

Abstract
Lopinavir is an antiretroviral drug used for the inhibition of HIV protease. Four related substances of lopinavir were observed during the manufacturing process of lopinavir in the laboratory and they were identified. The present work describes the origin, synthesis, characterization, and control of these related substances.
Keywords: Lopinavir, Related substances, Synthesis and Characterization of impurities
Introduction
Lopinavir is chemically known as (2S)-N-[(2S,4S,5S)-5-{[(2,6-dimethylphenoxy)acetyl]amino}-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide. Lopinavir is marketed by Abbott Laboratories under the brand name of Kaletra® with the combination of Ritonavir.
The presence of impurities in an Active Pharmaceutical Ingredient (API) will influence the quality and safety of the drug product. In the regulatory guidelines of the International Conference on Harmonization (ICH), it is recommended that impurities amounting to more than 0.1% [1] should be identified and characterized. Impurities are required in pure form to check the analytical performance characteristics such as specificity, linearity, range, accuracy, precision, limit of detection (LOD), limit of quantification (LOQ), robustness, system suitability testing, and relative retention factor [2].
During the process development of lopinavir 1 in our laboratory, we observed the formation of four substances that are structurally related to lopinavir. These unknown related substances were identified, monitored, and their structures were tentatively assigned on the basis of their fragmentation patterns in LC-MS [3]. In the present work, the identified related substances of lopinavir were synthesized and characterized by various spectroscopic techniques and further confirmed by co-injection studies using qualitative HPLC analysis. A few references [3–9] were found in the literature for related substances of lopinavir and its metabolites.
Results and Discussion
Lopinavir 1 has been synthesized by known literature methods [10–14]. Our route for the synthesis of lopinavir is shown in Scheme 1. (2S,3S,5S)-5-Amino-2-(dibenzylamino)-1,6-diphenylhexan-3-ol (2) was activated with N,N-carbonyldiimidazole followed by condensation with (2S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoic acid (3) to provide (2S)-N-[(2S,4S,5S)-5-(dibenzylamino)-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide (4). Compound 4, upon debenzylation with ammonium formate and palladium on charcoal, gave (2S)-N-[(2S,4S,5S)-5-amino-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide (5). Compound 5 was treated with L-pyroglutamic acid to give pure (2S)-N-[(2S,4S,5S)-5-amino-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide (S)-pyroglutamic acid salt 6. Condensation of compound 6 with (2,6-dimethylphenoxy)acetic acid gave lopinavir 1. Using the above-mentioned synthesis process, lopinair 1 was obtained with a purity of 99.5% and four impurities that are structurally related to lopinavir were identified. The chemical names and structures of these related substances of lopinavir 1 were identified as:
Sch. 1.
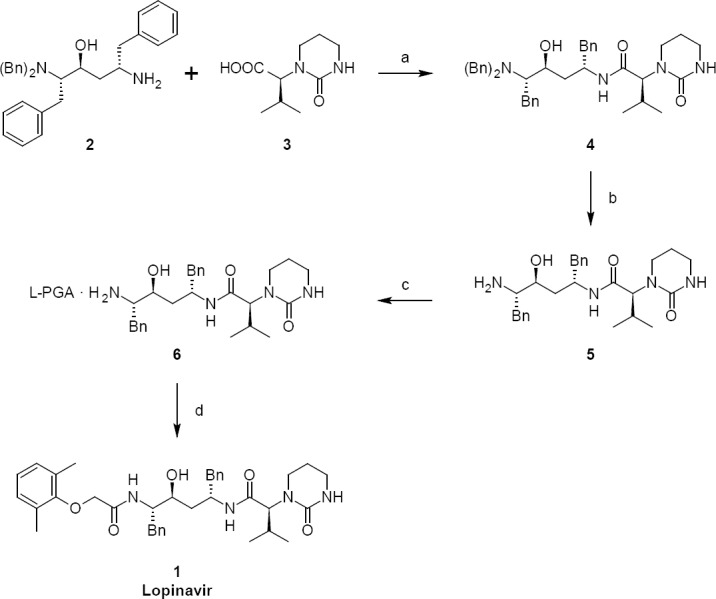
Synthesis of Lopinavir 1. Reagents and conditions: (a) N,N-carbonyldiimidazole, ethyl acetate; yield: 82.7%; (b) ammonium formate, palladium on charcoal, methanol; yield: 100%; (c) L-pyroglutamic acid, acetone, DMF; yield: 72.4%; (d) 2,6-dimethylphenoxyaceticacid, thionyl chloride, dichloromethane; yield: 85.7%
-
Lopinavir dimer, 7 (2S,2’S)-N,N’-[(3,3’,5,5’-tetramethylbiphenyl-4,4’-diyl)bis{oxy(1-oxoethane-2,1-diyl)imino[(2S,4S,5S)-4-hydroxy-1,6-diphenylhexane-5,2-diyl]}]bis[3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide]:
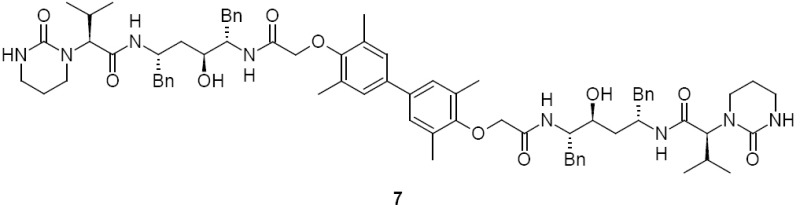
-
Lopinavir carboxymethyl analog, 8 (2S)-N-[(2S,4S,5S)-4-hydroxy-5-({[4-(2-{[(2S,3S,5S)-3-hydroxy-5-{[(2S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoyl]amino}-1,6-diphenylhexan-2-yl]amino}-2-oxoethoxy)-3,5-dimethylphenyl]acetyl}amino)-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide:
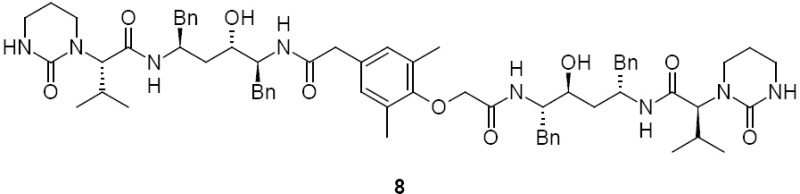
-
Lopinavir diamide, 9 N,N’-[(2S,3S,5S)-3-Hydroxy-1,6-diphenylhexane-2,5-diyl]bis[2-(2,6-dimethylphenoxy)acetamide]:
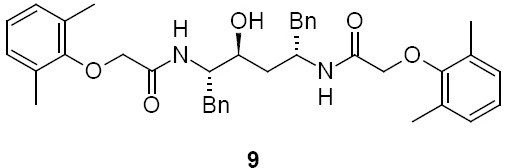
-
Diacylated, 10 (2S)-N-[(2S,4S,5S)-5-{[(2,6-dimethylphenoxy)acetyl]amino}-4-hydroxy-1,6-diphenylhexan-2-yl]-2-{3-[(2,6-dimethylphenoxy)acetyl]-2-oxotetrahydropyrimidin-1(2H)-yl}-3-methylbutanamide:
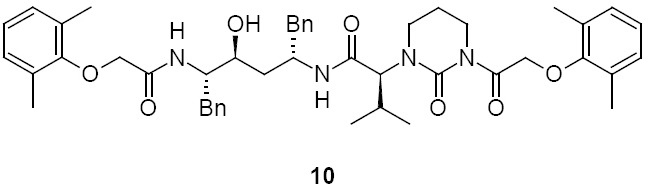
The origin, synthesis, characterization, and control of these related substances are described below. The synthetically prepared related substances were characterized by conventional spectroscopic studies and the presence of these related substances in the lopinavir batch was confirmed by spiking the related substances individually with a lopinavir sample from the manufactured batch. These studies confirmed the formation of related substances (7–10) during the manufacturing process of lopinavir 1.
Lopinavir Dimer 7
2,6-Dimethylphenol is a raw material used in the preparation of 2,6-dimethylphenoxyacetic acid (Scheme 2). 2,6-dimethylphenoxyacetic acid is a critical intermediate in the preparation of lopinavir 1 and is prepared from 2,6-dimethylphenol by acylation with chloroacetic acid by known literature methods [10–14].
Sch. 2.
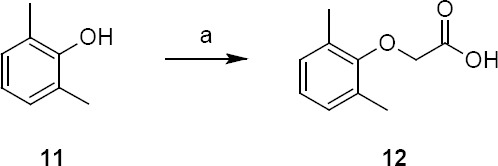
Synthetic scheme of 2,6-dimethylphenoxyacetic acid 12. Reagents and conditions: (a) chloroacetic acid, sodium hydroxide, water, reflux; yield: 90%
Fig. 1.
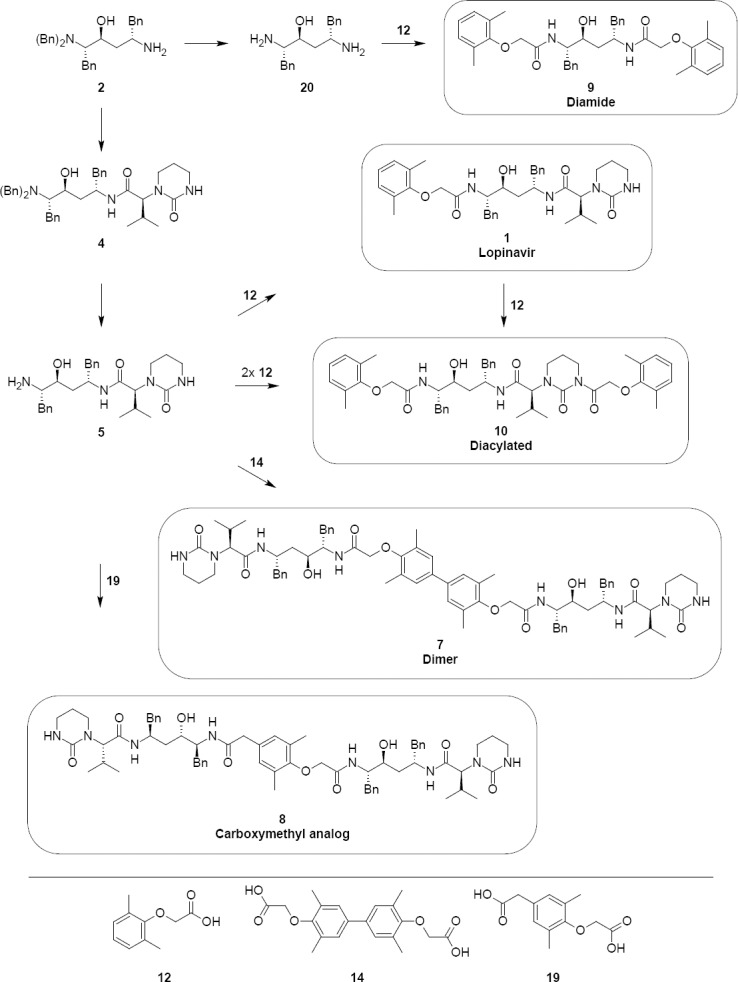
Possible synthetic pathways for lopinavir 1 and its related substances (7–10)
Phenols have a tendency to polymerize during storage; it is therefore necessary to study the dimers of phenols. We observed a dimer (13) at a level of ~0.3% in 2,6-dimethylphenol used in the laboratory and it was subsequently reacted to give lopinavir dimer 7. Lopinavir dimer 7 was independently prepared by the dimerization of 2,6-dimethylphenol to give compound 13. Compound 13 was acylated with chloroacetic acid to give compound 14. Compound 14 was condensed with compound 4 to produce lopinavir dimer 7 (as shown in Scheme 3).
Sch. 3.
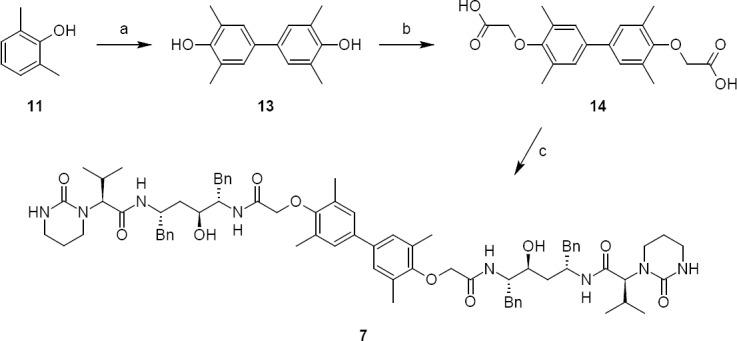
Synthetic scheme of lopinavir dimer 7. Reagents and conditions: (a) ferric chloride, water; yield: 25%; (b) chloroacetic acid, sodium hydroxide, water; yield: 50%; (c) 6, thionyl chloride, dichloromethane, sodium bicarbonate, water, ethyl acetate; yield: 40%
Lopinavir Carboxymethyl Analog 8
During the acylation of 2,6-dimethylphenol with chloroacetic acid (as shown in Scheme 2), the formation of ~0.2% of compound 19 was observed and was reacted in the subsequent steps to give lopinavir carboxymethyl analog 8.
Lopinavir carboxymethyl analog 8 was independently prepared from 2,6-dimethylphenol. 2,6-dimethylphenol reacted with acetic anhydride to give 2,6-dimethylphenyl acetate 15. 2,6-dimethylphenyl acetate 15 underwent fries rearrangement which gave para acetyl 2,6-dimethylphenol 16. Compound 16 reacted with chloroacetic acid to give compound 17. Compound 17 was treated with morpholin to give compound 18. Compound 18 was hydrolyzed with sodium hydroxide to give compound 19. Compound 19 condensed with compound 5 to give lopinavir carboxymethyl analog 8 (as shown in Scheme 4).
Sch. 4.
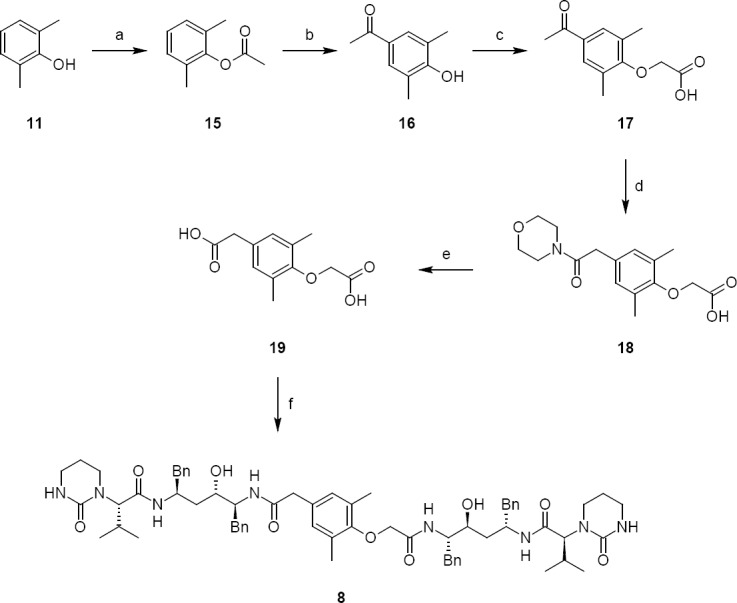
Synthetic scheme of lopinavir carboxymethyl analog 8. Reagents and conditions: (a) acetic anhydride, sulfuric acid; yield: 77%; (b) aluminium chloride, carbon disulfide, 120°C; yield: 81%; (c) chloroacetic acid, NaOH, water; yield: 70%; (d) morpholin, sulphur, NaOH; yield; 48%; (f) 5, thionyl chloride, dichloromethane, NaHCO3, water; yield; 38%
Lopinavir Diamide Impurity 9
Lopinavir diamide 9 originated from unreacted compound 2, present also in compound 4, underwent all the reaction sequences employed in the synthesis of lopinavir. Lopinavir diamide impurity 9 was formed by the condensation of diamine compound 20 with 2,6-dimethylphenoxyacetic acid 12.
Sch. 5.
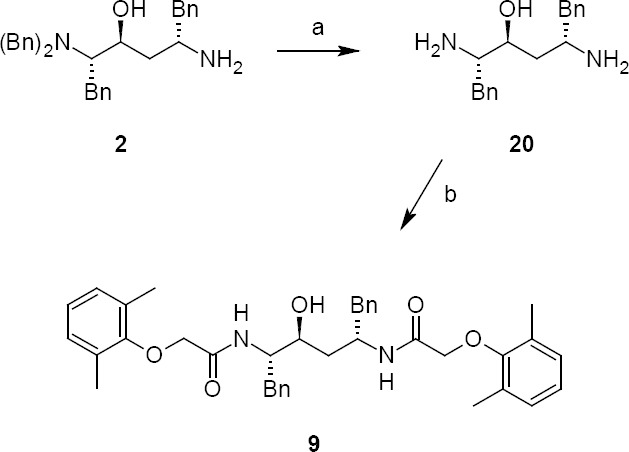
Synthetic scheme of lopinavir diamide impurity 9. Reagents and conditions: (a) ammonium formate, palladium on charcoal, methanol; yield: 100%; (b) (2,6-dimethylphenoxy)acetic acid, sodium bicarbonate; yield: 41%
Lopinavir Diacylated Impurity 10
Lopinavir diacylated impurity 10 was formed due to the acylation on the nitrogen presented in the pyrimidine ring of lopinavir with 2,6-dimethylphenoxy acetic acid during the synthesis of lopinavir. It was independently prepared by the condensation of compound 1 with compound 12.
Sch. 6.
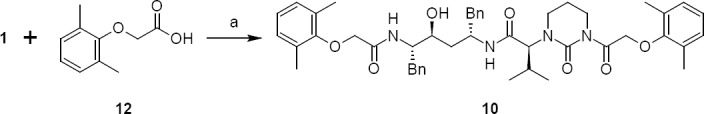
Synthetic scheme of lopinavir diacylated impurity 10. Reagents and conditions: (a) thionyl chloride, sodium bicarbonate; ethyl acetate; yield: 20%
Related substances 8–10 were eliminated during the purification of lopinavir 1. Related substance 7 originated due to the presence of dimer 13 in 2,6-dimethylphenol. It was controlled by keeping the limit in the specification of 2,6-dimethylphenol raw material.
Experimental
All melting points were determined with the Polmon melting point apparatus. 1H-NMR and 13C-NMR spectra were recorded on a Bruker 300 spectrometer. Chemical shifts are reported in ppm downfield from TMS as the internal standard. The mass spectra were measured on the Perkin Elmer PE SCIEX-API 2000 mass spectrometer. Elemental analyses were performed using a Heraeus CHN-O-Rapid instrument. Analytical HPLC were run with the Symmetry C18, 210 × 4.6 mm column at 290 nm. “RT” denotes room temperature. 2,6-dimethylphenol was purchased from Bluestar Newchemicals Materials Co., Ltd.
(2S,2’S)-N,N’-[(3,3’,5,5’-tetramethylbiphenyl-4,4’-diyl)bis{oxy(1-oxoethane-2,1-diyl)imino[(2S,4S,5S)-4-hydroxy-1,6-diphenylhexane-5,2-diyl]}]bis[3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide] (lopinavir dimer, 7):
Step I: 3,31,5,51-Tetramethyl-1,11-biphenyl-4,41-diol (13):
To a solution of 2,6-dimethylphenol 11 (10 g, 82 mmol) in DM water (400 mL) was added ferric chloride solution (22.15 g of ferric chloride·6 H2O dissolved in 100 mL DM water) in 30 min at 95–100°C. After stirring for 1 h at 95–100°C, the reaction mass cooled to room temperature and we extracted the product with dichloromethane (250 mL). The dichloromethane layer was concentrated below 40°C under reduced pressure to obtain a brown coloured residue, which was purified by silicagel (100–200 mesh) chromatography using a 1:9 v/v mixture of ethylacetate and hexane to obtain 5 g (25%) of 3,31,5,51-tetramethyl-1,11-biphenyl-4,41-diol (13) as an orange colored residue. IR (KBr, cm−1) 2883, 2855, 1633, 1595, 1462, 1364, 1343, 1296, 780, 766; 1H-NMR (300 MHz, DMSO-D6) δ 2.19 (S, 12H), 3.35 (S, 3H), 7.1 (S, 4H), 8.13 (S, 2H); MS (ESI, m/z): 237 [M-H]-.
Step II: 2,21-[(3,31,5,51-tetramethylbiphenyl-4,41-diyl)bis(oxy)]diacetic acid (14):
To a suspension of 3,31,5,51-tetramethyl-1,11-biphenyl-4,41-diol (13, 7.0 g, 28.92 mmol) and chloroacetic acid (5.46 g, 57.77 mmol) in DM water (50 mL) was added 10% aqueous sodium hydroxide (50 mL) in 10 min at 25–35°C. The reaction mass was heated and refluxed for ~2 h at 98-100°C. The reaction mass cooled to 25-30°C and we adjusted the pH of the reaction mass to 5.0 with concentrated hydrochloric acid (~6 mL) and it was then washed with toluene (50 mL). Thereafter, we precipitated the product by adjusting the pH to 2.0 with concentrated hydrochloric acid (~6 mL) at 25-30°C. After 1 h of stirring at 25-30°C, the product was filtered and dried to give 7.3 g (70%) of crude 2,21-[(3,31,5,51-tetramethyl biphenyl-4,41-diyl)bis(oxy)]diacetic acid 14, which was used as such in the next step without purification.
Step III: (2S,2’S)-N,N’-[(3,3’,5,5’-tetramethylbiphenyl-4,4’-diyl)bis{oxy(1-oxoethane-2,1-diyl)imino[(2S,4S,5S)-4-hydroxy-1,6-diphenylhexane-5,2-diyl]}]bis[3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide] (lopinavir dimer, 7):
To a suspension of 2,21-[(3,31,5,51-tetramethylbiphenyl-4,41-diyl)bis(oxy)]diacetic acid (14, 4.0 g, 11.66 mmol) in ethyl acetate (20 mL) was added thionyl chloride (3.32 g, 27.89 mmol) followed by N,N-dimethylformamide (0.1 mL) at 25–30°C. The reaction mass temperature was raised and it was stirred for 2 h at 50-55°C to complete the formation of acid chloride. Compound 6 (13.3 g, 22.35 mmol) was dissolved separately in a mixture of water (100 mL) and ethyl acetate (100 mL) at 20–25°C, then NaHCO3 was added (11.42 g, 135.95 mmol) portionwise at 20–25°C. Thereafter, we added the above-prepared acid chloride solution in ~15 min at 20–25°C. After 30 min of stirring at 20–25°C, we separated the aqueous layer from the reaction mass and washed the organic layer with 5% aqueous sodium bicarbonate (50 mL) followed by 10% aqueous sodium chloride (50 mL). The organic layer was concentrated at 50–60°C under reduced pressure to obtain 20 g of cream coloured powder, which was purified by flash chromatography using a 1:1 v/v mixture of hexane and ethyl acetate to obtain 14 g (40%) of pure lopinavir dimer (7). Melting range: 145–149°C; HPLC purity: 90.11%; IR (KBr, cm−1): 3839, 3746, 3407, 3318, 3061, 3028, 2961, 2927, 2870, 1651, 1516, 1452, 1348, 1308, 1195, 750, 701; 1H-NMR (300 MHz, DMSO-D6) δ 0.72 (d, 12H), 1.15 & 1.19 (2m, 8H), 1.96 & 2.11 (2m, 2H), 2.06 (s, 6H), 2.44 & 2.80 (2m, 10H), 2.9 (Abq, 4H), 3.02 (Abq, 2H), 3.4&3.5 (2d, 2H), 4.30 (d, 2H), 4.82 & 4.99 (2d, 2H), 6.30 (brs, 2H), 6.78 (S, 2H), 7.43 (d,2H), 7.57 & 7.71 (2d, 2H); 13C-NMR (75 MHz, DMSO-D6) δ: 16.1, 18.7, 19.6, 21.7, 25.5, 37.9, 38.7, 39.2, 39.5, 39.8, 40.1, 46.7, 52.6, 61.4, 68.4, 70.3, 125.6, 125.9, 126.9, 127.9, 129.1, 130.6, 135.6, 138.9, 139.1, 153.9, 155.4, 167.3, 169.3. HRMS: 1255.7251.
(2S)-N-[(2S,4S,5S)-4-hydroxy-5-({[4-(2-{[(2S,3S,5S)-3-hydroxy-5-{[(2S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoyl]amino}-1,6-diphenylhexan-2-yl]amino}-2-oxoethoxy)-3,5-dimethylphenyl]acetyl}amino)-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide (lopinavir carboxymethyl analog, 8):
Step I: 2,6-dimethylphenyl acetate (15):
To a solution of acetic anhydride (167 g, 1637 mmol) and concentrated sulfuric acid (5 g, 51 mmol) was added 2,6-dimethylphenol (11, 100 g, 819.6 mmol) in 30 min at 25–30°C. After stirring at 25–30°C for 1 h, we poured the reaction mass into cold water (500 mL) and extracted the product with dichloromethane. The organic layer was washed with 5% w/w aqueous sodium bicarbonate (500 mL) followed by water (500 mL). The organic layer was concentrated below 40°C under reduced pressure to obtain 103 g (77%) of 2,6-dimethylphenyl acetate (15) as an oily mass. IR (KBr, cm−1) 2950, 1590, 1494, 1439, 1381, 1350, 788, 764; 1H-NMR (300 MHz, CDCl3) δ: 2.15 (s, 3H), 2.26 (s, 6H), 7.05 (s, 3H); MS (ESI, m/z): 165 [M+H]+.
Step II: 1-(4-hydroxy-3,5-dimethylphenyl)ethanone (16):
To a suspension of aluminum chloride (105.6 g, 792.19 mmol) in carbon disulfide (200 mL) was added 2,6-dimethylphenyl acetate (15, 100 g, 609.75 mmol) in 20 min at 25–30°C. After 2 h of stirring at 39–40°C, carbon disulfide was distilled out and we raised the mass temperature to 120–125°C. After 6 h of stirring at 120-125°C, we cooled the mass to 20–25°C and added concentrated hydrochloric acid (50 mL) followed by water (500 mL) and stirred for 1 h at 30–50°C. Thereafter, the mass was cooled to 20–25°C and the product was extracted with dichloromethane (2 × 500 mL). The organic layer was concentrated below 40°C under reduced pressure to obtain a residue, further this residue crystallized from ethyl acetate (200 mL) to get 81 g (81%) of pure 1-(4-hydroxy-3,5-dimethylphenyl)ethanone (16). IR (KBr, cm−1) 2950, 1590, 1494, 1439, 1381, 1350, 788, 764; 1H-NMR (300 MHz, CDCl3) δ 2.22 (s, 6H), 2.39 (s, 3H), 7.58 (s, 2H), 9.15 (1H, OH); MS (ESI, m/z): 165 [M+H]+.
Step III: (4-Acetyl-2,6-dimethylphenoxy)acetic acid (17):
To a suspension of 1-(4-hydroxy-3,5-dimethylphenyl)ethanone (16, 60 g, 365.85 mmol) and chloroacetic acid (37.12 g, 392 mmol) in DM water (300 mL) was added 20% w/w aqueous sodium hydroxide (150 g, 750 mmol) in 10 min at 25–30°C. After stirring for 1 h at reflux temperature (98–100°C), the mass was cooled to 20–25°C and we adjusted the pH of the reaction mass to 5.0 with concentrated hydrochloric acid (40 mL, 430 mmol). The reaction mass was washed with toluene (200 mL) to remove the unreacted starting material and then we adjusted the aqueous layer pH to 2.0 with concentrated hydrochloric acid (35 mL, 370 mmol) at 20–25°C. The precipitated product was collected by filtration and dried at 45–50°C under reduced pressure to obtain 57 g (70%) of pure [4-acetyl-2,6-dimethylphenoxy)acetic acid (17). IR (KBr, cm−1) 2883, 2855, 1633, 1595, 1462, 1364, 1343, 1296, 780, 766; 1H-NMR (300 MHz, DMSO-D6) δ 2.29 (s, 6H), 2.46 (s, 3H), 4.45 (s, 2H), 7.65 (s, 2H); MS (ESI, m/z): 221 [M-H]-.
Step IV: [4-(Carboxymethoxy)-3,5-dimethylphenyl]acetic acid (19):
A mixture of (4-acetyl-2,6-dimethylphenoxy)acetic acid (17, 25 g, 112 mmol) and sulfur powder (5.4 g, 168 mmol) in morpholine (24.52 g, 281 mmol) was heated to 130°C and stirred for 12 h at 130°C. The reaction mass was cooled to 20–25°C and we added 10% w/w ethanolic sodium hydroxide solution (150 g, 375 mmol). Again, the mass temperature was raised to 85 °C and stirred for 10 h at 85°C. Thereafter, the reaction mass was concentrated at 85°C under reduced pressure, the obtained residue was dissolved in DM water (250 mL) and washed with ethyl acetate (250 mL). The aqueous layer was adjusted to pH 5.0 with concentrated hydrochloric acid (24 mL) at 20–25°C and again washed with ethyl acetate (125 mL) at 20–25°C. After that, the aqueous layer was adjusted to pH 1.0 with concentrated hydrochloric acid (12 mL) at 20–25°C and we extracted the product with dichloromethane (4X 100 mL). The combined dichloromethane layer was concentrated below 40°C under reduced pressure to obtain a gummy solid, which was crystallized from ethyl acetate and hexane to obtain 15 g (46%) of pure [4-(carboxymethoxy)-3,5-dimethylphenyl]acetic acid (19) as a pale yellow solid. IR (KBr, cm−1) 2883, 2855, 1633, 1595, 1462, 1364, 1343, 1296, 780, 766; 1H-NMR (300 MHz, DMSO-D6) δ 2.20 (s, 6H), 3.35 (s, 3H), 4.34 (s, 2H), 6.89 (s, 2H), 12.58 (brs 2H); MS (ESI, m/z): 237 [M-H]-.
Step V: (2S)-N-[(2S,4S,5S)-4-hydroxy-5-({[4-(2-{[(2S,3S,5S)-3-hydroxy-5-{[(2S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoyl]amino}-1,6-diphenylhexan-2-yl]amino}-2-oxoethoxy)-3,5-dimethylphenyl]acetyl}amino)-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide (lopinavir carboxymethyl analog, 8):
To a suspension of [4-(carboxymethoxy)-3,5-dimethylphenyl]acetic acid (18, 10 g, 42 mmol) in ethyl acetate (50 mL) was added thionyl chloride (11.0 g, 92.43 mmol) followed by N,N-dimethylformamide (0.1 mL) at RT. The reaction mass was heated to 45–50°C and stirred for 2 h at 45–50°C to complete the formation of acid chloride. Compound 6 (50 g, 84 mmol) was dissolved separately in a mixture of DM water (200 mL) and ethyl acetate (200 mL) at 20–25°C, then NaHCO3 (26.8 g, 319 mmol) was added portionwise at 20–25°C. Thereafter, we added the above-prepared acid chloride solution in ~30 min at 20–25°C. After 30 min of stirring at 20–25°C, the aqueous layer was separated from the reaction mass and the organic layer was washed with 3 N hydrochloric acid (200 mL) followed by DM water (200 mL). Finally, the organic layer was washed with 10% aqueous sodium chloride (200 mL). The organic layer was concentrated at 50–60°C under reduced pressure to obtain a cream coloured powder, which was purified by flash chromatography using a 1:1 v/v mixture of hexane and ethyl acetate to obtain 18 g (38%) of pure compound 8. Melting range: 140–145 °C; HPLC purity: 89.22%; IR (KBr, cm−1): 3856, 3410, 3326, 3062, 3028, 2963, 2931, 2871, 1644, 1514, 1452, 1307, 1209, 1180, 749, 701; 1H-NMR (300 MHz, DMSO-D6) δ 0.74 (d, 12H), 1.43 & 1.51 (2m, 8H), 1.96 & 2.11 (2m, 2H), 2.06 (s, 6H), 2.44-2.81(m, 8H), 2.91 & 3.01 (2m, 8H), 3.25 (m, 2H), 3.54 & 3.64 (2m, 2H), 3.93 & 4.00 (Abq, 2H), 4.17 & 4.25 (2m, 4H), 4.30 (d, 2H), 4.82 & 4.99 (2d, 2H), 6.30 (brs, 2H), 6.78 (s, 2H), 7.04-7.23 (m, 2H, Ar), 7.43 (d, 2H), 7.57 & 7.71 (2d, 2H); 13C-NMR (75 MHz, DMSO-D6) δ: 14.9, 16.7, 19.5, 20.4, 21.6, 22.5, 23.7, 26.3, 38.4, 38.8, 39.5, 39.8, 40.1, 40.4, 40.9, 41.2, 42.5, 47.6, 53.4, 53.7, 60.6, 62.3, 68.2, 69.3, 70.0, 71.1, 126.3, 126.8, 128.6, 129.9, 130.1, 130.8, 133.4, 139.7, 140.4, 153.7, 156.3, 168.1, 170.0, 170.2, 170.9. HRMS: 1135.6671.
N,N’-[(2S,3S,5S)-3-Hydroxy-1,6-diphenylhexane-2,5-diyl]bis[2-(2,6-dimethylphenoxy)acetamide] (lopinavir diamide, 9):
Step I: (2S,3S,5S)-2,5-diamino-1,6-diphenylhexan-3-ol (20):
To a solution of (2S,3S,5S)-5-amino-2-(dibenzylamino)-1,6-diphenylhexan-3-ol (2, 50 g, 108 mmol) in methanol was added ammonium formate (20.41 g, 320 mmol) followed by 5% palladium on charcoal (5 g, 50% wet) at room temperature. After raising the temperature to 50°C, the reaction was stirred for ~2 h at 50°C to complete the debenzylation. Thereafter, the reaction mass cooled to room temperature and we recovered the palladium on charcoal by filtration under a nitrogen atmosphere. The methanol filtrate was concentrated at 60°C under reduced pressure which resulted in a residue. This residue was dissolved in ethyl acetate (250 mL) and washed with water (100 mL), followed by 20% aqueous sodium chloride (100 mL). The ethyl acetate layer was concentrated at 60°C under reduced pressure to obtain 30 g (100%) of title compound 20 as a foamy solid.
Step II: N,N’-[(2S,3S,5S)-3-Hydroxy-1,6-diphenylhexane-2,5-diyl]bis[2-(2,6-dimethylphenoxy)acetamide] (lopinavir diamide, 9):
To a suspension of (2,6-dimethylphenoxy)acetic acid (12, 38 g, 211 mmol) in ethyl acetate (100 mL) was added thionyl chloride (31.40 g, 264 mmol) followed by N,N-dimethylformamide (0.1 mL) at 25–30°C. The reaction mass temperature was raised to 50–55°C and stirred for 2 h at 50–55°C to complete the formation of the acid chloride. We separately dissolved (2S,3S,5S)-2,5-diamino-1,6-diphenylhexan-3-ol (20, 30 g, 105 mmol) in a mixture of DM water (250 mL) and ethyl acetate (250 mL) at 20-25°C, then added NaHCO3 (55.4 g, 660 mmol) portionwise at 20–25°C. Thereafter, the above-prepared acid chloride solution was added in ~15 min at 20–25°C. After 30 min of stirring at 20–25°C, the aqueous layer was separated from the reaction mass and the organic layer was washed with 5% aqueous sodium bicarbonate solution (250 mL) followed by 10% aqueous sodium chloride solution (250 mL). The organic layer was concentrated at 50–60°C under reduced pressure to obtain 62.0 g of cream coloured powder, which was purified by flash chromatography using a 1:1 v/v mixture of hexane and ethyl acetate to obtain 27 g (41%) of title compound 9. Melting range: 158–162°C; HPLC purity: 95.01%; IR (KBr, cm−1): 3403, 3302, 3060, 3029, 2923, 2856, 1652, 1529, 1454, 1351, 1306, 1196, 740, 698; 1H-NMR (300 MHz, DMSO-D6) δ 1.65 (m, 2H), 2.14 & 2.16 (2s, 12H), 2.78 & 2.85 (2d, 4H), 3.70 (m, 1H), 3.95 & 4.05 (ABq, 2H), 4.11 (s, 2H), 4.35 (m, 2H), 5.08 (d, 1H), 6.93-7.27 (m, 16H, Ar), 7.52 & 7.86 (2d, 2H); HRMS:609.3342.
(2S)-N-[(2S,4S,5S)-5-{[(2,6-dimethylphenoxy)acetyl]amino}-4-hydroxy-1,6-diphenylhexan-2-yl]-2-{3-[(2,6-dimethylphenoxy)acetyl]-2-oxotetrahydropyrimidin-1(2H)-yl}-3-methylbutanamide (diacylated, 10):
To a suspension of (2,6-dimethylphenoxy)acetic acid (10 g, 55.55 mmol) in ethyl acetate (40 mL) was added thionyl chloride (7.93 g, 66.66 mmol) followed by N,N-dimethylformamide (0.1 mL) at 25–30°C. The reaction mass temperature was raised to 50–55°C and stirred for 2 h at 50–55°C to complete the formation of acid chloride. Compound 1 (34.88 g, 55.55 mmol) was dissolved separately in a mixture of water (100 mL) and ethyl acetate (100 mL) at 20–25°C, then we added sodium bicarbonate (27.20 g, 323.88 mmol) at 20–25°C. Thereafter, the above-prepared acid chloride solution was added in 10 min at 20–25°C. After 30 min of stirring, we separated the organic layer and washed it with 5% aqueous sodium bicarbonate (50 mL) followed by 10% aqueous sodium chloride (50 mL). The organic layer was concentrated at 55–60°C under reduced pressure to obtain 10 g of white coloured powder, which was purified by chromatography using a 1:1 v/v mixture of ethyl acetate and hexanes to obtain 2 g (20%) of compound 10. Melting range: 79-84 °C; HPLC purity: 88.26%; IR (KBr, cm−1): 3856, 3748, 3410, 3062, 3028, 2963, 2925, 2873, 1662, 1529, 1454, 1358, 1195, 750, 701; 1H-NMR (300 MHz, DMSO-D6) δ 0.74 (2d, 6H), 1.45-1.71 (2m, 4H), 2.02 (m, 1H), 2.15 & 2.24 (2s, 12H), 2.47, 2.97 & 3.53 (3m, 4H), 2.71 & 2.79 (2m, 4H), 3.62 (m, 1H), 4.05 & 4.15 (Abq, 2H), 4.24 (d, 1H), 4.28 & 4.37 (2m, 2H), 4.87 & 4.96 (ABq, 2H), 5.01 (d, 1H), 6.93-7.26 (m, 16H), 7.49-7.73 (2d, 2H, Ar); HRMS: 791.4399.
Fig. 1S.
1H-NMR spectrum of Lopinavir dimer 7
Fig. 2S.

13C-NMR spectrum of Lopinavir dimer 7
Fig. 3S.

MS spectrum of Lopinavir dimer 7
Fig. 4S.

IR spectrum of Lopinavir dimer 7
Fig. 5S.
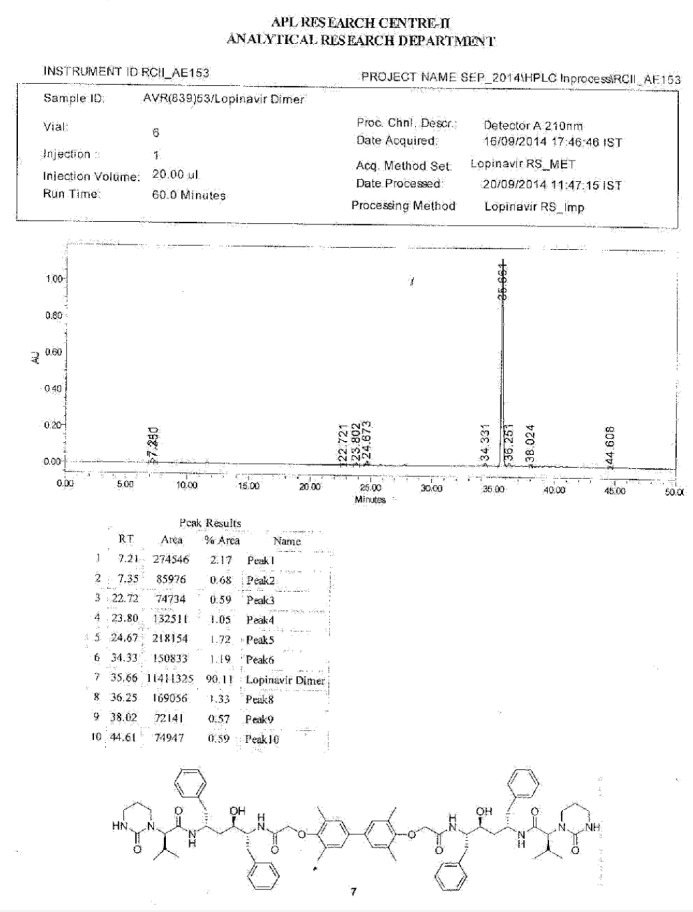
HPLC of Lopinavir dimer 7
Fig. 6S.

1H-NMR spectrum of Lopinavir carboxymethyl analog 8
Fig. 7S.

13C-NMR spectrum of Lopinavir carboxymethyl analog 8
Fig. 8S.

MS spectrum of Lopinavir carboxymethyl analog 8
Fig. 9S.
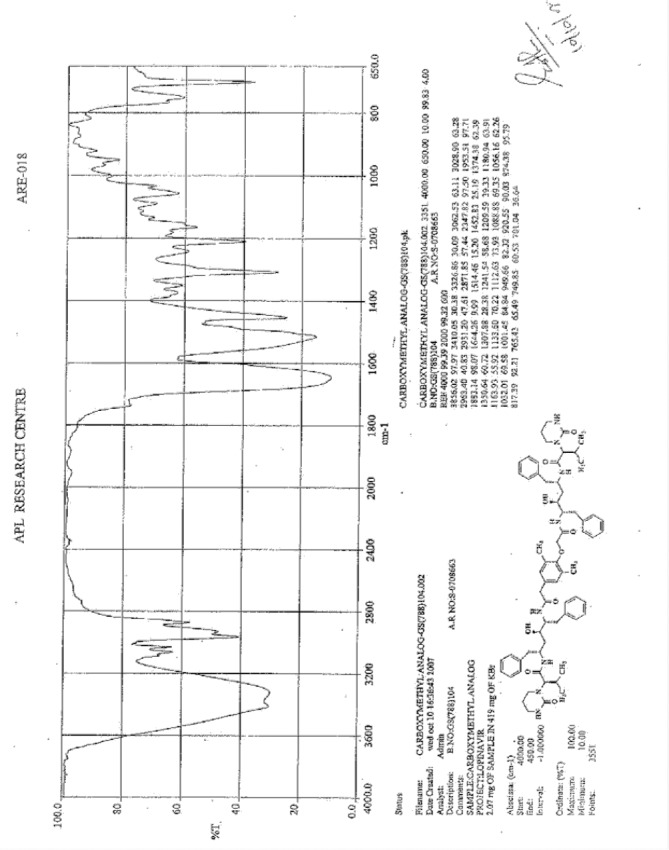
IR spectrum of Lopinavir carboxymethyl analog 8
Fig. 10S.
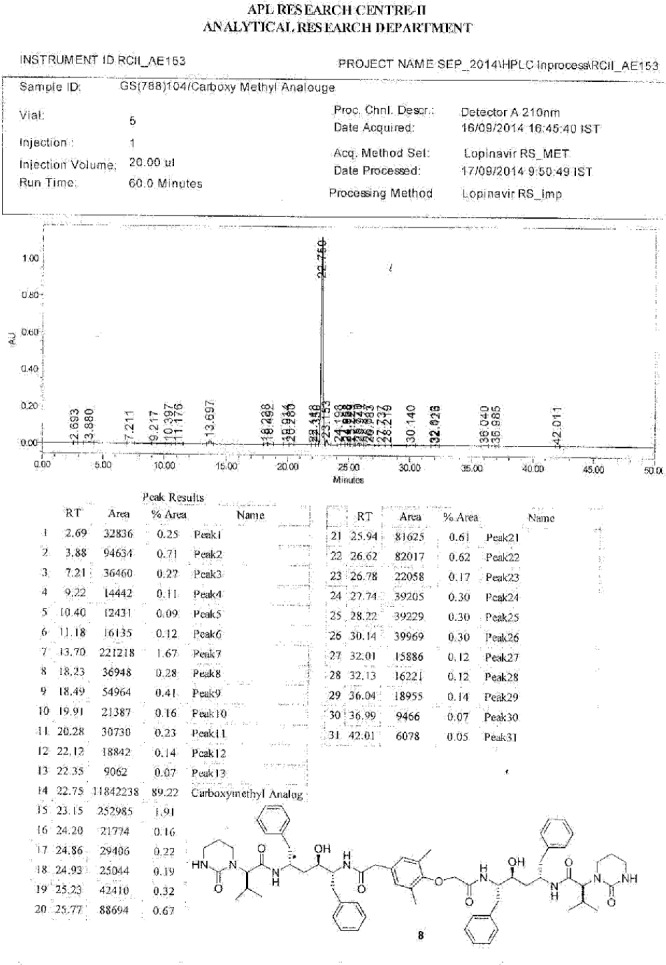
HPLC of Lopinavir carboxymethyl analog 8
Fig. 11S.

Data sheet Lopinavir diamide 9
Fig. 12S.

1H-NMR spectrum of Lopinavir diamide 9
Fig. 13S.

MS spectrum of Lopinavir diamide 9
Fig. 14S.

IR spectrum of Lopinavir diamide 9
Fig. 15S.
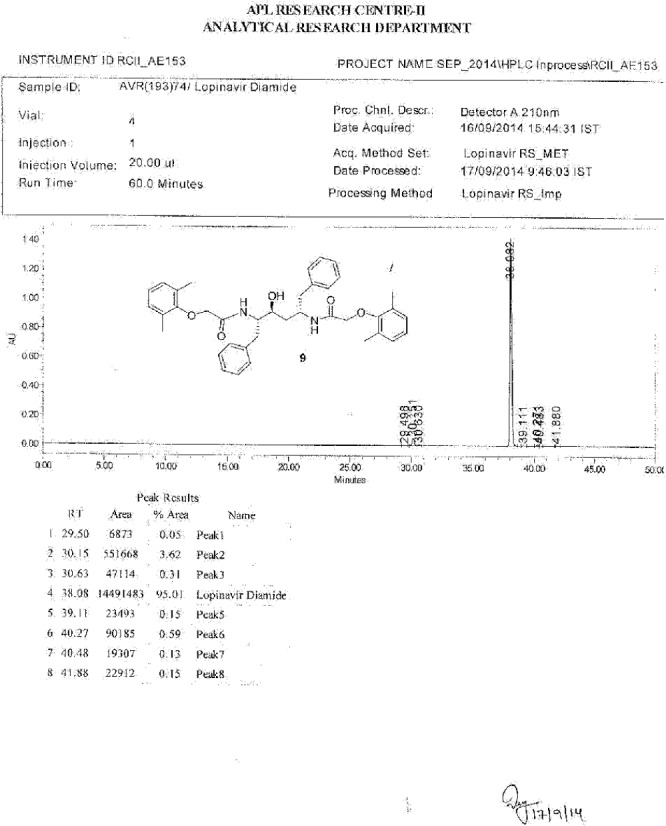
HPLC of Lopinavir diamide 9
Fig. 16S.

Data sheet of Diacylated Lopinavir 10
Fig. 17S.

1H-NMR spectrum of Diacylated Lopinavir 10
Fig. 18S.

MS spectrum of Diacylated Lopinavir 10
Fig. 19S.

IR spectrum of Diacylated Lopinavir 10
Fig. 20S.
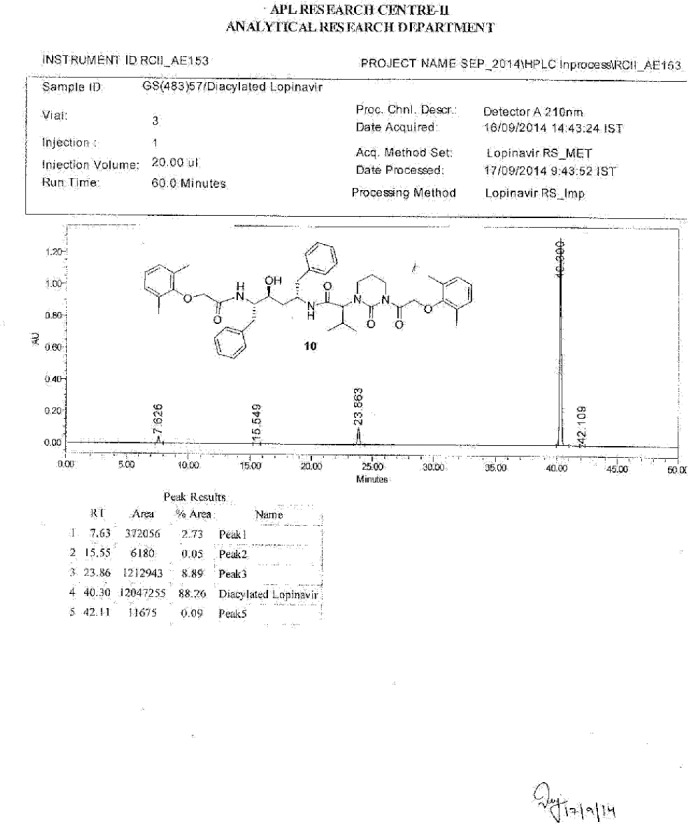
HPLC of Diacylated Lopinavir 10
Fig. 21S.
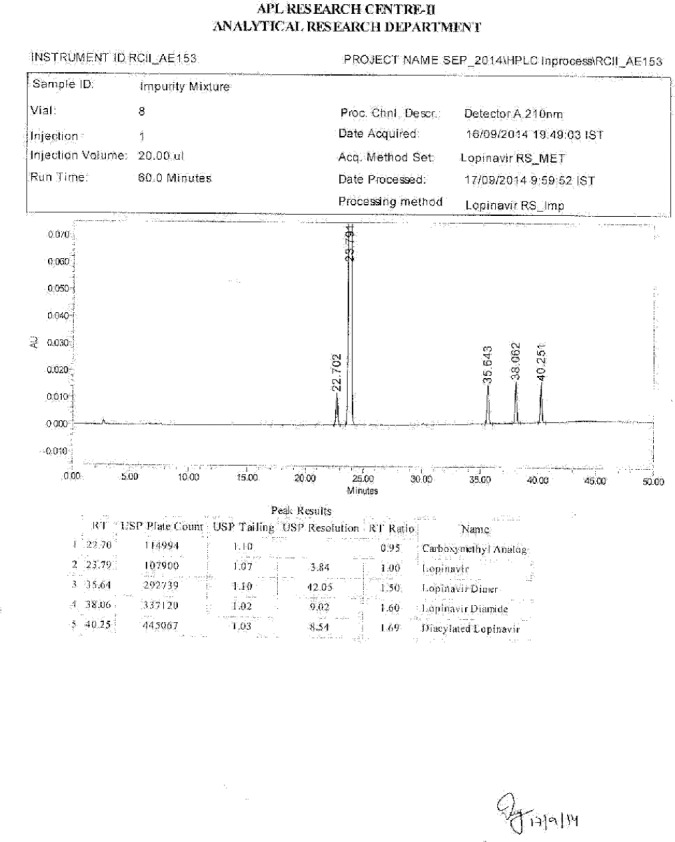
HPLC of Impurity mixture
Acknowledgement
Authors thank the management of Aurobindo Pharma Limited, Hyderabad for giving the permission to publish this work. Authors also thank the Analytical Research Department for their valuable contribution to this work.
Supporting Information
The scanned 1H-NMR spectra of compounds 9–10, the scanned 13C-NMR spectra of compounds 7 and 8, the scanned MS spectra of compounds 7–10, the scanned IR spectra of compounds 7–10, the scanned HPLC of compounds 7–10 and the impurity mixture are available in the online version (Type: PDF, Seize: ca. 3.2 MB): http://dx.doi.org/10.3797/scipharm.1407-14.
Authors’ Statement
Competing Interests
The authors declare no conflict of interest.
References
- [1].International Conference on Harmonization (ICH) guidelines. Q3A (R) impurities in New Drug Substances. Geneva, Switzerland: ICH guidelines; 2002. Feb, [Google Scholar]
- [2].International Conference on Harmonization (ICH) guidelines. Q2B validation of analytical procedure: Methodology. Geneva, Switzerland: ICH guidelines; 1996. Nov, [Google Scholar]
- [3].Chitturi SR, Bharathi Ch, Reddy AV, Reddy KC, Sharma HK, Handa VK, Dandala R, Bindu VH. Impurity profile study of lopinavir and validation of HPLC method for the determination of related substances in lopinavir drug substance. J Pharm Biomed Anal. 2008;48:1430–1440. doi: 10.1016/j.jpba.2008.09.015. http://dx.doi.org/10.1016/j.jpba.2008.09.015 . [DOI] [PubMed] [Google Scholar]
- [4].Sham HL, Betebenner DA, Herrin T, Kumar G, Saldivar A, Vasavanonda S, Molla A, Kempf DJ, Plattner JJ, Norbeck W. Synthesis and antiviral activities of the major metabolites of the HIV protease inhibitor ABT-378 (Lopinavir) Bioorg Med Chem Lett. 2001;11:1351–1353. doi: 10.1016/s0960-894x(01)00243-8. http://dx.doi.org/10.1016/S0960-894X(01)00243-8 . [DOI] [PubMed] [Google Scholar]
- [5].Sham HL, Zhao C, Li L, Betebenner DA, Saldivar A, Vasavanonda S, Kempf DJ, Plattner JJ, Norbeck DW. Novel lopinavir analogues incorporating non-Aromatic P-1 side chains--synthesis and structure--activity relationships. Bioorg Med Chem Lett. 2002;12:3101–3103. doi: 10.1016/s0960-894x(02)00643-1. http://dx.doi.org/10.1016/S0960-894X(02)00643-1 . [DOI] [PubMed] [Google Scholar]
- [6].Sham HL, Betebenner DA, Chen X, Saldivar A, Vasavanonda S, Kempf DJ, Plattner JJ, Norbeck DW. Synthesis and structure-activity relationships of a novel series of HIV-1 protease inhibitors encompassing ABT-378 (Lopinavir) Bioorg Med Chem Lett. 2002;12:1185–1187. doi: 10.1016/s0960-894x(02)00134-8. http://dx.doi.org/10.1016/S0960-894X(02)00134-8 . [DOI] [PubMed] [Google Scholar]
- [7].Sham HL, Betebenner DA, Rosenbrook W, Herrin T, Saldivar A, Vasavanonda S, Plattner JJ, Norbeck DW. Novel lopinavir analogues incorporating heterocyclic replacements of six-member cyclic urea--synthesis and structure-activity relationships. Bioorg Med Chem Lett. 2004;14:2643–2645. doi: 10.1016/j.bmcl.2004.02.089. http://dx.doi.org/10.1016/j.bmcl.2004.02.089 . [DOI] [PubMed] [Google Scholar]
- [8].Siva Lakshmi Devi A, Srinivasa Rao Y, Suresh Y, Yogeswar Reddy M, Jyothi G, Rajababu B, Prasad VS, Umamaheswar Rao V. Structural confirmation of regioisomers of Lopinavir impurities using MS and gradient COSY (1H and 13C NMR assignment of Lopinavir impurities) Magn Reson Chem. 2007;45:424–429. doi: 10.1002/mrc.1968. http://dx.doi.org/10.1002/mrc.1968 . [DOI] [PubMed] [Google Scholar]
- [9].Seshachalam U, Haribabu B, Chandrasekhar K. A novel validated LC method for quantitation of lopinavir in bulk drug and pharmaceutical formulation in the presence of its potential impurities and degradation products. Biomed Chromatogr. 2007;21:716–723. doi: 10.1002/bmc.810. http://dx.doi.org/10.1002/bmc.810 . [DOI] [PubMed] [Google Scholar]
- [10].Ambati VRR, Garaga S, Dandala R, Meenakshisunderam S. Process for the preparation of substantially pure (2S,3S,5S)-5-amino-2-N,N-dibenzylamino-3-hydroxy-1,6-diphenylhexane. 2007, US Patent No 8236991 B [Google Scholar]
- [11].Stoner EJ, Cooper AJ, Dickman DA, Kolacz Kowski L, Lallaman JE, Liu JH, Oliver-Shaffer PA, Patel KM, Paterson JB, Plata DJ, Riley DA, Sham HL, Stengel PJ, Tien JH. Synthesis of HIV Protease Inhibitor ABT-378 (Lopinavir) Org Process Res Dev. 2004;4:264–269. http://dx.doi.org/10.1021/op990202j . [Google Scholar]
- [12].Sham HL, Norbeck DW, Chen X, Betebenner DA. Retroviral protease inhibiting compounds 1995, US Patent No:5,914,332 [Google Scholar]
- [13].Stoner EJ, Cooper AH, Stengel PJ. Synthesis of ABT-378, an HIV Protease Inhibitor Candidate:? Avoiding the Use of Carbodiimides in a Difficult Peptide Coupling. Org Process Res Dev. 1999;3:145–148. http://dx.doi.org/10.1021/op980214p . [Google Scholar]
- [14].Ramu E, Venkateswara Rao B. short approach to the synthesis of the ritonavir and lopinavir core and its C-3 epimer via cross metathesis. Tetrahedron Asymmetry. 2009;20:2201–2204. http://dx.doi.org/10.1016/j.tetasy.2009.09.003 . [Google Scholar]
- [15].Pandey G, Muralikrishna C, Bhalerao UT. Mushroom tyrosinase catalysed coupling of hindered phenols: A novel approach for the synthesis of diphenoquinones and bisphenols. Tetrahedron Lett. 1990;31:3771–3771. http://dx.doi.org/10.1016/S0040-4039(00)97467-7 . [Google Scholar]
- [16].Liao D, Li H, Lei X. Efficient generation of ortho-quinone methide: application to the biomimetic syntheses of (±)-schefflone and tocopherol trimers. Org Lett. 2012;14:18–21. doi: 10.1021/ol202641y. http://dx.doi.org/10.1021/ol202641y . [DOI] [PubMed] [Google Scholar]
- [17].Singh AP, Gupta R. Copper(I) in the Cleft: Syntheses, Structures and Catalytic Properties of {Cu+-Co3+-Cu+}and {Cu+-Fe3+-Cu+}Heterobimetallic Complexes. Eur J Inorg Chem. 2010. pp. 4546–4554. http://dx.doi.org/10.1002/ejic.201000547 .
- [18].Sentürk M, Gülçin I, Daştan A, Küfrevioğlu OI, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem. 2009;17:3207–3211. doi: 10.1016/j.bmc.2009.01.067. http://dx.doi.org/10.1016/j.bmc.2009.01.067 . [DOI] [PubMed] [Google Scholar]