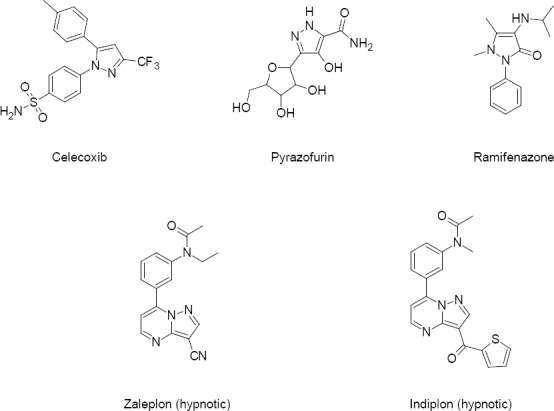
The structures of some drugs bearing the pyrazole and pyrazolopyrimidine moiety
In light of these facts and as a continuation of our previous work in the synthesis of novel compounds with promising biological applications [14–16], we report herein the synthesis of new 5-aminopyrazole and pyrazolo[1,5-a]pyrimidine derivatives and an examination of their cytotoxic activity. The structures of the synthesized compounds were established based on elemental analysis and spectral data (IR, MS, 1H-NMR, and 13C-NMR).
Results and Discussion
Chemistry
N-substituted cyanoacetamide derivatives 1a–c [17] were utilized as key starting materials in the synthesis of novel heterocyclic compounds (Schemes 1, 3, and 4). Cyanoacetamide derivatives 1a–c were reacted with 4-methoxyphenylisothiocyanate in absolute ethanol in the presence of an equimolar amount of potassium hydroxide to give the corresponding intermediates (2a–c); when the latter was alkylated with methyl iodide in ethanol, it afforded the novel ketene N,S-acetals 3a–c. The structures of 3a–c were established on the basis of their elemental analysis and spectral data (MS, IR, 1H–NMR, and 13C-NMR). As an example, the mass spectrum of compound 3b (m/z 353) [M+] revealed the molecular formula C19H19N3O2S. Its IR spectrum (KBr/cm−1) showed a band at 3357 corresponding to an NH group, a band at 2189 for a C≡N group, and a band at 1629 for a C=O group. Its 1H-NMR spectrum (DMSO-d6, δ ppm) revealed three singlets at 2.17, 2.21, and 3.72 representing -SCH3, -CH3, and -OCH3 protons, respectively, four doublets at 6.92, 7.05, 7.25, and 7.37 corresponding to the aromatic protons (AB system, each with JHH=8.4 Hz), and two singlets at 9.37 and 11.82 assigned to two NH groups which were D2O exchangeable. Its 13C-NMR spectrum (DMSO-d6, δ ppm) was characterized by signals at 17.0, 119.2, and 165.3 assigned to -SCH3, -C≡N, and C=O carbons, respectively. The reaction of the compounds 3a–c with hydrazine hydrate in refluxing ethanol gave the corresponding 5-amino-N-aryl-1H-pyrazole-4-carboxamides 4a–c (Scheme 1). The structures of 4a–c were established on the basis of their elemental analysis and spectral data (MS, IR, 1H–NMR, and 13C-NMR). Structure 4c was supported by its mass spectrum (m/z 358) [M+], which agrees with its molecular formula C17H16ClN5O2. Its IR spectrum (KBr/cm−1) showed a band at 3347 and 3031 corresponding to NH and NH2 groups and a band at 1642 for a C=O group. Its 1H-NMR spectrum (DMSO-d6, δ ppm) displayed a singlet at 3.64 representing -OCH3 protons, a broad signal at 6.00 corresponding to the NH2 group which was D2O exchangeable, a multiplet at 6.77–7.51 related to the aromatic protons, and another three singlets at 8.30, 8.82, and 11.20 assignable to the three NH groups which were D2O exchangeable. Its 13C-NMR spectrum (DMSO-d6, δ ppm) was characterized by signals at 55.7, 87.9, and 163.6 assigned to -OCH3, C4 of the pyrazole moiety, and C=O carbons, respectively.
Sch. 1.
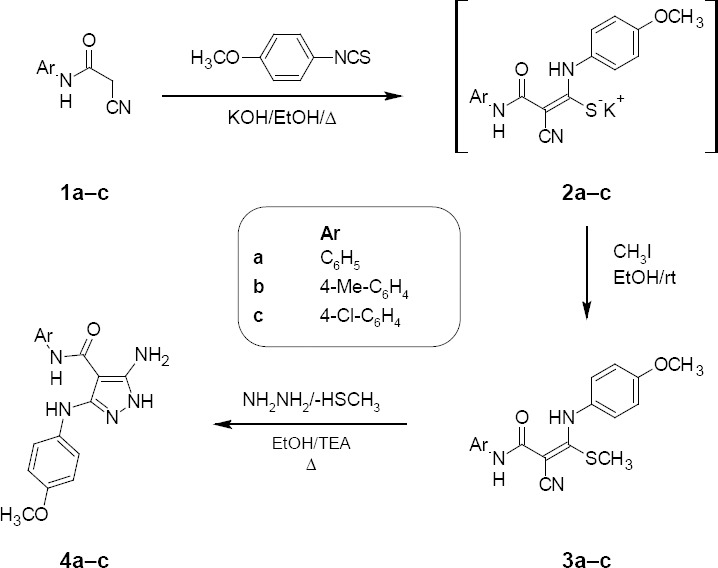
Synthesis of N-(aryl)-2-cyano-3-(4-methoxyphenylamino)-3-(methylthio)acrylamides 3a–c and 5-amino-N-aryl-3-(4-methoxyphenylamino)-1H-pyrazole-4-carboxamides 4a–c
Sch. 2.
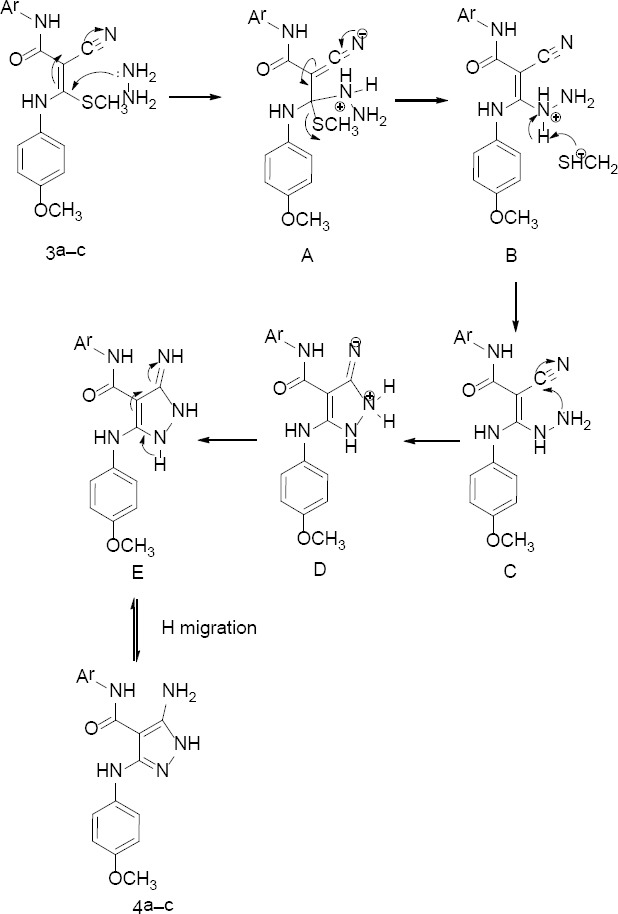
Mechanism for the formation of 5-amino-N-aryl-3-[(4-methoxyphenyl)amino]-1H-pyrazole-4-carboxamides 4a–c
Sch. 3.
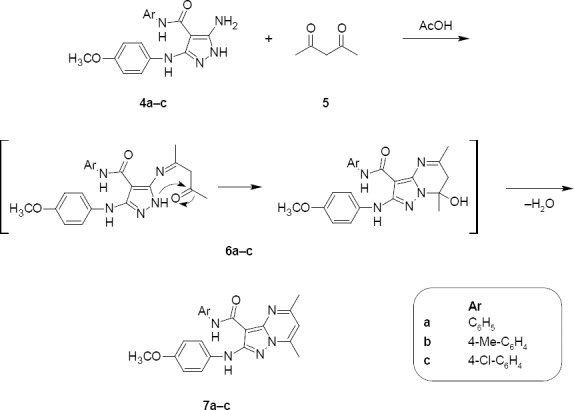
Synthesis of N-aryl-2-[(4-methoxyphenyl)amino]-5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxamides 7a–c
Sch. 4.
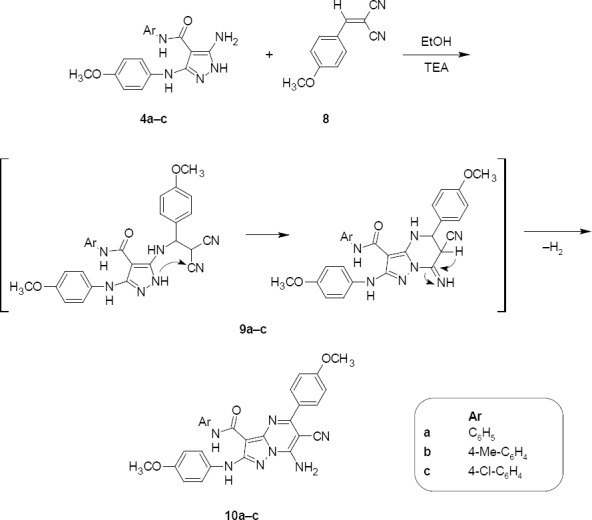
Synthesis of 7-amino-N-aryl-6-cyano-5-(4-methoxyphenyl)-2-[(4-methoxyphenyl)amino]pyrazolo[1,5-a]pyrimidine-3-carboxamides 10a–c
The possible formation of 5-aminopyrazoles 4a–c is shown in Scheme 2. First, Michael addition to ketene N,S-acetals 3a–c occurs with the lone pair of the NH2 group in hydrazine to form intermediate adduct A. Then, the methylthio ion is removed, which results in the formation of intermediate B. This methylthio ion abstracts the proton of the ammonium ion to produce an intermediate C. Subsequently, intermolecular cyclization occurs by the lone pair on the NH2 group, attacking the cyano group to produce intermediate D. The ammonium proton abstraction in D occurs to form E, followed by aromatic-driven 1,5-hydrogen migration to yield products 4a–c [18].
Compounds 4a–c were reacted with acetylacetone 5 in boiling glacial acetic acid to afford the corresponding new pyrazolo[1,5-a]pyrimidines 7a–c. The formation of compounds 7a–c was therefore assumed to proceed via an initial attack of the exocyclic amino group of 4a–c on the keto group of the 1,3-dicarbonyl compound 5, followed by intramolecular cyclization via the elimination of water (Scheme 3). The structures of 7a–c were established on the basis of their elemental analysis and spectral data (MS, IR, 1H–NMR, and 13C-NMR). Structure 7c was supported by its mass spectrum (m/z 421) [M+], which agrees with its molecular formula C22H20ClN5O2. Its IR spectrum (KBr/cm−1) showed a band at 3314 corresponding to two NH groups and a band at 1664 for a C=O group. Its 1H-NMR spectrum displayed three singlets at 2.55, 2.62, and 3.70 due to two -CH3 and -OCH3 protons, respectively, a signal at 6.92 corresponding to the H-6 proton of the pyrimidine nucleus, two doublets at 6.88 and 7.36 related to the four aromatic protons (AB system, JHH=7.8 Hz), another two doublets at 7.61 and 7.68 corresponding to the four aromatic protons in the other ring (AB system, JHH=6.7 Hz), and two singlets at 9.06 and 10.03 assignable to the two NH groups which were D2O exchangeable. Its 13C-NMR spectrum (CDCl3, δ ppm) was characterized by signals at 17.3, 24.6, 55.6, 108.5, and 163.0 assigned to two -CH3, -OCH3, C6 of the pyrazolopyrimidine moiety and C=O carbons, respectively.
2-(4-Methoxybenzylidene)malononitrile 8 was reacted with 4a–c in refluxing ethanol in the presence of triethylamine to give 7-amino-6-cyano-5-aryl-2-(arylamino)pyrazolo[1,5-a]pyrimidine-3-carboxamides 10a–c. The formation of compounds 10a-c is assumed to proceed via an initial attack of the exocyclic amino function of compounds 4a–c on the α,β-unsaturated system in compound 8 followed by the intramolecular cyclization and spontaneous autooxidation through the loss of a hydrogen molecule [19] (Scheme 4). The structures of 10a–c were established on the basis of their elemental analysis and spectral data (MS, IR, 1H–NMR, and 13C-NMR). As an example, structure 10b was supported by its mass spectrum (m/z 520) [M++1], which agrees with its molecular formula C29H25N7O3. Its IR spectrum (KBr/cm−1) showed an absorption band at 3447, 3296 which corresponds to the NH and NH2 groups, a band at 2211 due to C≡N, and a band at 1653 due to C=O. Its 1H-NMR spectrum displayed three singlets at 2.24, 3.71, and 3.84 due to -CH3 and two -OCH3 protons, respectively, six doublets at 6.85, 7.13, 7.16, 7.46, 7.51, and 7.97 related to the aromatic protons (AB system, JHH=9.2, 7.5, 8.5, 8.4, 8.2, and 8.0 Hz, respectively), and three singlets at 8.70, 9.78, and 12.76 assignable to NH2 and two NH groups, respectively, which were D2O exchangeable. Its 13C-NMR spectrum (CDCl3, δ ppm) was characterized by signals at 55.4, 55.6, 114.8, and 163.6 assigned to two -OCH3, -C≡N, and C=O carbons, respectively.
Biological Evaluation
In Vitro Cytotoxic Screening
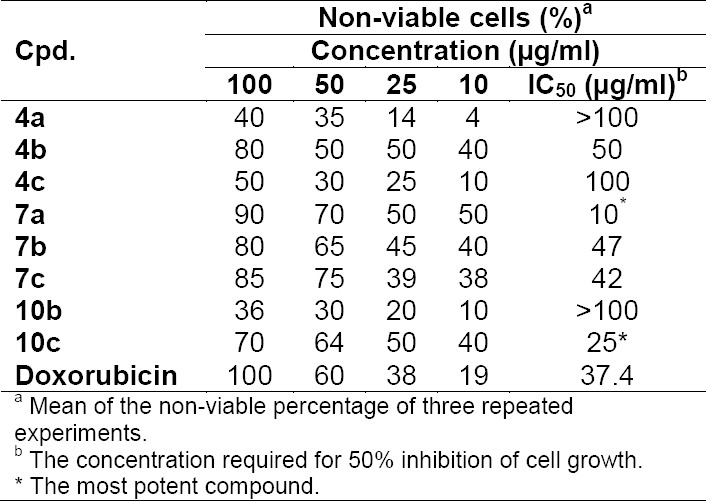
In the present study, some of the newly synthesized compounds were evaluated in vitro for their cytotoxic activities against the Ehrlich Ascites Carcinoma cells (EAC) where Doxorubicin was used as a standard drug. The results were expressed as the IC50 value, which corresponds to the concentration required for 50% inhibition of cell growth of the treated cells when compared to that of the control cells. From the results in Table 1, it was found that compounds 7a (IC50=10 µg/ml) and 10c (IC50=25 µg/ml) exhibited the highest cytotoxic activity compared to the reference drug, Doxorubicin (IC50=37.4 µg/ml). The compounds 4b (IC50=50 µg/ml), 7b (IC50=47µg/ml), and 7c (IC50=42 µg/ml) displayed moderate cytotoxic activity against EAC cells, while compounds 4a, 4c, and 10b showed lower activity than the reference drug.
Tab. 1.
Cytotoxicity of the tested compounds against Ehrlich Ascites Carcinoma (EAC) cells
Experimental
All melting points were measured on a Gallenkamp melting point apparatus and are uncorrected. The IR spectra were recorded (KBr disk) on a Perkin Elmer 1650 FTIR instrument. The 1H-NMR (500 MHz) and 13C-NMR (125 MHz) spectra were recorded on a Varian spectrometer using DMSO-d6 or CDCl3 as the solvent and TMS as an internal standard. Chemical shifts are reported in ppm. Mass spectra were recorded on a Varian MAT 112 spectrometer at 70 eV. Elemental analyses were performed at the Microanalytical Center, Cairo University, Egypt.
The progress of the reactions was monitored by thin-layer chromatography (TLC) using aluminum sheets coated with silica gel F254 (Merck) with viewing under short-wavelength UV lamp detection. All evaporations were carried out under reduced pressure at 40°C.
Reagents and solvents used in the synthesis were purchased from Sigma-Aldrich. Compounds 1a–c were prepared according to the reported procedure [17].
N-(Aryl)-2-cyano-3-[(4-methoxyphenyl)amino]-3-(methylthio)acrylamides (3a–c)
A mixture of N-substituted cyanoacetamide derivatives 1a-c (0.01 mol) and 4-methoxyphenylisothiocyanate (0.01 mol) was heated for 5-10 min in ethanol (25 ml) containing potassium hydroxide (0.01 mol). After cooling, methyl iodide (0.01 mol) was added. The reaction mixture was stirred at room temperature for 1 h and then poured onto ice water. The precipitated product was filtered and recrystallized from ethanol.
2-Cyano-3-[(4-methoxyphenyl)amino]-3-(methylthio)-N-phenylacrylamide (3a)
Light yellow prisms, m.p. 102–104°C, yield (71%). IR (KBr) νmax/cm−1 3382 (NH), 2186 (C≡N), 1633 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.18 (s, 3H, SCH3), 3.72 (s, 3H, OCH3), 6.91 (d, 2H, aromatic, JHH=9.2 Hz), 7.02 (t, 1H, aromatic, JHH=7.7 Hz), 7.23 (t, 2H, aromatic, JHH=8.4 Hz), 7.25 (d, 2H, aromatic, JHH=8.4 Hz), 7.48 (d, 2H, aromatic, JHH=7.7 Hz), 9.46 (s, 1H, NH, D2O exchangeable), 11.78 (s, 1H, NH, D2O exchangeable). Anal. Calcd. (%) for C18H17N3O2S (339.41): C, 63.70; H, 5.05; N, 12.38. Found: C, 63.50; H, 5.20; N, 12.20%.
2-Cyano-3-[(4-methoxyphenyl)amino]-N-(4-methylphenyl)-3-(methylthio)acrylamide (3b)
Colorless prisms, m.p. 175–176°C, yield (76%). IR (KBr) νmax/cm−1 3357 (NH), 2189 (C≡N), 1629 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.17 (s, 3H, SCH3), 2.21 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 6.92 (d, 2H, aromatic, JHH=8.4 Hz), 7.05 (d, 2H, aromatic, JHH=8.4 Hz), 7.25 (d, 2H, aromatic, JHH=8.4 Hz), 7.37 (d, 2H, aromatic, JHH=8.4 Hz), 9.37 (s, 1H, NH, D2O exchangeable), 11.82 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, δ ppm) 17.0 (-SCH3), 20.9 (-CH3), 55.8 (-OCH3), 78.6 (C2, acrylamide), 114.9 (2C, aromatic), 119.2 (C≡N), 121.8, 126.3, 129.3, 131.5, 133.4, 136.1, 158.1 (10C, aromatic), 165.3 (C=O), 168.1 (C3, acrylamide). MS m/z: 353 [M+]. Anal. Calcd. (%) for C19H19N3O2S (353.44): C, 64.57; H, 5.42; N, 11.89. Found: C, 64.40; H, 5.64; N, 12.00%.
N-(4-Chlorophenyl)-2-cyano-3-[(4-methoxyphenyl)amino]-3-(methylthio)acrylamide (3c)
White crystals, m.p. 156–158°C, yield (70%). IR (KBr) νmax/cm−1 3310 (NH), 2193 (C≡N), 1635 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.19 (s, 3H, SCH3), 3.71 (s, 3H, OCH3), 6.91 (d, 2H, aromatic, JHH=6.9 Hz), 7.25 (d, 2H, aromatic, JHH=6.9 Hz), 7.30 (d, 2H, aromatic, JHH=6.9 Hz), 7.52 (d, 2H, aromatic, JHH=6.9 Hz), 9.62 (s, 1H, NH, D2O exchangeable), 11.68 (s, 1H, NH, D2O exchangeable). Anal. Calcd. (%) for C18H16ClN3O2S (373.86): C, 57.83; H, 4.31; N, 11.24. Found: C, 58.00; H, 4.10; N, 11.00%.
5-Amino-N-aryl-3-[(4-methoxyphenyl)amino]-1H-pyrazole-4-carboxamides (4a–c)
A mixture of compounds 3a–c (0.01 mol), hydrazine hydrate (0.01 mol), and few drops of triethylamine in ethanol (30 ml) was refluxed for 4 h and then the solvent evaporated under reduced pressure. The resulting solid product was collected by filtration and recrystallized from ethanol.
5-Amino-3-[(4-methoxyphenyl)amino]-N-phenyl-1H-pyrazole-4-carboxamide (4a)
White crystals, m.p. 175–177°C, yield (79%). IR (KBr) νmax/cm−1 3354, 3030 (NH, NH2), 1645 (C=O). 1H-NMR (DMSO-d6, δ ppm) 3.63 (s, 3H, OCH3), 5.98 (s, 2H, NH2, D2O exchangeable), 6.76 (d, 2H, aromatic, JHH=7.7 Hz), 6.98 (t, 1H, aromatic, JHH=7.7 Hz), 7.18 (t, 2H, aromatic, JHH=8.4 Hz), 7.25 (d, 2H, aromatic, JHH=7.7 Hz), 7.45 (d, 2H, aromatic, JHH=7.7 Hz), 8.30 (s, 1H, NH, D2O exchangeable), 8.74 (s, 1H, NH, D2O exchangeable), 11.22 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, δ ppm) 55.6 (-OCH3), 88.3 (C4, pyrazole), 114.7, 117.5, 120.3, 123.3, 129.1, 137.6, 139.4 (11C, aromatic), 149.1 (C5, pyrazole), 150.9 (C3, pyrazole), 153.1 (C, aromatic), 163.6 (C=O). Anal. Calcd. (%) for C17H17N5O2 (323.35): C, 63.15; H, 5.30; N, 21.66. Found: C, 63.35; H, 5.15; N, 21.50%.
5-Amino-3-[(4-methoxyphenyl)amino]-N-(4-methylphenyl)-1H-pyrazole-4-carboxamide (4b)
White crystals, m.p. 198–200°C, yield (82%). IR (KBr) νmax/cm−1 3358, 3047 (NH, NH2), 1640 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.21 (s, 3H, CH3), 3.64 (s, 3H, OCH3), 5.95 (s, 2H, NH2, D2O exchangeable), 6.76 (d, 2H, aromatic, JHH=8.4 Hz), 7.04 (d, 2H, aromatic, JHH=8.4 Hz), 7.16 (d, 2H, aromatic, JHH=8.4 Hz), 7.33 (d, 2H, aromatic, JHH=8.4 Hz), 8.30 (s, 1H, NH, D2O exchangeable), 8.66 (s, 1H, NH, D2O exchangeable), 11.21 (s, 1H, NH, D2O exchangeable). Anal. Calcd. (%) for C18H19N5O2 (337.38): C, 64.08; H, 5.68; N, 20.76. Found: C, 64.25; H, 5.50; N, 20.90%.
5-Amino-N-(4-chlorophenyl)-3-[(4-methoxyphenyl)amino]-1H-pyrazole-4-carboxamide (4c)
White crystals, m.p. 190–192°C, yield (80%). IR (KBr) νmax/cm−1 3347, 3031 (NH, NH2), 1642 (C=O). 1H-NMR (DMSO-d6, δ ppm) 3.64 (s, 3H, OCH3), 6.00 (s, 2H, NH2, D2O exchangeable), 6.77–7.51 (m, 8H, aromatic), 8.30 (s, 1H, NH, D2O exchangeable), 8.82 (s, 1H, NH, D2O exchangeable), 11.20 (s, 1H, NH, D2O exchangeable). 13C-NMR (DMSO-d6, δ ppm) 55.7 (-OCH3), 87.9 (C4, pyrazole), 114.7, 117.7, 121.9, 126.8, 128.9, 137.4, 138.4 (11C, aromatic), 149.1 (C5, pyrazole), 151.1 (C3, pyrazole), 153.2 (C, aromatic), 163.6 (C=O). MS m/z: 358 [M+]. Anal. Calcd. (%) for C17H16ClN5O2 (357.79): C, 57.07; H, 4.51; N, 19.57. Found: C, 57.30; H, 4.40; N, 19.75%.
Synthesis of N-Aryl-2-[(4-methoxyphenyl)amino]-5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxamide (7a–c)
A mixture of compounds 4a–c (0.01 mol) with acetylacetone 5 (0.01 mol) in glacial acetic acid (20 ml) was refluxed for 6 h, then poured onto crushed ice, and the separated solid was filtered, dried well, and recrystallized from ethanol to afford compounds 7a–c.
2-[(4-Methoxyphenyl)amino]-5,7-dimethyl-N-phenylpyrazolo[1,5-a]pyrimidine-3-carboxamide (7a)
White crystals, m.p. 215–217°C, yield (85%). IR (KBr) νmax/cm−1 3305 (NH), 1653 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.50 (s, 3H, CH3), 2.56 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 6.85 (d, 2H, aromatic, JHH=6.7 Hz), 6.89 (s, 1H, pyrimidine H-6), 7.06 (t, 1H, aromatic, JHH=7.4 Hz), 7.33 (t, 2H, aromatic, JHH=7.8 Hz), 7.59 (d, 2H, aromatic, JHH=8.8 Hz), 7.63 (d, 2H, aromatic, JHH=7.8 Hz), 9.07 (s, 1H, NH, D2O exchangeable), 9.94 (s, 1H, NH, D2O exchangeable). MS m/z: 387 [M+]. Anal. Calcd. (%) for C22H21N5O2 (387.43): C, 68.20; H, 5.46; N, 18.08. Found: C, 68.28; H, 5.40; N, 18.00%.
2-[(4-Methoxyphenyl)amino]-5,7-dimethyl-N-(4-methylphenyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (7b)
White crystals, m.p. 260–261°C, yield (88%). IR (KBr) νmax/cm−1 3317 (NH), 1663 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.25 (s, 3H, CH3), 2.61 (s, 3H, CH3), 2.69 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 6.90 (d, 2H, aromatic, JHH=8.4 Hz), 7.00 (s, 1H, pyrimidine H-6), 7.16 (d, 2H, aromatic, JHH=8.3 Hz), 7.59 (d, 2H, aromatic, JHH=6.3 Hz), 7.66 (d, 2H, aromatic, JHH=6.3 Hz), 9.28 (s, 1H, NH, D2O exchangeable), 10.01 (s, 1H, NH, D2O exchangeable). MS m/z: 401 [M+]. Anal. Calcd. (%) for C23H23N5O2 (401.46): C, 68.81; H, 5.77; N, 17.44. Found: C, 68.75; H, 5.81; N, 17.50%.
N-(4-Chlorophenyl)-2-[(4-methoxyphenyl)amino]-5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxamide (7c)
White crystals, m.p. 254–256°C, yield (83%). IR (KBr) νmax/cm−1 3314 (NH), 1664 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.55 (s, 3H, CH3), 2.62 (s, 3H, CH3), 3.70 (s, 3H, OCH3), 6.88 (d, 2H, aromatic, JHH=7.8 Hz), 6.92 (s, 1H, pyrimidine H-6), 7.36 (d, 2H, aromatic, JHH=7.8 Hz), 7.61 (d, 2H, aromatic, JHH=6.7 Hz), 7.68 (d, 2H, aromatic, JHH=6.7 Hz), 9.06 (s, 1H, NH, D2O exchangeable), 10.03 (s, 1H, NH, D2O exchangeable). 13C-NMR (CDCl3, δ ppm) 17.3, 24.6 (-2CH3), 55.6 (-OCH3), 86.7 (C3, pyrazolopyrimidine), 108.5 (C6, pyrazolopyrimidine), 114.2, 118.7, 120.5, 128.0 (8C, aromatic), 134.0 (C3a, pyrazolopyrimidine), 137.5, 145.7, 146.0 (3C, aromatic), 154.2 (C7, pyrazolopyrimidine), 156.8 (C, aromatic), 156.9 (C2, pyrazolopyrimidine), 160.1 (C5, pyrazolopyrimidine), 163.0 (C=O). MS m/z: 421 [M+]. Anal. Calcd. (%) for C22H20ClN5O2 (421.88): C, 62.63; H, 4.78; N, 16.60. Found: C, 62.70; H, 4.75; N, 16.50%.
Synthesis of 7-amino-N-aryl-6-cyano-5-(4-methoxyphenyl)-2-[(4-methoxyphenyl)amino]pyrazolo[1,5-a]pyrimidine-3-carboxamides (10a–c)
A mixture of compounds 4a-c (0.01 mol) with 2-(4-methoxybenzylidene)malononitrile 8 (0.01 mol) and a catalytic amount of triethylamine (four drops) in absolute ethanol (30 ml) was refluxed for 6 h. The solvent was concentrated under reduced pressure and the solid obtained was collected and recrystallized from ethanol to give 10a-c.
7-Amino-6-cyano-5-(4-methoxyphenyl)-2-[(4-methoxyphenyl)amino]-N-phenyl-pyrazolo[1,5-a]pyrimidine-3-carboxamide (10a)
Yellow crystals, m.p. > 300ºC, yield (83%). IR (KBr) νmax/cm−1 3440, 3356 (NH, NH2), 2210 (C≡N), 1650 (C=O). 1H-NMR (DMSO-d6, δ ppm) 3.71 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.86 (d, 2H, aromatic, JHH=8.9 Hz), 7.06 (t, 1H, aromatic, JHH=8.6 Hz), 7.15 (d, 2H, aromatic, JHH=8.6 Hz), 7.34 (t, 2H, aromatic, JHH=7.7 Hz), 7.58 (d, 2H, aromatic, JHH=7.9 Hz), 7.82 (d, 2H, aromatic, JHH=8.9 Hz), 7.95 (d, 2H, aromatic, JHH=8.8 Hz), 9.02 (s, 2H, NH2, D2O exchangeable), 9.21 (s, 1H, NH, D2O exchangeable), 10.12 (s, 1H, NH, D2O exchangeable). Anal. Calcd. (%) for C28H23N7O3 (505.53): C, 66.52; H, 4.59; N, 19.39. Found: C, 66.60; H, 4.55; N, 19.45%.
7-Amino-6-cyano-5-(4-methoxyphenyl)-2-[(4-methoxyphenyl)amino]-N-(4-methylphenyl)-pyrazolo[1,5-a]pyrimidine-3-carboxamide (10b)
Yellow crystals, m.p. 248–250°C, yield (78%). IR (KBr) νmax/cm−1 3447, 3296 (NH, NH2), 2211 (C≡N), 1653 (C=O). 1H-NMR (DMSO-d6, δ ppm) 2.24 (s, 3H, CH3), 3.71 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 6.85 (d, 2H, aromatic, JHH=9.2 Hz), 7.13 (d, 2H, aromatic, JHH=7.5 Hz), 7.16 (d, 2H, aromatic, JHH=8.5 Hz), 7.46 (d, 2H, aromatic, JHH=8.4 Hz), 7.51 (d, 2H, aromatic, JHH=8.2 Hz), 7.97 (d, 2H, aromatic, JHH=8.0 Hz), 8.70 (s, 2H, NH2, D2O exchangeable), 9.78 (s, 1H, NH, D2O exchangeable), 12.76 (s, 1H, NH, D2O exchangeable). 13C-NMR (CDCl3, δ ppm) 21.0 (-CH3), 55.4, 55.6 (-2OCH3), 77.0 (C6, pyrazolopyrimidine merged with the peak of CDCl3), 92.2 (C3, pyrazolopyrimidine), 114.4, 144.6 (4C, aromatic), 114.8 (C≡N), 119.6, 121.9, 127.8, 129.3, 129.6 (9C, aromatic), 131.5 (C3a, pyrazolopyrimidine), 132.1, 133.1, 136.4, 150.6 (4C, aromatic), 156.1 (C2, pyrazolopyrimidine), 162.3 (C, aromatic), 163.2 (C5, pyrazolopyrimidine), 163.6 (C=O), 167.4 (C7, pyrazolopyrimidine). MS m/z: 520 [M++1]. Anal. Calcd. (%) for C29H25N7O3 (519.55): C, 67.04; H, 4.85; N, 18.87. Found: C, 67.15; H, 4.80; N, 18.90%.
7-Amino-N-(4-chlorophenyl)-6-cyano-5-(4-methoxyphenyl)-2-[(4-methoxyphenyl)amino]pyrazolo[1,5-a]pyrimidine-3-carboxamide (10c)
Yellow crystals, m.p. 240–242°C, yield (80%). IR (KBr) νmax/cm−1 3433, 3297 (NH, NH2), 2215 (C≡N), 1660 (C=O). 1H-NMR (DMSO-d6, δ ppm) 3.71 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.86 (d, 2H, aromatic, JHH=9.0 Hz), 7.14 (d, 2H, aromatic, JHH=8.8 Hz), 7.39 (d, 2H, aromatic, JHH=6.7 Hz), 7.48 (d, 2H, aromatic, JHH=8.0 Hz), 7.63 (d, 2H, aromatic, JHH=8.6 Hz), 7.98 (d, 2H, aromatic, JHH=10.0 Hz), 8.63 (s, 2H, NH2, D2O exchangeable), 9.87 (s, 1H, NH, D2O exchangeable), 12.78 (s, 1H, NH, D2O exchangeable). Anal. Calcd. (%) for C28H22ClN7O3 (539.97): C, 62.28; H, 4.11; N, 18.16. Found: C, 62.20; H, 4.15; N, 18.20%.
Biological Experiments
In Vitro Cytotoxic Activity
Doxoroubicin, the reference drug which was used in this study, is one of the most effective antitumor agents used to produce regressions in acute leukemia, Hodgkin disease, and other lymphoma. The relationship between the survival ratio and drug concentration was plotted to obtain the survival curve of the Ehrlich Ascites Carcinoma (EAC) cells. The parameter IC50 is the concentration of the drugs inducing 50% inhibition of cell growth of the treated cells in comparison with the growth of the control cells.
Procedure
The EAC cells were obtained by needle aspiration of the ascetic from preinoculated mice under aseptic conditions. The tumor cells suspension (2.5×106 cells/ml) was prepared in RPMI-1640 media. The tested compounds were prepared with various dilutions by dissolving: 100, 50, 25, and 10 µg in DMSO (1 ml). In a set of sterile test tubes, to 0.8 ml of RPMI-1640 media containing (glutamine, fetal calf serum as the nutrient, streptomycin, and pencillin) 0.1 ml of each tested compound was added. The test tube was then incubated at 37°C for 2 h. The trypan blue exclusion test was carried out to calculate the percentage of non-viable cells after 2 h of incubation:
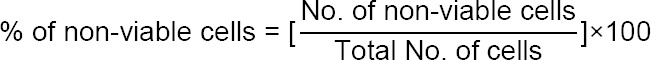
Conclusion
In conclusion, N-substituted cyanoacetamide derivatives 1a-c were used as starting materials for the synthesis of some new N-(aryl)-2-cyano-3-(methylthio)acrylamides 3a–c, 5-amino-1H-pyrazoles 4a–c, and pyrazolo[1,5-a]pyrimidines 7a–c and 10a–c. The new synthesized compounds were characterized by analytical and spectroscopic data. Some selected new compounds were screened for their potential cytotoxic activity. The results of the cytotoxicity for the tested compounds against Ehrlich Ascites Carcinoma (EAC) cells indicated that the pyrazolo[1,5-a]pyrimidine derivatives 7a (IC50=10 µg/ml) and 10c (IC50=25 µg/ml) were found to have the most potent growth inhibitory activity against EAC cells in comparison with the reference drug, Doxorubicin (IC50=37.4 µg/ml). Accordingly, this class of compounds could be considered as useful templates for future development, derivatization, or modification to obtain more potent and selective antitumor agents.
Acknowledgement
The authors wish to express their thanks to the National Research Centre for the facilities provided.
Authors’ Statement
Competing Interests
The authors declare no conflict of interest.
References
- [1].Ningaiah S, Bhadraiah UK, Doddaramappa SD, Keshavamurthy S, Javarasetty C. Novel pyrazole integrated 1,3,4-oxadiazoles: Synthesis, characterization and antimicrobial evaluation. Bioorg Med Chem Lett. 2014;24:245–248. doi: 10.1016/j.bmcl.2013.11.029. http://dx.doi.org/10.1016/j.bmcl.2013.11.029 . [DOI] [PubMed] [Google Scholar]
- [2].Murlykina MV, Sakhno YI, Desenko SM, Konovalova IS, Shishkin OV, Sysoiev DA, Kornet MN, Chebanov VA. Features of switchable multicomponent heterocyclizations of salicylic aldehydes and 5 aminopyrazoles with pyruvic acids and antimicrobial activity of the reaction products. Tetrahedron. 2013;69:9261–9269. http://dx.doi.org/10.1016/j.tet.2013.08.055 . [Google Scholar]
- [3].El-Sayed MA-A, >Abdel-Aziz NI, Abdel-Aziz AA-M, El-Azab AS, ElTahir KEH. Synthesis, biological evaluation and molecular modeling study of pyrazolo and pyrazoline derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Part 2. Bioorg Med Chem. 2012;20:3306–3316. doi: 10.1016/j.bmc.2012.03.044. http://dx.doi.org/10.1016/j.bmc.2012.03.044 . [DOI] [PubMed] [Google Scholar]
- [4].Alcaro S, Artese A, Botta M, Zizzari AT, Orallo F, Ortuso F, Schenone S, Brullo C, Yáñez M. Hit identification and biological evaluation of anticancer pyrazolopyrimidines endowed with anti-inflammatory activity. ChemMedChem. 2010;5:1242–1246. doi: 10.1002/cmdc.201000165. http://dx.doi.org/10.1002/cmdc.201000165 . [DOI] [PubMed] [Google Scholar]
- [5].Balbi A, Anzaldi M, Macciò C, Aiello C, Mazzei M, Gangemi R, Castagnola P, Miele M, Rosano C, Viale M. Synthesis and biological evaluation of novel pyrazole derivatives with anticancer activity. Eur J Med Chem. 2011;46:5293–5309. doi: 10.1016/j.ejmech.2011.08.014. http://dx.doi.org/10.1016/j.ejmech.2011.08.014 . [DOI] [PubMed] [Google Scholar]
- [6].Kamal A, Tamboli JR, Nayak VL, Adil SF, Vishnuvardhan MVPS, Ramakrishna S. Synthesis of pyrazolo[1,5-a]pyrimidine linked aminobenzothiazole conjugates as potential anticancer agents. Bioorg Med Chem Lett. 2013;23:3208–3215. doi: 10.1016/j.bmcl.2013.03.129. http://dx.doi.org/10.1016/j.bmcl.2013.03.129 . [DOI] [PubMed] [Google Scholar]
- [7].Bondock S, Adel S, Etman HA, Badria FA. Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles. Eur J Med Chem. 2012;48:192–199. doi: 10.1016/j.ejmech.2011.12.013. http://dx.doi.org/10.1016/j.ejmech.2011.12.013 . [DOI] [PubMed] [Google Scholar]
- [8].Abd El Razik HA, Abdel Wahab AE. Synthesis and biological evaluation of some novel fused pyrazolopyrimidines as potential anticancer and antimicrobial agents. Arch. Pharm. 2011;344:184–196. doi: 10.1002/ardp.201000188. http://dx.doi.org/10.1002/ardp.201000188 . [DOI] [PubMed] [Google Scholar]
- [9].Ranatunge RR, Garvey DS, Janero DR, Letts LG, Martino AM, Murty MG, Richardson SK, Young DV, Zemetseva IS. Synthesis and selective cyclooxygenase-2 (COX-2) inhibitory activity of a series of novel bicyclic pyrazoles. Bioorg Med Chem. 2004;12:1357–1366. doi: 10.1016/j.bmc.2004.01.012. http://dx.doi.org/10.1016/j.bmc.2004.01.012 . [DOI] [PubMed] [Google Scholar]
- [10].Bekhit AA, Ashour HMA, Abdel Ghany YS, Bekhit AEA, Baraka A. Synthesis and biological evaluation of some thiazolyl and thiadiazolyl derivatives of 1H-pyrazole as anti-inflammatory antimicrobial agents. Eur J Med Chem. 2008;43:456–463. doi: 10.1016/j.ejmech.2007.03.030. http://dx.doi.org/10.1016/j.ejmech.2007.03.030 . [DOI] [PubMed] [Google Scholar]
- [11].Fioravanti R, Bolasco A, Manna F, Rossi F, Orallo F, Ortuso F, Alcaro S, Cirilli R. Synthesis and biological evaluation of N-substituted-3,5-diphenyl-2-pyrazoline derivatives as cyclooxygenase (COX-2) inhibitors. Eur J Med Chem. 2010;45:6135–6138. doi: 10.1016/j.ejmech.2010.10.005. http://dx.doi.org/10.1016/j.ejmech.2010.10.005 . [DOI] [PubMed] [Google Scholar]
- [12].George CFP. Pyrazolopyrimidines. Lancet. 2001;358:1623–1626. doi: 10.1016/s0140-6736(01)06656-9. http://dx.doi.org/10.1016/S0140-6736(01)06656-9 . [DOI] [PubMed] [Google Scholar]
- [13].Wegner F, Deuther-Conrad W, Scheunemann M, Brust P, Fischer S, Hiller A, Diekers M, Strecker K, Wohlfarth K, Allgaier C, Steinbach J, Hoepping A. GABAA receptor pharmacology of fluorinated derivatives of the novel sedative-hypnotic pyrazolopyrimidine indiplon. Eur J Pharm. 2008;580:1–11. doi: 10.1016/j.ejphar.2007.10.016. http://dx.doi.org/10.1016/j.ejphar.2007.10.016 . [DOI] [PubMed] [Google Scholar]
- [14].Hafez TS, Osman SA, Yosef HAA, Abd El-All AS, Hassan AS, El-Sawy AA, Abdallah MM, Youns M. Synthesis, Structural Elucidation and In Vitro Antitumor Activities of Some Pyrazolopyrimidines and Schiff Bases derived from 5-Amino-3-(arylamino)-1H-pyrazole-4-carboxamides. Sci Pharm. 2013;81:339–357. doi: 10.3797/scipharm.1211-07. http://dx.doi.org/10.3797/scipharm.1211-07 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Osman SA, Yosef HAA, Hafez TS, El-Sawy AA, Mousa HA, Hassan AS. Synthesis and antibacterial activity of some novel chalcones, pyrazoline and 3-cyanopyridine derivatives based on khellinone as well as Ni(II), Co(II) and Zn(II) complexes. Aust J Basic Appl Sci. 2012;6:852–863. [Google Scholar]
- [16].Osman SA, Mousa HA, Yosef HAA, Hafez TS, El-Sawy AA, Abdallah MM, Hassan AS. Synthesis, characterization and cytotoxicity of mixed ligand Mn(II), Co(II) and Ni(II) complexes. J Serb Chem Soc. 2014;79:953–964. http://dx.doi.org/10.2298/JSC130813134O . [Google Scholar]
- [17].Eremeev AV, Piskunova IP, El’kinson RS. Synthesis of 2-amino-1-azirines and their reactions with carboxylic acids. Chem Heterocycl Comp. 1985;21:998–1002. http://dx.doi.org/10.1007/BF00515022 . [Google Scholar]
- [18].Al-Adiwish WM, Tahir MIM, Siti-Noor-Adnalizawati A, Hashimc SF, Ibrahim N, Yaacob WA. Synthesis, antibacterial activity and cytotoxicity of new fused pyrazolo[1,5-a]pyrimidine and pyrazolo[5,1-c][1,2,4]triazine derivatives from new 5-aminopyrazoles. Eur J Med Chem. 2013;64:464–476. doi: 10.1016/j.ejmech.2013.04.029. http://dx.doi.org/10.1016/j.ejmech.2013.04.029 . [DOI] [PubMed] [Google Scholar]
- [19].Ghozlan SAS, Hassanien AA. β-Amino-β-(pyrid-4-yl)acrylonitrile in heterocyclic synthesis: synthesis of some new pyridine, pyridone, pyrazole, thiophene, fused pyrimidine and triazine derivatives. Tetrahedron. 2002;58:9423–9429. http://dx.doi.org/10.1016/S0040-4020(02)01220-6 . [Google Scholar]