

Abstract
Langerhans cells (LCs) are bone marrow (BM)–derived epidermal dendritic cells (DCs) that represent a critical immunologic barrier to the external environment, but little is known about their life cycle. Here, we show that in lethally irradiated mice that had received BM transplants, LCs of host origin remained for at least 18 months, whereas DCs in other organs were almost completely replaced by donor cells within 2 months. In parabiotic mice with separate organs, but a shared blood circulation, there was no mixing of LCs. However, in skin exposed to ultraviolet light, LCs rapidly disappeared and were replaced by circulating LC precursors within 2 weeks. The recruitment of new LCs was dependent on their expression of the CCR2 chemokine receptor and on the secretion of CCR2-binding chemokines by inflamed skin. These data indicate that under steady-state conditions, LCs are maintained locally, but inflammatory changes in the skin result in their replacement by blood-borne LC progenitors.
Langerhans cells (LCs) are members of a family of highly specialized antigen-presenting cells called dendritic cells (DCs)1–3. They are typically localized in the basal and suprabasal layers of the epidermis and represent the principal hematopoietic barrier to the external environment. LCs are well equipped to ingest foreign antigens that breach the skin. Upon activation, LCs increase their expression of major histocompatibility complex (MHC) class II and costimulatory molecules and migrate to the T cell areas of regional lymph nodes (LNs) where they initiate a systemic immune response by presenting processed antigens to T cells1,4.
Given the importance of LCs in skin immunity, the mobilization of LCs to regional lymph nodes as well as the recruitment of LC precursors from the circulation into the skin must be tightly regulated events. Although we are beginning to understand the mechanisms that regulate the migration of LCs from the epidermis to regional LNs during inflammatory conditions, far less is known about the migration of LCs during steady-state conditions or the recruitment of LC precursors into the epidermis.
LCs have been found in skin-draining lymphatics during steady-state conditions in both animals and humans5,6, and their number increases dramatically after local exposure to ultraviolet (UV) irradiation or contact allergens; this suggests that both in steady-state and inflammatory conditions, migratory LCs are replaced. Earlier studies show that after allogeneic bone marrow (BM) transplants, LCs are completely replaced by donor cells within a few weeks; this has provided the basis for the concept that LCs are derived from a mobile pool of BM-derived pre-cursors that are constantly recruited to the skin2,7. In contrast, in skin xenograft experiments, donor LCs persisted in the grafted skin for more than 9 weeks8. Therefore, the rate at which LCs are normally replaced by BM-derived precursors is unclear.
The migration of leukocytes to inflammatory sites depends on a cascade of discrete events mediated, in part, by chemokines and their receptors9–11. During skin inflammation, numerous chemokines are secreted in the skin, including RANTES, a ligand for CCR1 and CCR512; CCL2 (also known as MCP-1), a ligand for CCR213; CCL22 (MDC) and CCL17 (TARC), ligands for CCR414,15; CCL20 (MIP-3α), the ligand for CCR616,17; CCL9 (MIG), CCL10 (IP10) and CCL11 (ITAC), ligands for CXCR318,19. In addition, several chemokines are made constitutively in normal skin, including CXCL12 (SDF)20 and CCL27 (CTACK)21. Which, if any, of these chemokines are involved in attracting LC precursors to the skin has not been defined.
Here, we analyzed the recruitment of circulating LC precursors to the skin of congenic parabiotic mice and lethally irradiated mice transplanted with congenic BM. Our results show that under steady-state conditions and after injuries that were only weakly inflammatory, BM precursors were not recruited to the skin and LCs were maintained by a stable renewable population present in the skin. However, as a consequence of UV light–induced skin inflammation, blood-borne LC precursors were actively recruited to the skin. The recruitment of such precursors was dependent on their expression of the CCR2 chemokine receptor.
Results
Origin of LCs after BM transplantation
To explore the homeostasis of LCs in steady-state conditions, we reconstituted lethally irradiated 8-week-old C57BL/6 mice that expressed the CD45.2 allele of CD45 with unfractionated BM cells isolated from CD45 congenic (C57BL/6 CD45.1+) donor mice. Eight weeks after transplantation, >90% of spleen, liver and kidney DCs as well as peritoneal macrophages and blood monocytes were of donor origin (Fig. 1a). In contrast, <3% of LCs in the skin were of donor (CD45.1+) origin 8 weeks after transplantation; this percentage did not increase over 18 months (Fig. 1b–f). With respect to morphology and distribution, the host LCs that persisted in chimeric animals were indistinguishable from those of naïve animals (Fig. 1d,e). Although the frequency of host DCs in the chimeric animals was low, skin-draining (inguinal) LNs from chimeric animals showed a higher proportion of host DCs compared to non-skin–draining (mesenteric) LNs and spleen (Fig. 1g).
Figure 1. LC chimerism after congenic BM transplantation.

Eight-week-old CD45.2-expressing C57BL/6 mice were lethally irradiated and injected intravenously with 5 × 105 BM cells isolated from congenic CD45.1-expressing mice. (a) Contour plots show the expression of CD45.1 and CD45.2 on gated I-Ab+CD11c+ spleen and kidney DCs, CD11b+Gr-1−B220−NK1.1− peritoneal macrophages and CD11b+Gr-1− blood monocytes isolated 8 weeks after transplantation. The percentages of CD45.1+- and CD45.2+-gated cells are indicated. (b,c) Epidermal cells were isolated before and at different times after congenic BM transplantation; three to six mice were analyzed independently for each time point. (b) One representative experiment showing the expression of CD45.1 and CD45.2 on gated I-Ab+CD11c+ epidermal LCs. (c) Bar graphs show the mean percentages of LCs that were donor (CD45.1)– or host (CD45.2)–derived at different times after transplantation. (d–f) Epidermal sheets were obtained from (d) nonreconstituted (naïve) C57BL/6 mice or (e,f) chimeric animals 18 months after congenic BM transplantation and stained with (d,e) CD45.2 or (f) CD45.1 mAbs. (g) DC chimerism in inguinal (skin-draining), mesenteric and spleen LNs was determined at 6 months after congenic BM transplantation in four different mice. Bar graphs show the percentages of I-Ab+CD11c+ DCs that expressed CD45.2 in different secondary lymphoid organs in each reconstituted animal.
Origin of LCs in parabiotic mice
The above results suggest that under steady-state conditions, LCs are not replaced by circulating precursors. To further evaluate this possibility, we analyzed pairs of parabiotic mice that shared a single blood circulation for prolonged periods. Each parabiotic pair reached a complete mixing of B cells, T cells and granulocytes within 10 days after the parabiotic procedure (data not shown). In contrast, no mixing of LCs was detectable for at least 6 months after parabiosis, which indicated that LCs were not replaced by circulating precursors (Fig. 2). These results were consistent with the radiation chimera studies and indicated that during steady-state conditions blood-borne progenitors are not recruited to the skin and do not give rise to LCs.
Figure 2. Origin of LCs in parabiotic mice.

Parabiotic mice consisted of one CD45.1+ (mouse A) and one CD45.2+ mouse (mouse B) or one green fluorescent protein (GFP)–transgenic mouse (GFP+, mouse A) and one non-GFP–transgenic mouse (GFP−,mouse B) sharing the same blood circulation. Bar graphs show the mean percentages of gated LCs that were CD45.1+ and CD45.2+ in recipient parabiotic mice 7 and 14 weeks after parabiosis or GFP+ and GFP− in recipient parabiotic mice 6 months after parabiosis. Data represent the means of two separate experiments.
Proliferative capacity of LCs in chimeric mice
To address the possibility that LCs are renewed by proliferating precursors present in the skin, we administered 5-bromodeoxyuridine (BrdU) to nonreconstituted mice and to mice that had been lethally irradiated and reconstituted with congenic BM cells 3 months earlier so that they were fully chimeric at the time of BrdU administration. The rate of BrdU incorporation by host LCs was similar in chimeric and naïve animals (Fig. 3). Because in chimeric animals the majority (>95%) of circulating cells were of donor origin (CD45.1), whereas all BrdU+ LCs were of host origin (CD45.2), these results suggested that during adult life, LCs either proliferate or derive from proliferating precursors present in the skin.
Figure 3. Rate of LC proliferation in chimeric mice.

Groups of five CD45.2+ C57BL/6 mice were reconstituted with congenic CD45.1+ BM. Sixteen weeks later, reconstituted and nonreconstituted CD45.2+ mice (controls) received BrdU in their drinking water for 4 weeks.The percentage of CD45.2+ (host) LCs that had incorporated BrdU was determined by flow cytometry at different times after BrdU administration.
LC replacement during skin inflammation
Although LCs were not replaced by circulating precursors under steady-state conditions, LC homeostasis might be altered after skin injury. To address this question, chimeric animals that had been reconstituted with congenic BM after lethal x-irradiation were left untreated, exposed to UV light or painted with a mixture of acetone and oil that induced a similar amount of local inflammation and LC migration as fluorescein isothiocyanate (FITC) painting22 (data not shown). Each of these injuries induced marked reduction of LCs, which was followed by their recovery in 4–5 weeks (Fig. 4a,b). Recruitment of donor LCs was detected 2–3 weeks after UV exposure, but not after x-irradiation or treatment with acetone and oil and increased in animals that were exposed to UV light for a longer period (Fig. 4c,d). Such donor-derived LCs constituted the predominant LC population for at least 12 weeks after UV exposure (Fig. 4e–g), which suggested that they permanently replaced resident host LCs. The recruitment of donor-derived LCs did not seem to be dependent only on available sites in the epidermis because both topically administered acetone and oil as well as x-irradiation resulted in substantial depletion of host LCs followed by their recovery without recruitment of blood-derived LC precursors.
Figure 4. LC homeostasis during skin inflammation.

CD45.2+ mice were lethally irradiated and reconstituted with 5 × 105 CD45.1+ BM cells and either left untreated or exposed 8 weeks later to UV light for 10 or 30 min or to a mixture of oil and acetone applied to each ear. (a,b) Three ears from three different animals were collected at different times after skin injury and analyzed to determine (a) the total number of LCs per ear and (b) the percentage of epidermal cells that were host LCs. (c) Dot plots show the expression of CD45.1 and CD45.2 on gated I-Ab+CD11c+PI− LCs 3 weeks after x-irradiation alone or x-irradiation followed by skin irritation or UV exposure. (d) Bar graphs show the mean percentages of total epidermal cells that were host- or donor-derived LCs analyzed 3 weeks after skin injury in three different animals. (e–g) Images of epidermal sheets obtained from C57BL/6 animals reconstituted with CD45.1+ BM 12 weeks after exposure to UV for 30 min and stained with (e) CD45.1, (f) I-Ab or (g) CD45.2 mAbs. Magnification: ×20.
Role of CCR2 in the recruitment of LC precursors
Because blood-borne LC precursors were recruited during skin inflammation, we wanted to know whether this recruitment was regulated by pro-inflammatory chemokines and their receptors11. Although blood-borne LC precursors have not been clearly identified, different studies suggest that monocytes23, circulating MHC class II−(I-Ab−)CD11c+ cells24, dermal CD14+cells25 and CD34+ cell–derived CD1a+ cells26 may give rise to LCs. Monocytes express a variety of chemokine receptors, including CCR1, CCR2, CCR5 and CXCR4, whereas dermal CD14+ cells and CD34+ cell–derived CD1a+ cells do not express CCR1, CCR2 or CCR5 but express CCR625,26. We found that freshly isolated LCs as well as circulating I-Ab−CD11c+ cells expressed CCR2 (Fig. 5a). Although analysis of mice deficient in CCR2 has not revealed any deficiency in LCs27, we tested the importance of this receptor in LC replacement during inflammation using CCR2−/− BM chimeras28. We found that 14 days after UV exposure, mice reconstituted with CCR2−/− BM had 75% fewer LCs compared to mice reconstituted with wild-type BM, which indicated that CCR2 is important in the recruitment of LC precursors to inflamed skin (Fig. 5b). The frequency of CCR2−/− LCs increased progressively; after 4–8 weeks the numbers of these cells were similar to those in control animals (Fig. 5c,d). Therefore, although CCR2 plays a critical role in the rapid recruitment of LC precursors after skin inflammation, other chemoattractants appear to be able to contribute to LC recruitment, allowing a slow replenishment of the compartment.
Figure 5. Role of CCR2 in the recruitment of circulating LC precursors to inflamed skin.

(a) cDNA was obtained from various samples and analyzed by PCR for the expression of CCR2. Blood I-Ab−CD11c+ (common DC precursor population), purified blood I-Ab−CD11c+CD3−CD4−CD8−Gr-1− cells; I-Ab− CD11c+ BM, purified I-Ab−CD11c+ BM cells; BM-DC, purified I-Ab+CD11c+ cells differentiated from BM cells in the presence of GM-CSF and IL-4; LC, purified I-Ab+CD11c+ epidermal cells; mono, purified circulating blood CD11b+Gr-1− cells; B cells, purified B220+ blood cells. β-actin primers acted as controls for each sample of cDNA.(b–e) Groups of ten C57BL/6 CD45.1+ mice were reconstituted with CCR2−/− or CCR2+/+ congenic BM alone, a 1:1 mixture of CCR2−/− congenic and syngeneic CCR2+/+ BM or with 1:1 CCR2+/+ congenic and syngeneic CCR2+/+ BM and exposed 3 weeks later to UV light for 20 min. Bar graphs show the percentage of PI−-gated epidermal cells that were CD45.1+ or CD45.2+ LCs (b) 14, (c) 21 and (d) 56 days after UV exposure. (e) Reconstitution of spleen and mesenteric LN–derived DCs and monocytes analyzed 1 day before UV exposure. One representative experiment is shown (n = 5).
Because CCR2 is expressed by many cell types that may infiltrate inflamed skin, including macrophages and T cells29, the LC deficiency that was detected early after UV-irradiation may reflect a requirement for CCR2 in other hematopoietic cell types involved in LC development. To test whether the requirement for CCR2 was intrinsic to the LC precursor, we followed the reconstitution of mutant and wild-type LCs generated from mixed BM cells (1:1 CCR2−/−:CCR2+/+) in the same UV-irradiated animals. Analysis of these chimeric mice 14 days after UV irradiation established that the CCR2 requirement was intrinsic to the LC precursors: CCR2−/− cells were severely compromised in their ability to reconstitute the LC population, whereas there was efficient reconstitution by wild-type cells (Fig. 5b–d). This effect was limited to the LC population because CCR2−/− cells contributed to spleen and mesenteric LN DC populations, blood monocytes (Fig. 5e) and blood T and B cells (data not shown) with the same efficiency as the wild-type cells. In contrast to the 100% CCR2−/− chimeras, the deficiency in CCR2−/− LCs in the mixed chimeras persisted for at least 8 weeks after UV irradiation (Fig. 5d). This suggested that CCR2−/− cells are unable to compete with the wild-type cells in order to occupy available LC sites in the epidermis.
Chemokine expression in inflamed skin
The observation that BM-derived LC precursors were recruited to the skin in a CCR2-dependent manner after UV irradiation, but not after exposure to acetone and oil or x-irradiation, led us to ask whether CCR2 ligands were selectively induced in the skin after UV-irradiation. We tested CCL2 and the related CCR2 ligand, CCL7 (MCP-3), by reverse-transcribed-polymerase chain reaction (RT-PCR) using RNA isolated from normal skin or skin that had been exposed to x-irradiation, oil and acetone or UV light. Low amounts of CCL2 and CCL7 transcripts were present in normal skin, which supported published observations13,30,31, but they were markedly increased after skin exposure to UV light (Fig. 6). In contrast, CCL2 and CCL7 transcripts were reduced after exposure to oil and acetone or x-irradiation (Fig. 6a). CCL27 was constitutively present in all samples and appeared to be weakly induced after skin exposure to UV light. In agreement with the RT-PCR analysis, immunohistochemical analysis demonstrated that CCL2 and CCL7 were abundant in UV-exposed skin (Fig. 6b-d), whereas they were undetectable in normal skin (Fig. 6e and data not shown).
Figure 6. MCP chemokines and injured skin.
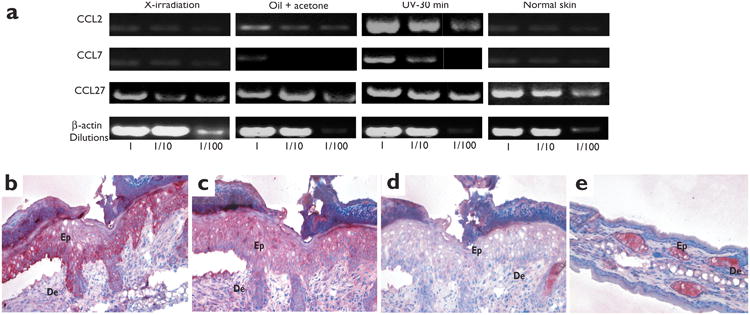
(a) cDNA was prepared from the epidermis of chimeric animals 4 days after lethal irradiation, skin irritation or UV exposure. Serial dilutions of each cDNA sample were analyzed by PCR for the presence of CCL2, CCL7 and CCL27. β-actin primers acted as controls for each sample of cDNA. (b,e) Skin sections were stained in red with CCL2 mAb, (c) CCL7 mAb or (d) an isotype mAb control. (b–d) Serial skin sections obtained from ears isolated 4 days after UV treatment of mice. (e) Noninflamed skin isolated before UV exposure. Ep, epidermis; De, dermis. Magnification: ×20. One representative experiment is shown (n = 3).
Discussion
We demonstrated here the failure of circulating LC precursors to enter noninflamed skin in two independent experimental models. First, for at least 18 months after congenic BM transplantation, the majority of LCs were still of host origin, whereas nearly all circulating leukocytes—including monocytes and DCs—were of donor origin by 8 weeks after transplantation. Second, there was no mixing of LCs in congenic parabiotic mice that shared a single blood circulation. In additional studies, when newborn or adult mice were transplanted with congenic fetal liver or BM cells, no LC chimerism was detectable in the skin for more than 6 months, despite rapid chimerism in BM, spleen and LNs (data not shown). This suggested that as soon as 1 day after birth, LCs become autonomous in the skin. This was consistent with reports of the presence of differentiated LCs in the epidermis of fetal mice, rats and humans32–34. In combination, these results suggest that the seeding of LC precursors occurs mainly during gestational life, probably from the fetal liver, and it will be interesting to determine whether in utero transplantation of hematopoietic cells results in the migration of LC precursors into fetal skin.
These findings contrast with earlier studies in models of allogeneic transplantation, which showed that LCs are rapidly replaced by BM-derived precursors7,35. A major difference between these studies and our work is that we transplanted mice with syngeneic rather than allogeneic BM. Allogeneic BM transplantation induces graft versus host disease (GvHD) and it seems likely that, like the effect of UV-irradiation, GvHD-associated skin inflammation promotes recruitment of BM-derived LC precursors. It will be interesting to assess this possibility by performing allogeneic BM transplantation with T cell-depleted BM, an approach that would avoid the induction of GvHD.
Migratory LCs have been found in skin-draining lymphatic vessels under steady-state conditions in both humans and mice5,6, which suggests that there is a constant turnover of LCs even in the absence of inflammatory signals. Our findings provide further evidence for constitutive LC migration, as the frequency of host-derived DCs in the skin-draining LNs of BM chimeras 6 months after transplantation was greater than in any of the other lymphoid tissues examined. In contrast to the skin, immature DCs in other epithelia, such as in the gut and the conducting airways, are replaced by BM-derived precursors36–38 (and data not shown).
The results of BrdU studies in chimeric mice support the view that LCs are continuously turning over in noninflamed skin. In these studies, BrdU was administered to BM-transplanted mice 8 weeks after complete chimerism was established (as assessed in the blood, BM and spleen), so that the majority of circulating cells were of donor origin at the time of BrdU administration. Host LCs that persisted in these animals incorporated BrdU at the same rate as LCs of nontransplanted animals, which suggested that LCs proliferate locally. In one study, in which a 1-h intravenous labeling with [3H]thymidine was used, no proliferating LCs were detected in the skin39. However, because we found that only 5–10% of LCs incorporate BrdU 10–12 days after systemic administration of BrdU, it is probable that the duration of labeling used in this study was too short to detect proliferating LC precursors. The capacity of LCs to divide in the skin is also suggested by earlier studies that demonstrated human and murine LCs in metaphase in the skin40–42. However, in these studies it was unclear whether the rate of LC division was sufficient to continually replenish the cells that are lost during homeostatic LC trafficking. Our findings establish that under steady-state conditions, LCs are capable of maintaining themselves in the skin for months or years without the need of new blood-borne precursors. It is not known whether LC replenishment during steady-state conditions may be mediated by epidermal LCs or a subpopulation of such cells or by a distinct LC precursor population present in the skin. Experiments that can distinguish a proliferative population of LCs from differentiated LCs present in the epidermis will be useful.
Circulating LC precursors were recruited to the skin under conditions that resulted in an exaggerated LC loss. The recruitment of blood-borne LCs was not dependent on available LC sites in the skin because both local skin irritation and x-irradiation induced marked reduction of host LCs followed by their recovery without the participation of blood-borne LC precursors. These results were consistent with studies showing that partial depletion of LCs from xenografted skin is followed by their recovery within a few weeks to pretreatment numbers41,43. In contrast, there was substantial recruitment of blood-borne LCs within 2 weeks of UV exposure of the skin. This recruitment appeared related to the degree of inflammation and was increased with dose and time of exposure to UV light. We found that UV treatment of mice reconstituted with fetal liver cells rather than BM cells also led to substantial recruitment of LCs, which indicated that fetal liver contains LC precursors (data not shown). Like UV light, heat applied to skin can deplete LCs from a treated area, followed by full LC recovery in 2 weeks44. It is not known whether LC repopulation in this system is also mediated by circulating precursors, although this would be predicted by our findings.
It will be useful to know whether circulating LCs that are recruited after UV exposure have the capacity to repopulate the skin after subsequent skin inflammatory injury. We found that a mixture of oil plus acetone applied to the skin of chimeric mice (CD45.2 recipients of CD45.1 BM) 3 months after their exposure to UV light induced a 50% decrease of LCs followed by a complete recovery in 1 week, as would be seen in normal mice (data not shown). This result suggested that LCs acquired from circulating precursors have the capacity to proliferate in the skin and replenish the LC population. However, without a marker that distinguishes circulating precursors from those in the skin, we cannot rule out the possibility that in this experiment, circulating precursors rather than LCs in the skin were the immediate source of new LCs. A definitive answer to the question will require the use of a congenic transplantation model with additional cellular markers.
Our finding that MCP gradients play a major role in attracting CCR2+ LC precursors to the skin is consistent with the finding that transgenic overexpression of CCL2 in the basal layer of the epidermis causes an increase in LC frequency31. The ability of CCR2−/− cells, after a 4–8 week delay, to give rise to normal numbers of LCs indicates that other chemokines may also contribute to LC precursor recruitment. However, when CCR2−/− BM cells were transplanted with an equal number of wild-type BM cells, the CCR2−/− cells consistently failed to give rise to LCs. Our findings in mice reconstituted with 100% CCR2−/− BM indicated that sufficient LC precursors are produced to fully populate the epidermis over time. Thus, results obtained with the mixed BM chimeras suggest that LC precursors compete to occupy a limited number of available LC sites or “niches” in the skin and that when these are occupied, further LC recruitment is inhibited.
Our observation that CCR2 is required for efficient repopulation of inflamed skin with LCs extends the previously described role for CCR2 in the recruitment of monocytes to sites of inflammation28,45–48. Similarly, CCL2−/− mice have reduced numbers of macrophages in the skin 24 h after challenge with a contact sensitizer45. We also found reduced numbers of dermal macrophages in the skin of UV-irradiated mice that had been reconstituted with CCR2−/− BM cells (data not shown). Taken together, these results support a fundamental role for MCP chemokines in the recruitment of antigen-presenting cells to sites of inflammation, which may explain the impaired antigen-specific T helper type 1 responses observed in CCR2−/− mice28,47. The accumulation of MCP proteins at the dermal-epidermal junction in proximity to blood vessels that we observed here and that also occurs in human inflamed skin49, together with the reduced recruitment of circulating monocytes to the dermis in mice reconstituted with CCR2−/− BM cells (data not shown), suggested that MCP chemokines act mainly in the recruitment of circulating precursors from the blood to the skin.
In conclusion, we propose that a pool of hematopoietic LC precursors seeds the skin during embryonic life. This seeding of LCs appears to be independent of MCP chemokines because CCR2−/− mice have normal numbers of LCs27. As early as 1 day after birth, the seeded pool of LCs becomes autonomous in the skin and able to compensate for the low LC loss that occurs during steady-state turnover and minor injuries. However, once an inflammatory threshold is reached, multiple changes occur in the skin, including the secretion of CCL2 and CCL7 by inflamed epithelial cells, allowing a marked loss of LCs and the recruitment of blood-borne CCR2+ LC progenitors.
Methods
Animals
Five- to eight-week-old female C57BL/6 (CD45.1) and congenic C57BL/6 mice (CD45.2) were from Jackson Laboratory (Bar Harbor, ME) or the Stanford University animal facility. C57BL/6 CCR2−/− mice were generated as described28. The Stanford University Committee on Animal Welfare approved all animal protocols.
Cytokines and media
Murine granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor-α (TNF-α) (Peprotech, Rocky Hill, NJ) were reconstituted in PBS. Cells were cultured in vitro in the presence of complete medium (CM), which included RPMI supplemented with 10% fetal calf serum (FCS) (Gibco-BRL, Gaithersburg, MD), penicillin G (100 U/ml) and streptomycin (100 μg/ml) (Bio-Whitaker, Walkersville, MD). Cell staining and sorting were done in PBS with FCS (2%) and sodium azide (0.1%).
Antibodies, cell staining and sorting
Three-color analyses were done on a FACSCalibur and cell sorting was done on a FACSVantage (both from Becton Dickinson, Mountain View, CA). Monoclonal antibodies (mAbs) to I-Ab, B220, CD3ε, CD4, CD8, CD11b, CD11c, CD19, CD45.1, CD45.2, Gr-1, TCRγδ, TCRαβ, Ter-119, isotypes controls and the second-step antibodies (allophycocyanin- and peridinine chlorophyll protein–conjugated streptavidin) were from Pharmingen (San Diego, CA).
Transplantation of congenic BM cells
Seven- to eight-week-old recipient CD45.2+ C57BL/6 mice were lethally irradiated with 950 rad by an x-ray source operated at 200 kV which delivered 85 rad/min. Mice were irradiated with two doses of 475 rad each, 3 h apart and injected intravenously with BM cells (5 × 105) obtained from adult mice. Transplanted BM was isolated from congenic CD45.1+ C57BL/6 animals to allow identification of donor-derived cells. The presence of donor- and host-derived LCs in the epidermis was evaluated before and at different times after transplantation.
LC isolation
LCs were obtained from epidermal sheets of mouse ears following a slightly modified protocol4. Ears split with the aid of forceps were incubated in 0.5% trypsin (Gibco-BRL) and EDTA (5 mM) in PBS for 60 to 90 min at 37 °C to allow separation of epidermal sheets from the dermis. The epidermal sheets were incubated in CM for 48 h in the presence of GM-CSF (10 ng/ml) and TNF-α (10 ng/ml). Migratory cells present in the supernatant were collected, counted and the percentages of host and donor LCs was determined by flow cytometry by gating on propidium iodide (PI)–negative cells. The total number of cells (5 × 105–6 × 105) obtained per ear included 6–12% hematopoietic cells comprised of 3–6% LCs and 3–6% γδ T cells in wild-type mice. To confirm that this method reliably resulted in the release of LCs from the epidermis, epidermal sheets isolated from either injured or uninjured mice (see below) were analyzed by immunofluorescence at the end of the incubation period. We found that the remaining I-Ab+CD11c+ LCs never exceeded 10% of total migratory LCs, which indicated that the vast majority of LCs were consistently removed from the cultured epidermal sheets and available for analysis.
Isolation of spleen, kidney and LN DCs and peritoneal macrophages
Spleen, kidney, inguinal and mesenteric LNs were collected and injected with collagenase D (1 mg/ml) (Boehringer-Mannheim, Mannheim, Germany) in CM for 60–90 min at 37 °C. Digested LNs and splenocytes were resuspended in CM and layered above a 14.5% Metrizamide solution (Sigma, St. Louis, MO) in CM. Digested kidney cells were resuspended in CM and layered over lympholyte-M solution (Cederlane Laboratories Ltd., Hornby, Canada). The enriched DCs were collected at the interface, washed twice and analyzed by flow cytometry for the presence of host and donor DCs. Peritoneal macrophages were isolated as described50 and analyzed by flow cytometry for the presence of donor or host CD11b+Gr-1− macrophages.
Parabiosis
Pairs of parabiotic mice that consisted of one CD45.1 and one CD45.2 mouse or one GFP-transgenic mouse and one non-GFP–transgenic mouse were generated following a protocol described elsewhere51. The CD45.1–CD45.2 parabiotic pair were analyzed 7 and 14 weeks after parabiosis, whereas the GFP–non-GFP parabiotic pair were maintained for 6 months before being analyzed.
BrdU labeling and analysis
CD45.2+ mice were lethally irradiated and reconstituted with congenic CD45.1+BM. Sixteen weeks later the chimeric and nonchimeric CD45.2+ mice (controls) were injected intraperitoneally with 1 mg of BrdU (Sigma), resuspended in PBS to ensure immediate availability, then given BrdU (0.4 mg/ml) in sterile drinking water that was changed daily for 4 weeks. LCs were isolated at different times after BrdU administration and stained with anti-CD45.2 together with monoclonal anti–I-Ab and anti-CD11c. Surface-stained LCs were subsequently analyzed for the presence of BrdU in CD45.1 and CD45.2 LC subsets with the BrdU Flow Kit (Pharmingen), according to the manufacturer's instructions.
Skin injury
Sex- and age-matched CD45.2+ mice were lethally irradiated and reconstituted with CD45.1+BM. Transplanted mice were either left untreated or after 8 weeks were exposed to UV (wavelength: 254 nm; voltage: 8 W; source: 38 cm from the target) for 10 or 30 min or painted on both ears with 200 μl of a mixture of 1:1 acetone: dibutylphthalate (D-2270, Sigma). Non-BM–transplanted age- and sex-matched CD45.2+ mice acted as controls. The percentages of remaining host as well as recruited donor LCs were measured by flow cytometry at different times after lethal irradiation, UV exposure or skin painting with acetone and dibutylphthalate.
Chemokine receptor expression and chemokine secretion in normal and inflamed skin
Total epidermal cells were isolated 1, 4 and 7 days after UV exposure, skin painting or x-irradiation (950 rad) and analyzed for the presence of CCL2, CCL7 and CCL27. LCs were isolated from epidermal cells based on their expression of I-Ab+CD11c+ with a FACSVantage sorter (Becton Dickinson). BM cells were cultured in the presence of GM-CSF (10 ng/ml) and interleukin 4 (IL-4, 10 ng/ml) for 7 days, and BM-derived DCs were purified with CD11c-conjugated microbeads (Miltenyi Biotech Inc, Auburn, CA). Circulating monocytes were purified from the blood by FACS based on their phenotype (CD11b+Gr-1−). Circulating DC precursors were isolated as described24. Briefly, the cells were double sorted on a FACSVantage based on their expression of CD11c and the absence of I-Ab, CD3, CD4, CD8 and Gr-1 antigens. Similarly, CD11c+I-Ab− cells were double sorted from whole BM cells. The absence of contaminating cells including CD19+, NK1.1+ or Ter119+ cells in purified CD11c+I-Ab– BM and CD11c+I-Ab− blood cells was confirmed by flow cytometry. LCs, BM-derived DCs, I-Ab−CD11c+ blood DC precursors and I-Ab−CD11c+ BM cells were then analyzed by RT-PCR for expression of the chemokine receptor CCR2. Total RNA was extracted from bulk epidermal cells and purified cells with RNeasy Kit (Qiagen Inc, Valencia, CA) and then treated with RNAse-free DNAse (Gibco-BRL). RNA was quantified by spectrophotometry. First-strand cDNA was synthesized from total RNA with the Superscript II RNase H-reverse transcriptase (Gibco-BRL) with random primers, as described by the manufacturer. Primer pairs used for PCR were as follows: CCL2 (sense: 5′–GGCATCACAGTCCGAGTCACAC-3′; antisense: 5′–GCTCTCTCTTCCACCAC-CA–3′; expected size: 500 bp); CCL7 (sense: 5′–CACTCTCTTTCTCCACCATG–3′; anti-sense: 5′–GCTAACACAACGTTAAAGTGAC–3′; expected size: 550 bp); CCL27 (sense: 5′–GCGGTTCTGGGGATGAACACAG–3′; antisense: 5′–AGCAGCCTCCCGCTGT-TACTG–3′; expected size: 220bp); CCR2 primers were used as described28. For all samples, cDNA synthesis was controlled by RT-PCR with β-actin primers. Where indicated, semi-quantification was conducted by analyzing the PCR product of tenfold serial dilutions of each cDNA sample.
Immunofluorescence analysis of normal and inflamed skin
To obtain epidermal sheets, shaved ears were split with the aid of forceps into dorsal and ventral halves and both halves were incubated in 0.5 M ammonium acetate (Sigma) reconstituted in distilled water for 60–90 min at 37 °C to allow separation of epidermal sheets from the dermis. The sheets were blocked with FcγRII-III (CD16-CD32) mAb for 30 min, stained with 5–10 μg/ml of FITC-conjugated CD45.1, phycoerythrin (PE)-conjugated CD45.2 or PE-conjugated I-Ab mAb overnight at 4 °C and then washed for 30 min at room temperature before being mounted in mounting media (Vector Laboratories, Burlingame, CA). Images were viewed with a fluorescence microscope (DMIRB, Leica, Wetzlar, Germany) and processed with Adobe Photoshop software (Adobe, Mountain View, CA).
Immunohistochemistry
Ears were collected and frozen in liquid nitrogen before and 4 and 8 days after UV exposure of the skin. Cryostat-cut skin sections (5 μm) were air-dried for 1 h, fixed in acetone for 20 min and then washed in PBS. Tissue sections were blocked with FcγRII-III (CD16-CD32) mAb for 30 min, then stained with 15 μg/ml of CCL2 and CCL7 mAb (R&D Systems, Minneapolis, MN) for 2 h at room temperature followed by streptavidin-conjugated rabbit anti–goat IgG (Caltag, Burlingame, CA) for 1 h at room temperature. Biotinylated reagents were detected with alkaline phosphatase–avidin (Vector Laboratories). Enzyme reactions were developed with conventional substrates for alkaline phosphatase (Vector Red, Vector Laboratories). Endogenous alkaline-phosphatase activity was blocked with levamisole (Vector Laboratories). Sections were counterstained with hematoxylin (Vector Laboratories) and mounted with mounting media (Vector Laboratories). Images were viewed with a bright-field microscope (DMIRB, Leica) and processed with Adobe Photoshop software (Adobe).
Transplantation of CCR2−/−BM cells
Eight-week-old recipient CD45.1+ C57BL/6 mice were lethally irradiated and injected intravenously with 5 × 105 BM cells isolated from CCR2−/− or wild-type CD45.2+ mice or with a 1:1 mixture of CCR2−/−:CD45.2+ BM and syngeneic wild-type BM or 1:1 CCR2+/+:CD45.2+ BM and syngeneic wild-type BM. All transplanted animals were exposed 3 weeks after transplantation to UV for 20 min. The chimerism of blood cells, spleen and mesenteric LN DCs was determined 1 day before UV exposure and the LC chimerism was determined at different times after UV exposure.
Acknowledgments
We thank C. Benike for critical review of the manuscript, M. Katics for excellent animal care and D.Jones for formatting the manuscript. Supported by Ernst Schering Research Foundation (H. K.) and by grants from the National Institutes of Health (HL57443) and the Tobacco-Related Disease Research Program (9RT-0229) (to E. G.E.).
Footnotes
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.Schuler G, et al. Murine epidermal Langerhans cells as a model to study tissue dendritic cells. Adv Exp Med Biol. 1993;329:243–249. doi: 10.1007/978-1-4615-2930-9_41. [DOI] [PubMed] [Google Scholar]
- 2.Stingl G, Tamaki K, Katz SI. Origin and function of epidermal Langerhans cells. Immunol Rev. 1980;53:149–174. doi: 10.1111/j.1600-065x.1980.tb01043.x. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 4.Schuler G, Steinman RM. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J Exp Med. 1985;161:526–546. doi: 10.1084/jem.161.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jakob T, Ring J, Udey MC. Multistep navigation of Langerhans/dendritic cells in and out of the skin. J Allergy Clin Immunol. 2001;108:688–696. doi: 10.1067/mai.2001.118797. [DOI] [PubMed] [Google Scholar]
- 6.Hemmi H, et al. Skin antigens in the steady state are trafficked to regional lymph nodes by transforming growth factor-β1-dependent cells. Int Immunol. 2001;13:695–704. doi: 10.1093/intimm/13.5.695. [DOI] [PubMed] [Google Scholar]
- 7.Katz SI, Tamaki K, Sachs DH. Epidermal Langerhans cells are derived from cells originating in bone marrow. Nature. 1979;282:324–326. doi: 10.1038/282324a0. [DOI] [PubMed] [Google Scholar]
- 8.Krueger GG, Daynes RA, Emam M. Biology of Langerhans cells: selective migration of Langerhans cells into allogeneic and xenogeneic grafts on nude mice. Proc Natl Acad Sci USA. 1983;80:1650–1654. doi: 10.1073/pnas.80.6.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 10.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 11.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12127:121. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 12.Sebastiani S, et al. The role of chemokines in allergic contact dermatitis. Arch Dermatol Res. 2002;293:552–559. doi: 10.1007/s00403-001-0276-9. [DOI] [PubMed] [Google Scholar]
- 13.Barker JN, et al. Monocyte chemotaxis and activating factor production by keratinocytes in response to IFN-γ. J Immunol. 1991;146:1192–1197. [PubMed] [Google Scholar]
- 14.Campbell JJ, et al. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature. 1999;400:776–780. doi: 10.1038/23495. [DOI] [PubMed] [Google Scholar]
- 15.Katou F, et al. Macrophage-derived chemokine (MDC/CCL22) and CCR4 are involved in the formation of T lymphocyte-dendritic cell clusters in human inflamed skin and secondary lymphoid tissue. Am J Pathol. 2001;158:1263–1270. doi: 10.1016/S0002-9440(10)64077-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dieu-Nosjean MC, et al. Macrophage inflammatory protein 3α is expressed at inflamed epithelial surfaces and is the most potent chemokine known in attracting Langerhans cell precursors. J Exp Med. 2000;192:705–718. doi: 10.1084/jem.192.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakayama T, et al. Inducible expression of a CC chemokine liver- and activation-regulated chemokine (LARC)/macrophage inflammatory protein (MIP)-3α/CCL20 by epidermal keratinocytes and its role in atopic dermatitis. Int Immunol. 2001;13103:95. doi: 10.1093/intimm/13.1.95. [DOI] [PubMed] [Google Scholar]
- 18.Tensen CP, et al. Human IP-9:A keratinocyte-derived high affinity CXC-chemokine ligand for the IP-10/Mig receptor (CXCR3) J Invest Dermatol. 1999;112:716–722. doi: 10.1046/j.1523-1747.1999.00581.x. [DOI] [PubMed] [Google Scholar]
- 19.Flier J, et al. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J Pathol. 2001;194:398–405. doi: 10.1002/1096-9896(200108)194:4<397::aid-path899>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 20.Pablos JL, et al. Stromal-cell derived factor is expressed by dendritic cells and endothelium in human skin. Am J Pathol. 1999;155:1577–1586. doi: 10.1016/S0002-9440(10)65474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morales J, et al. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc Natl Acad Sci USA. 1999;96:14470–14475. doi: 10.1073/pnas.96.25.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cumberbatch M, Illingworth I, Kimber I. Antigen-bearing dendritic cells in the draining lymph nodes of contact sensitized mice: cluster formation with lymphocytes. Immunology. 1991;74:139–145. [PMC free article] [PubMed] [Google Scholar]
- 23.Geissmann F, et al. Transforming growth factor β1, in the presence of granulocyte/macrophage colony-stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J Exp Med. 1998;187:961–966. doi: 10.1084/jem.187.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.del Hoyo GM, et al. Characterization of a common precursor population for dendritic cells. Nature. 2002;415:1043–1047. doi: 10.1038/4151043a. [DOI] [PubMed] [Google Scholar]
- 25.Larregina AT, et al. Dermal-resident CD14+ cells differentiate into Langerhans cells. Nature Immunol. 2001;2:1151–1158. doi: 10.1038/ni731. [DOI] [PubMed] [Google Scholar]
- 26.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-α cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 27.Sato N, et al. CC chemokine receptor (CCR)2 is required for langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J Exp Med. 2000;192:205–218. doi: 10.1084/jem.192.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boring L, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nieto M, et al. Polarization of chemokine receptors to the leading edge during lymphocyte chemotaxis. J Exp Med. 1997;186:153–158. doi: 10.1084/jem.186.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ying S, Taborda-Barata L, Meng Q, Humbert M, Kay AB. The kinetics of allergen-induced transcription of messenger RNA for monocyte chemotactic protein-3 and RANTES in the skin of human atopic subjects: relationship to eosinophil, T cell, and macrophage recruitment. J Exp Med. 1995;181:2153–2159. doi: 10.1084/jem.181.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura K, Williams IR, Kupper TS. Keratinocyte-derived monocyte chemoattractant protein 1 (MCP-1): analysis in a transgenic model demonstrates MCP-1 can recruit dendritic and Langerhans cells to skin. J Invest Dermatol. 1995;105:635–643. doi: 10.1111/1523-1747.ep12324061. [DOI] [PubMed] [Google Scholar]
- 32.Foster CA, Holbrook KA, Farr AG. Ontogeny of Langerhans cells in human embryonic and fetal skin: expression of HLA-DR and OKT-6 determinants. J Invest Dermatol. 1986;86:240–243. doi: 10.1111/1523-1747.ep12285201. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi M, Asano H, Fujita Y, Hoshino T. Development of ATPase-positive, immature Langerhans cells in the fetal mouse epidermis and their maturation during the early postnatal period. Cell Tiss Res. 1987;248:315–322. doi: 10.1007/BF00218198. [DOI] [PubMed] [Google Scholar]
- 34.Hsiao L, Takeya M, Arao T, Takahashi K. An immunohistochemical and immunoelectron microscopic study of the ontogeny of rat Langerhans cell lineage with anti-macrophage and anti-Ia monoclonal antibodies. J Invest Dermatol. 1989;93:780–786. doi: 10.1111/1523-1747.ep12284420. [DOI] [PubMed] [Google Scholar]
- 35.Tamaki K, Stingl G, Katz SI. The origin of Langerhans cells. J Invest Dermatol. 1980;74:309–311. doi: 10.1111/1523-1747.ep12543533. [DOI] [PubMed] [Google Scholar]
- 36.Pugh CW, MacPherson GG, Steer HW. Characterization of nonlymphoid cells derived from rat peripheral lymph. J Exp Med. 1983;157:1758–1779. doi: 10.1084/jem.157.6.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fossum S. Dendritic leukocytes: features of their in vivo physiology. Res Immunol. 1989;140:883–891. doi: 10.1016/0923-2494(89)90048-5. [DOI] [PubMed] [Google Scholar]
- 38.Holt PG, Haining S, Nelson DJ, Sedgwick JD. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J Immunol. 1994;153:256–261. [PubMed] [Google Scholar]
- 39.Ghaznawie M, Papadimitriou JM, Heenan PJ. The steady-state turnover of murine epidermal Langerhans cells. Br J Dermatol. 1999;141:57–61. doi: 10.1046/j.1365-2133.1999.02921.x. [DOI] [PubMed] [Google Scholar]
- 40.Czernielewski J, Vaigot P, Prunieras M. Epidermal Langerhans cells–a cycling cell population. J Invest Dermatol. 1985;84:424–426. doi: 10.1111/1523-1747.ep12265523. [DOI] [PubMed] [Google Scholar]
- 41.Czernielewski JM, Demarchez M. Further evidence for the self-reproducing capacity of Langerhans cells in human skin. J Invest Dermatol. 1987;88:17–20. doi: 10.1111/1523-1747.ep12464659. [DOI] [PubMed] [Google Scholar]
- 42.Miyauchi S, Hashimoto K. Epidermal Langerhans cells undergo mitosis during the early recovery phase after ultraviolet-B irradiation. J Invest Dermatol. 1987;88:703–708. doi: 10.1111/1523-1747.ep12470379. [DOI] [PubMed] [Google Scholar]
- 43.Krueger GG, Emam M. Biology of Langerhans cells: analysis by experiments to deplete Langerhans cells from human skin. J Invest Dermatol. 1984;82:613–617. doi: 10.1111/1523-1747.ep12261453. [DOI] [PubMed] [Google Scholar]
- 44.Ghaznawie M, Papadimitriou JM, Heenan PJ. The repopulation of murine Langerhans cells after depletion by mild heat injury. Br J Dermatol. 1999;141:206–210. doi: 10.1046/j.1365-2133.1999.02966.x. [DOI] [PubMed] [Google Scholar]
- 45.Lu B, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters W, Dupuis M, Charo IF. A mechanism for the impaired IFN-γ production in C-C chemokine receptor 2 (CCR2) knockout mice: role of CCR2 in linking the innate and adaptive immune responses. J Immunol. 2000;165:7072–7077. doi: 10.4049/jimmunol.165.12.7072. [DOI] [PubMed] [Google Scholar]
- 48.Belperio JA, et al. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. J Clin Invest. 2001;108:547–556. doi: 10.1172/JCI12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanbervliet B, et al. Sequential involvement of CCR2 and CCR6 ligands for immature dendritic cell recruitment: possible role at inflamed epithelial surfaces. Eur J Immunol. 2002;32:231–242. doi: 10.1002/1521-4141(200201)32:1<231::AID-IMMU231>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 50.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. 1968;128:415–435. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]