

Abstract
Oxons are bioactive metabolites of organophosphorus insecticides (OPs) that covalently inactivate serine hydrolases. KIAA1363 is one of the most abundant serine hydrolases in mouse brain. Although the physiological consequences related to the inhibition of KIAA1363 due to environmental exposures to OPs are poorly understood, the enzyme was previously shown to have a role in the detoxification of oxons. Here, we overexpressed human KIAA1363 and CES1 in COS7 cells and compared the potency of inhibition (IC50s, 15 min) of KIAA1363 and CES1 by chlorpyrifos oxon (CPO), paraoxon (PO), and methyl paraoxon (MPO). The order of potency was CPO > PO ≫ MPO for both enzymes. We also determined the bimolecular rate constants (kinact/Ki) for reactions of CPO and PO with KIAA1363 and CES1. KIAA1363 and CES1 were inactivated by CPO at comparable rates (4.4 × 106 s−1M−1 and 6.7 × 106 s−1M−1, respectively), whereas PO inactivated both enzymes at slower rates (0.4 × 106 s−1M−1 and 1.5 × 106 s−1M−1, respectively). Finally, the reactivation rate of KIAA1363 following inhibition by CPO was evaluated. Together, the results define the kinetics of inhibition of KIAA1363 by active metabolites of agrochemicals and indicate that KIAA1363 is highly sensitive to inhibition by these compounds.
Keywords: carboxylesterase, oxon, bimolecular rate constant, enzyme inhibition, chlorpyrifos oxon, paraoxon
Graphical Abstract
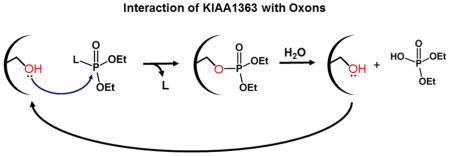
1. Introduction
Organophosphorus insecticides (OPs) are used in significant quantities in agriculture to prevent pest infestation. Approximately 73 million pounds of OP insecticides were used in the United States in 2001 [1]. Human exposure to these compounds is widespread; for example, dialkylphosphate metabolites of OP insecticides were detected in urine samples of roughly half the NHANES study population (http://www.cdc.gov/exposurereport/pdf/FourthReport.pdf). Toxicity of OPs is due to the in vivo generation of oxons, which are formed mainly in liver via cytochrome P450-catalyzed oxidation of the parent OP. The oxons exert their acute toxicity by inhibiting the activity of acetylcholinesterase, a member of the serine hydrolase superfamily, resulting in cholinergic overload [2–4]. The oxons are also capable of reacting with several other serine hydrolases [5, 6]. For example, oxons can covalently modify the catalytic serine residue in carboxylesterase 1 (CES1), which is a serine hydrolase found in large quantities in human liver and other tissues. The reaction with CES1 is an important mechanism by which oxons are detoxified [6]. We have previously defined the kinetic constants (bimolecular rate constant, kinact/Ki) for the covalent reaction of several oxons and carbamates with recombinant CES1 [7], [8]. CES1 was an extremely effective scavenger of oxons, exhibiting a kinact/Ki of ~107 s−1M−1 for chlorpyrifos oxon (CPO). In addition, the phosphorylated adduct was relatively stable (t1/2 = 31 h at 37°C) [7]. Thus, CES1 appears to act as a covalent sink that functionally removes these toxic compounds from the body.
KIAA1363 is a 45–50-kDa glycosylated enzyme that is expressed in most tissues, including macrophages [9], and is one of the most abundant serine hydrolases in mouse brain membranes [10]. The physiological role of KIAA1363 is still not completely understood, although it is highly expressed in several invasive cancer cell lines [11]. One biochemical pathway regulated by KIAA1363 is the removal of an acetyl moiety from the sn-2 hydroxyl group of the glycerol backbone of acetyl monoacylglycerol ethers (AcMAGEs) [12]. AcMAGEs are lipid mediators that contribute to the invasive phenotype of certain cancer cells. In addition, KIAA1363 was reported to have cholesteryl ester hydrolase activity, leading it to be renamed neutral cholesteryl ester hydrolase (nCEH) [13], although this particular biochemical activity and designation for KIAA1363 remains highly controversial [14, 15]. With respect to its toxicological properties, murine KIAA1363 was shown to have a high affinity for CPO and was the predominant CPO binding and metabolizing enzyme in the mouse brain [9, 10]. In vitro studies using brain membranes from wildtype mice showed that 1 nM [3H]-CPO could be completely hydrolyzed after a 60 min incubation, whereas brain membranes from KIAA1363-null mice were 13-fold less efficient at hydrolyzing CPO. Furthermore, on the basis of SDS-PAGE analysis, KIAA1363 was the most prominent protein in brain membranes radiolabeled by 1 nM [3H]-CPO [10]. These results implied that KIAA1363 could be covalently modified by CPO and its enzyme activity transiently inhibited during the course of the 60-min reaction, but that the enzyme was reactivated to its functional state by the end of 60 min, i.e. CPO is in effect a slowly turned over substrate. Importantly, KIAA1363−/− mice were found to be more susceptible to OP-induced acute intoxication than wildtype mice [16], indicating a protective role for this enzyme. However, whether transient inhibition of KIAA1363 by long-term exposure to low concentrations of CPO might cause adverse toxicological effects due to changes in key lipid metabolism pathways is unclear. Furthermore, current physiologically-based pharmacokinetic/pharmacodynamic (PBPK/PD) models of OPs do not incorporate KIAA1363-dependent detoxication pathways [17]. These are important gaps that need to be addressed.
In this study, we evaluated the kinetic parameters that describe the interaction of oxons with KIAA1363. We overexpressed human KIAA1363 in COS7 cells to examine its sensitivity to OP oxons and determined the half-maximal inhibitory concentrations (IC50s) when treated by CPO, paraoxon (PO), and methyl paraoxon (MPO). These oxons are derived from chlorpyrifos, parathion, and methyl parathion, respectively, and are common pesticides used in agricultural settings. In addition, we determined the bimolecular rate constants for the reaction of CPO and PO with KIAA1363, and compared them to the rate constants obtained with CES1, a highly efficient enzyme at detoxifying oxons [7]. Finally we evaluated the reactivation rate constant of KIAA1363 phosphorylated by reaction with CPO, i.e. the rate constant for the hydrolysis of the KIAA1363-diethylphosphate adduct.
The recombinant proteins in COS-7 cell lysates were used without further purification. The justification for this is as follows. Fluorescent reporter probes indicate that there is substantial co-localization of KIAA1363 and endoplasmic reticulum membranes in cells [18], indicating that KIAA1363 is embedded within this membrane. As compared to cytosolic proteins, integral membrane proteins are challenging to purify for structural and functional studies. The KIAA1363 protein that we overexpressed in COS7 cells also lacked a histidine tag, making its purification even more challenging. There are advantages to studying a protein in a cell lysate versus studying the pure protein. Protein activity can often be lost when proteins are examined in their purified, isolated forms [19]. Requisite co-factors, protein partners, and small molecule modulators might be lost when the protein is purified and studied in isolation. Using cell lysates, rather than an isolated protein, allows the protein under examination to be studied in an environment closer to its native physiological environs [20]. There are, however, disadvantages to studying a protein in a cell lysate. Only by examining a specific reaction using a purified, isolated protein can one determine the intrinsic kinetic parameters (kcat and Km) of the enzyme. In cell lysates, these values are “apparent” under the specific conditions employed.
2. Materials and methods
2.1. Cells, Chemicals, Reagents, and Transfections
COS-7 cells, Dulbecco’s Modified Eagle’s Medium (DMEM), gentamicin sulfate solution (50 mg/ml), and Hanks’ balanced salt solution without calcium, magnesium or phenol red were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Fetal bovine serum (FBS) was purchased from Invitrogen (Carlsbad, CA). pCMV6-XL5 expression vectors containing either human KIAA1363 cDNA [also called arylacetamide deacetylase like 1 (AADACL1) or neutral cholesteryl ester hydrolase (nCEH)] (REFSEQ accession number: NM_001146276.1) or human CES1 cDNA (isoform c) (REFSEQ accession number: NM_001266.4) were purchased from Origene (Rockville, MD). KIAA1363 and CES1 plasmids were transfected into COS-7 cells in 60-mm plates using the manufacturer’s instructions (FuGene 6, Invitrogen). Transfections of COS-7 cells with an empty vector were performed in parallel. After forty-eight hours, the cells were washed with PBS, harvested by scraping, and lysed in ice-cold 50 mM Tris-HCl (pH 7.4) buffer by sonication. The recombinant proteins in COS-7 cell lysates were used without further purification.
O,O′-Diethyl 3,5,6-trichloro-2-pyridyl phosphate (chlorpyrifos oxon) and O,O′-diethyl p-nitrophenyl phosphate (paraoxon) were kind gifts from Dr. Howard Chambers, Department of Entomology, Mississippi State University. Oxons were >99% pure when assessed by thin-layer chromatography [21]. para-Nitrophenyl valerate (pNPV), 4-methylumbelliferyl acetate (4-MUBA), and all general reagents and buffers were purchased from Sigma (St. Louis, MO). The activity-based serine hydrolase probe, fluorophosphonate-biotin (FP-biotin), was from Toronto Research Chemicals (North York, Ontario). Avidin-horse radish peroxidase (HRP) was from Sigma.
2.2. Protein Assays
The protein concentration of cell lysates was measured using the BCA reagent (Pierce, Rockford, IL) according to the manufacturer’s instructions
2.3. Enzyme Assays
Hydrolysis reactions using the pro-fluorogenic substrate 4-MUBA (final concentration 250 μM) were performed at 37°C in a 96-well plate format in a total volume of 300 μL/well in 50 mM Tris-HCl (adjusted to pH 7.4 at room temperature). COS-7 cell lysates containing overexpressed KIAA1363 or CES1 proteins were diluted to a final protein concentration of 0.1 mg/ml per well. In parallel, equivalent amounts of mock-transfected cell lysate protein were also assayed in order to subtract the intrinsic 4-MUBA hydrolytic activity of the COS-7 lysate/buffer mix.
Working solutions of the oxons were diluted in ethanol and added to the reaction mixture to give the desired concentrations. The final volume of ethanol in the wells was 1.5% (v/v); this amount of ethanol did not affect enzymatic activity. KIAA1363 and CES1 enzymatic reaction rates were corrected by subtracting the mock-transfected COS-7 lysate hydrolysis rate. For IC50 measurements, the enzyme and oxon were incubated for either 0 min or 15 min at 37°C, followed by addition of 4-MUBA (final concentration 250 μM). Reaction progress was monitored by measuring the fluorescence of the hydrolysis product 4-methyumbelliferone (ex. 302 nm, em. 355 nm) for 5 min at 37°C. The initial slopes of the progress curves were determined and used to calculate enzymatic activities. The curves were linear during the 5 min reaction period. IC50 values were determined by plotting the % inhibition of enzyme activity versus the oxon concentration. % inhibition was defined as: (rate of reaction with no oxon – rate of reaction with oxon) / rate of reaction with no oxon × 100. IC50 values were interpolated from the curves.
2.4. Kinetic Studies
A competitive kinetic scheme describing the covalent inhibition of serine hydrolases (E) by oxons (I) (and its reactivation) in the presence of ester substrate (S) is shown in Fig. 5A. To determine the bimolecular rate constants for enzyme inactivation, the oxon (at various concentrations) and 4-MUBA (250 μM) were added to reaction buffer and prewarmed at 37°C (5 min). Cell lysate containing overexpressed KIAA1363 or CES1 protein (0.1 mg/ml final concentration) was added to initiate the reaction. Progress of the reaction was monitored for 45 min.
Fig. 5. Effect of the substrate on the progressive inhibition of KIAA1363 by oxon.

(A) General kinetic scheme that describes the inhibition and reactivation of serine hydrolases (E) by oxons (I) in the presence of an ester substrate (S). The turnover number kcat for the ester substrate (S) is a function of the rates of acylation (k2) and deacylation (k3) [i.e., kcat = k2 × k3/(k2+ k3)] [24]; however, these steps are not explicitly shown for simplicity. (B) Inhibition of KIAA1363 by varying concentrations of CPO. The progress of the 4-MUBA hydrolysis reaction was followed by measuring the fluorescence of 4-methylumbelliferone produced during the 45 min monitoring period. Inset, representative experiment describing the inhibition of KIAA1363-catalyzed 4-MUBA hydrolysis by CPO. KIAA1363-transfected cell lysate was added to a prewarmed mixture of CPO and 4-MUBA (i.e., pre-incubation time was 0 min between CPO and KIAA1363 prior to the addition of substrate). Reaction progress was monitored by fluorescence for 5 min at 37°C.
For inhibition of CES1, the reaction curves were fit to Eqn. (1), which assumes that no reactivation of enzyme occurs during the 45 min time period. This assumption is supported by the half-life of reactivation of pure CES1-diethylphosphate protein (t1/2 = 31 h) [7].
| (1) |
Data were fitted using SigmaPlot 8.0, and the value for the apparent first-order rate constant of enzyme inactivation (kobs) was determined at each oxon concentration. F0 is fluorescence at t = 0, Ft is fluorescence at time t, F∞is fluorescence at time infinity, t is time in s, and kobs is the observed rate constant in s−1.
Evidence exists that KIAA1363 can completely turnover 1 nM CPO in 60 min (100% hydrolysis) [9]. Thus, we fitted the kinetic data for KIAA1363 to a model that includes enzyme reactivation [22], as given in Eqn. (2).
| (2) |
F0 is fluorescence at t = 0, kobs is the observed first-order inhibition rate constant, v0 is the initial velocity of the 4-MUBA hydrolysis reaction, and vss is the steady-state velocity. The non-linear pre-steady state phase of the reaction depicts the progressive inhibition of enzyme by oxon, which is represented by the time-dependent decrease in the slope of the progress curve. In the linear steady-state phase of the reaction, during which the reaction velocity equals vss, the rates of enzyme phosphorylation and dephosphorylation are considered to be equal [22].
The kobs values obtained were then plotted against the oxon concentration and fitted to Eqn. (3) [23].
| (3) |
kinact is the first-order rate constant for the inactivation (phosphorylation) of the enzyme by the oxon, Ki′ is the apparent dissociation constant for the reversible E·I (enzyme-oxon) complex, and [I] is the inhibitor (oxon) concentration. Because KIAA1363 and CES1 were not purified from transfected COS-7 cell lysates, the true bimolecular rate constant is not known and therefore the apparent bimolecular rate constant is kinact/Ki′. However, the apparent dissociation constant (Ki′) between enzyme and oxon can be corrected by Eqn. (4), which takes the reporter substrate 4-MUBA into account. The presence of 4-MUBA in the enzyme-oxon reaction effectively reduces the affinity of oxon for the enzyme (Fig. 5A); therefore, Ki is the corrected dissociation constant for E·I and is calculated as follows.
| (4) |
Ki′ is the apparent dissociation constant for E·I, [S] is the 4-MUBA concentration (250 μM), and Km is the Michaelis constant for 4-MUBA (227 μM, determined in the absence of oxon). Thus, the corrected bimolecular rate constant can be expressed as: kinact/[Ki × (1 + [S]/Km)].
In the linear steady-state phase of the reaction of 4-MUBA hydrolysis, the rates of enzyme phosphorylation and dephosphorylation are equal [22]. Thus, the term vss is a measure of the steady-state turnover of oxon and a function of [I], as shown in Eqn. (5).
| (5) |
v0 is the phosphorylation rate extrapolated to zero inhibitor concentration, vmin is the asymptotic rate at infinite inhibitor concentration, and KmI is the Michaelis constant for KIAA1363-catalyzed hydrolysis of the OP oxon [24]. Values for v0, vmin, and KmI were obtained from least-squares fits of the data to Eqn. (5).
The reactivation (dephosphorylation) rate constant (kreact) was calculated using Eqn. (6), which can be rearranged to give Eqn. (7) [25], where VS is the velocity of the reporter substrate (4-MUBA, 250 μM) hydrolysis reaction in the absence of inhibitor. It was assumed that the rate of the “aging” reaction was insignificant (see Discussion for more explanation).
| (6) |
| (7) |
2.5. Inhibition of KIAA1363 or CES1 determined using the activity-based protein probe FP-biotin
KIAA- or CES1-transfected COS7 cell lysates (1 mg/ml protein, 25 μl reaction volume, 50 mM Tris-HCl, pH 7.4) were preincubated for 15 min with varying concentrations of CPO, then treated with FP-biotin (2 μM final; 1 μl of a 25× stock dissolved in DMSO) for 1 h at room temperature. Reactions were quenched by adding 10 μL of 6× SDS-PAGE loading buffer (reducing) and heating the samples for 5 min (95°C). Biotinylated proteins were resolved by SDS-PAGE (10%), transferred to a PVDF membrane, and detected with avidin-HRP as described in detail in [26]. Densitometry of bands was used to estimate IC50 values.
2.6. Reactivation of oxon-inhibited KIAA1363 activity
KIAA-transfected COS7 cell lysate (1 mg/ml protein, 50 μl reaction volume, 50 mM Tris-HCl, pH 7.4) was pre-incubated with CPO (1 μM final concentration; ethanol vehicle, 2% v/v) for 15 min at 37°C. A parallel control reaction containing KIAA-transfected COS7 cell lysate was treated with ethanol (2% v/v). The reactions were then diluted 1:100 (v/v) with 50 mM Tris-HCl (pH 7.4) buffer and incubated for an additional 60 min. Aliquots were removed every 10 min to assay the residual 4-MUBA hydrolysis activity.
2.7. Statistical Analysis
SigmaStat was used to perform Student’s t-test and linear regression analysis.
3. Results
3.1. High-throughput biochemical assay of KIAA1363 activity
Overexpression of KIAA1363 protein in COS-7 cells was verified by serine hydrolase activity profiling (Fig. 1A,B) and enzyme activity assays (Fig. 1C). Following reaction with FP-biotin, KIAA1363-transfected COS-7 cell lysate exhibited a doublet band on the blot (~45–50 kDa), which was absent when the lysate was denatured prior to adding FP-biotin (Fig. 1A). The ester-containing reporter substrates 4-MUBA and pNPV were hydrolyzed by KIAA1363; 4-MUBA was cleaved ~5× faster than pNPV (Fig. 1C). Because the putative physiological substrates for KIAA1363 (AcMAGE and cholesteryl esters) do not lend themselves to a high-throughput assay format, we designed a 96-well spectrofluorometric plate assay using the substrate 4-MUBA to evaluate the potency of inhibition of KIAA1363 by OP oxons, as measured by IC50s (Fig. 2).
Fig. 1. Expression of recombinant human KIAA1363 in COS7 cells.

(A) COS7 cells were mock-transfected or transfected with plasmid containing human KIAA1363 cDNA. After cell lysis, serine hydrolases were labeled with the activity probe FP-biotin (2 μM, 1 h, room temperature), followed by SDS-PAGE and avidin-HRP blotting to detect biotinylated proteins. KIAA1363 doublet bands are indicated. * indicates an endogenous biotinylated protein in COS7 cell lysates. Negative control reactions included cell lysates that were preheated (pre-heat) before addition of FP-biotin. (B) Concentration dependence of the FP-biotin reaction with KIAA1363-transfected COS7 cell lysates. The indicated concentration of FP-biotin was incubated with cell lysate for 1 h at room temperature. * indicates an endogenous biotinylated protein in COS7 cell lysates. (C) Hydrolysis activity of KIAA1363-(KIAA1363) and mock-transfected (control) COS7 cell lysates using the substrates pNPV and 4-MUBA. Data represent mean ± standard deviation (n=3). *, p<0.05, KIAA1363 versus control, Student’s t-test.
Fig. 2. Format to measure KIAA1363 or CES1 activities using a high-throughput fluorescence assay.

(A) Inhibitor (oxon, variable concentrations) and substrate (4-MUBA, 250 μM final concentration) were added to each well of a 96-well plate and pre-warmed for 5 min (37°C), followed by addition of the serine hydrolase (KIAA1363- or CES1-transfected or mock transfected cell lysate) to initiate the hydrolytic reaction. (B) Progress of the hydrolysis reaction catalyzed by recombinant serine hydrolase was monitored by fluorescence (F). (C) Each progress curve represents the corrected F, as indicated by the red solid curve (no oxon) and red dashed curve (1 nM oxon), and was monitored for 45 min. Values from triplicate wells were averaged and the mock-lysate wells were subtracted from the hydrolase-lysate wells. The rate of reaction between oxon and enzyme is slowed by the presence of 4-MUBA, thereby enabling kobs (i.e., first-order rates of enzyme inactivation) to be obtained by fitting the progress curves to Eqns. 1 or 2.
3.2. Inhibitor potency: IC50 determination
IC50s for CPO and PO toward KIAA1363 were compared with those for CES1, a related serine hydrolase that was also expressed in COS-7 cells. Two approaches were used to estimate the IC50 values of the oxons. First, the spectrofluorometric method was used to evaluate the inhibition of either KIAA1363- or CES1-transfected cell lysates by the oxons following either a 0 or 15 min pre-incubation period prior to the addition of substrate. The 0 min pre-incubation indicates that the oxon and ester substrate (4-MUBA) see the enzyme at the same time (i.e., simultaneously), whereas the 15 min pre-incubation indicates that the enzyme is treated with oxon for 15 min prior to addition of 4-MUBA. The oxons potently inhibited KIAA1363 activity with IC50 values in the low nanomolar range (Fig. 3). On the basis of IC50s, CPO was a more potent inhibitor than PO of KIAA1363 and CES1. The IC50 values obtained following a 15 min incubation of enzyme and inhibitor were lower than IC50 values obtained following the 0 min incubation (Fig. 3), which is consistent with a time-dependent covalent reaction between the catalytic serine residue of KIAA1363 and oxon. Moreover, CES1 was slightly more sensitive than KIAA1363 to the inhibitory effects of both oxons. In contrast to CPO and PO, MPO was a relatively poor inhibitor of KIAA1363 (IC50 > 100 nM) and was not further studied.
Fig. 3. Potency of inhibition of CES1 and KIAA1363 by CPO and PO determined by fluorescence assay.

IC50 values were determined with (+) or without (−) a pre-incubation period with oxon (37°C, 15 min) prior to the addition of substrate (4-MUBA). Data represent the means ± standard deviation of 2–3 independent experiments. Data are presented in both graphical (left) and tabular (right) forms.
In the second approach, KIAA1363- or CES1-transfected cell lysates were treated with CPO for 15 min, followed by addition of the activity probe FP-biotin (Fig. 4). Using the competitive activity-based protein profiling (ABPP) assay, somewhat higher IC50 values were obtained compared with the spectrofluorometric method (Fig. 3). Nevertheless, both approaches indicated that KIAA1363 was potently inhibited by nanomolar concentrations of CPO.
Fig. 4. Potency of inhibition of KIAA1363 and CES1 by CPO determined using the serine hydrolase activity-based probe, FP-biotin.

(A) Scheme describing the strategy to characterize CPO-mediated inactivation of KIAA1363 (and CES1) by activity-based protein profiling (ABPP). The activity probe, FP-biotin, is shown in simplified form with a reactive warhead (black circle) attached to a biotin (B) tag (green star). Recombinant CES1 (B) and KIAA1363 (C) proteins were incubated with the indicated concentrations of CPO for 15 min (37°C), followed by addition of FP-biotin (2 μM, 1 h, room temperature). After separation of proteins by SDS-PAGE and avidin-HRP blotting, the intensity of the biotin-labeled CES1 and KIAA1363 proteins were quantified and plotted versus CPO concentration. IC50 values were interpolated from the curves and are indicated on the graphs. Data are representative of two independent experiments.
3.3. Corrected bimolecular rate constants (kinact/Ki) of KIAA1363 inhibition by CPO and PO
The corrected bimolecular rate constants were determined for the oxons and KIAA1363. Using the approach outlined by Crow et al. [7], the bimolecular rate constant for either CPO or PO and KIAA1363 was determined by titrating KIAA1363-transfected COS-7 cell lysate with oxon in the presence of 4-MUBA. The fluorescence of 4-methylumbelliferone produced by the hydrolysis of 4-MUBA in mock-transfected COS-7 cell lysates was determined in parallel on the same multi-well plate and subtracted from the KIAA1363 lysate-catalyzed reactions (Fig. 2A).
The kinetic scheme describing the inhibition of a serine hydrolase by oxon, which can undergo reactivation, in the presence of an ester-containing reporter substrate is shown in Fig. 5A. Progress curves for KIAA1363-catalyzed 4-MUBA hydrolysis in the presence of increasing concentrations of CPO are shown in Fig. 5B. Each progress curve was fit to an equation that describes the inactivation and spontaneous reactivation of the enzyme (Eqn. 2) to obtain the apparent first-order rate constant of inactivation (kobs). kobs values determined at each CPO concentration were corrected by subtracting the apparent kobs determined in the absence of inhibitor, which represents the progressive inactivation of enzyme activity in the absence of oxon (possibly due to slight denaturation of KIAA1363 over the 45 min duration of the assay). This correction yields the kobs (corrected) values.
Values for kobs (corrected) were plotted against the appropriate concentration of oxon and fit to Eqn. 3 to obtain kinact and Ki (Fig. 6A), and the corrected bimolecular rate constants (kinact/Ki) for the reaction of KIAA1363 with either CPO or PO could be calculated. The kinetic values are reported in Table 1. On the other hand, data obtained with CES1 and oxon inhibitors did not fit a hyperbolic equation; instead, a linear equation was used to fit the data (Fig. 6B; data for CPO shown). The bimolecular rate constants for the oxons and CES1 are also reported in Table 1. Although the kinact/Ki for CPO/CES1 overexpressed in COS7 cells (6.7 × 106 s−1 M−1) is slightly lower than that previously reported for CPO and purified CES1 [7], this is not surprising given that our current data was obtained using CES1 protein in a crude cell lysate. We used CES1 in a crude lysate to compare results directly with KIAA1363, because it was also in a crude lysate (i.e., not purified).
Fig. 6. Concentration-dependent inactivation of either KIAA1363 or CES1 by CPO.

The observed first-order inhibition rate constant (kobs) was plotted against CPO concentration. For KIAA1363 (A), data were fitted with Eqn. (3): kinact = 0.0062 s−1; Ki = 0.76 nM. For CES1 (B), data were fitted with Eqn. (3), assuming that Ki (CPO) ≫ [I]: kinact/Ki = 2.5 × 106 s−1M−1. The graphs are representative of the type of data used to determine the kinetic parameters.
Table 1.
Corrected Kinetic Constants for Reactions of KIAA1363 and CES1 with CPO or PO
| Kinetic parameters a | |||||
|---|---|---|---|---|---|
| Enzyme | Oxon | kinact (s−1) | Ki c (M) | kinact/Ki (s−1M−1) | kreact (s−1) |
| KIAA1363 | CPO | 0.0069 ± 0.0007 | 1.6 × 10−9 | 4.4(±0.4) × 106 | 0.00043 ± 0.00019 |
| CES1 | CPO | — b | — b | 6.7(±4.2) × 106 | — b |
| KIAA1363 | PO | 0.00092 | 2.9 × 10−9 | 0.4 × 106 | 0.0003 |
| CES1 | PO | — b | — b | 1.5 × 106 | — b |
Values are means ± SD of 2–4 independent experiments, each with triplicate reactions for each oxon concentration.
Not determined because curves were linear, not hyperbolic (see Fig. 6B for CPO data).
Corrected Ki = Ki′/(1 + [S]/Km). [S] = 250 μM; Km4-MUBA = 227 μM
In addition to determining kinact/Ki, fitting the progress curves for CPO-inhibited KIAA1363 activity to Eqn. 2 permitted estimates of the steady-state velocity (vss) to be obtained. The various values of vss were then plotted against the corresponding CPO concentration (I) and the data fit to Eqn. 5 to obtain estimates for v0, vmin, and KmI, as described in the Methods section. The reactivation rate constant, kreact, could then be calculated using Eqn. 7. This value is also reported in Table 1. The ratio kinact/kreact (phosphorylation/dephosphorylation) for CPO and KIAA1363 was 16, which indicated that the rate of inactivation of KIAA1363 by CPO was 16× faster than the rate of reactivation.
As shown in Fig. 7, differences in the shapes of the inhibition progress curves for KIAA1363 and CES1 in the presence of a common oxon (paraoxon) were apparent. It is known that CES1 is essentially irreversibly inactivated by paraoxon in the time frame considered here [7], which is exemplified by the time-dependent decrease in the slopes (deceleration) for each curve seen in Fig. 7A. In contrast to CES1, the slope of the reaction curves for KIAA1363 do not approach zero but instead have a more linear appearance with a positive slope (Fig. 7B), even at high PO concentrations (100 nM), suggesting a steady-state turnover of the oxon by KIA1363. Thus, a constant (steady state) amount of free enzyme remains available to catalyze the hydrolysis of 4-MUBA even at high PO concentrations, unlike what is seen for CES1 which is inactivated. Nevertheless, we estimated the corrected bimolecular rate constant for the reaction between PO and KIAA1363 (Table 1). The kinact for PO was 7.5-fold lower than kinact for CPO (Table 1), indicating CPO is more potent than PO at inhibiting KIAA1363. In addition, by analyzing the data using Eqns. 5 and 6, the ratio kinact/kreact for the reaction between PO and KIAA1363 was calculated to be 3.4, whereas (as mentioned above) this ratio was 16 for the reaction of CPO with KIAA1363. Using the empirical kinact value for the PO and KIAA1363 reaction (Table 1), the kreact for the reaction was calculated to be 0.0003 s−1 (kreact = kinact/3.4). Because both CPO and PO produce the same modification on the active-site serine residue (diethylphosphorylation) in KIAA1363, then the “true” kreact must be identical in each case. The calculated kreact values obtained for CPO-KIAA1363 (0.0004 s−1) and PO-KIAA1363 (0.0003 s−1) are reasonably similar to one another, which is consistent with this premise.
Fig. 7. Effect of the substrate on the progressive inhibition of CES1 and KIAA1363 by oxon.

Inhibition of CES1- (A) and KIAA1363-catalyzed (B) hydrolytic activity by varying concentrations of PO. The progress of the 4-MUBA hydrolysis reaction was followed by measuring fluorescence for 45 min.
Attempts to directly measure the rate of KIAA1363 reactivation (kreact), as was previously done for CES1 by treating the KIAA1363 with excess oxon followed by removal of the unreacted oxon using a desalting spin filter [7], were stymied by the fact that control KIAA1363 activity was lost during the desalting process suggesting either enzyme instability or adsorption to the spin filter. Therefore, an alternative approach was used: KIAA1363 was treated with excess CPO (1 μM) or vehicle (control) for 15 min, then the reaction mixtures were immediately diluted 100-fold and incubated for up to 60 min (at 37°C). Aliquots were removed every 10 min and KIAA1363 activity determined using the substrate 4-MUBA. The kreact value determined by this approach was 0.00038 s−1 (Fig. 8). This result supported the notion that KIAA1363 can be reactivated (t1/2 of reactivation = 30.4 min) following its inhibition by oxons. It also validated the calculated kreact in Table 1. It should be noted, however, that this approach does not remove unreacted oxon; although the oxon is diluted by a factor of 100, it is possible that it will re-inactivate KIAA1363. Further, the substantial dilution of enzyme also causes a technical issue to arise with regard to the ability to measure its activity (i.e., the signal-to-noise ratio of the spectrofluorometric method is reduced). These are two technical limitations of this method and why more emphasis should be given to the calculated kreact values in Table 1.
Fig. 8. Reactivation of CPO-inhibited KIAA1363.

KIAA-transfected COS7 cell lysate (1 mg/ml protein) was pre-incubated with CPO (1 μM final concentration) for 15 min at 37°C. A parallel control reaction containing KIAA-transfected COS7 cell lysate was treated with ethanol vehicle. Reactions were then diluted 1:100 (v/v) with 50 mM Tris-HCl (pH 7.4) and aliquots removed at the indicated times to assay the residual 4-MUBA hydrolysis activity for a total incubation time of 60 min.
4. Discussion
A possible limitation of obtaining kinetic parameters for a specific reaction in cell lysates is that the measured values, i.e. Vmax for a particular enzyme, will be the sum of activities for all isoenzymes of that particular enzyme. This is a major issue for drug metabolizing enzymes such as cytochrome P450s (CYPs) that have multiple isozymes that catalyze the conversion of a specific substrate to a particular product. Therefore, using isolated enzymes for the CYPs is important for determining the intrinsic kinetic parameters for each isoform. However, in the case of KIAA1363, there are no other known isozymes for this enzyme. In addition, on the basis of the gel-based ABPP profiling (Figure 1), we have verified that the native COS7 cells (the recipient cells of the KIAA1363-containing plasmid) do not express KIAA1363. Thus, our “background” cell lysate yields low level non-KIAA1363-derived signals that can be subtracted from the KIAA1363-transfected cell lysate. As shown in Figure 2, the intrinsic hydrolysis activity in the control (“background”) cell lysates was subtracted from the hydrolysis activity in the KIAA1363-transfected cell lysates. Thus, the corrected hydrolysis activity we measured using the substrate 4-MUBA can be attributed to the activity of KIAA1363. It should be noted that other studies of esterase inhibition by organophosphorus compounds have been done in crude tissue lysates. For example, Estevez et al. used the cytosolic (soluble) fraction from either chicken brain or sciatic nerve to examine the inhibition of neuropathy target esterase (NTE) by paraoxon [27]. Ki and IC50 values were derived using this crude mixture of enzymes. In addition, Quistad et al. used a variety of sources, including bovine pancreas, rat serum, and bovine milk, to study the inhibition of several esterases by OP poisons [28]. Thus, our approach using an enzyme that was overexpressed in cell lysates was appropriately controlled by the use of control cell lysates and has precedence in the literature.
Pharmacokinetic modeling of OP insecticides requires kinetic characterization of the interaction of the OP with enzymes responsible for its metabolism. Here, we determined the potencies and rate constants of CPO and PO with respect to their ability to inhibit KIAA1363, which is one of the most abundant serine hydrolases found in mouse brain and an important detoxication enzyme of OPs [10]. We found that KIAA1363 was highly sensitive to inhibition by the oxons, exhibiting IC50s between 1–17 nM, depending on the experimental approach used. These data are in line with IC50s previously reported for KIAA1363 and oxons when using mouse brain membranes and 1 nM [3H]-CPO to radiolabel serine hydrolases [10]. In addition, we determined the corrected bimolecular rate constants for the reactions of the oxons with both KIAA1363 and CES1, which is another detoxification enzyme. The corrected kinact/Ki for CPO and KIAA1363 was ~10-fold higher than for PO and KIAA1363, whereas the bimolecular rate constants for the reaction of CPO with either KIAA1363 or CES1 were similar to each other (Table 1). These results indicated that KIAA1363 and CES1 are both equally sensitive to CPO. Although kinetic parameters for the interaction of CPO with CES1 have been reported [7], they have not been for CPO and KIAA1363. Assuming that kcat ≈ kreact for the catalytic turnover of CPO by KIAA1363, i.e., the rate limiting step is the reactivation of the inhibited enzyme, which is a reasonable assumption for esterase-oxons because typically kreact ≪ kinact for this type of reaction, and KmCPO = 4.7 nM (which was obtained by fitting data to Eqn. 5), the apparent catalytic efficiency (kcat/Km) of CPO turnover by KIAA1363 was calculated to be 9.1 × 104 M−1s−1. Interestingly, the catalytic efficiency for CPO hydrolysis by KIAA1363 is similar to human paraoxonase (PON1), an important enzyme that detoxifies CPO [29]. For example, catalytic efficiencies for the hydrolysis of CPO by recombinant PON1Q192 and PON1R192 isoforms were 9 × 104 M−1s−1 and 15 × 104 M−1 s−1, respectively [30]. Whereas PON1 is abundantly expressed in liver and blood, KIAA1363 is not [9], thus KIAA1363 will not impact the pharmacokinetics of oxons to the extent that PON1 does [30]. Therefore, it can be concluded that PON1 (and probably CES1 due to its abundant levels in liver) is a more important factor in overall CPO metabolism in vivo than KIAA1363. Nevertheless, on the basis of its robust expression in brain and sensitivity to oxons, it is likely that KIAA1363 has an important protective function in this organ [10]. Our results lend further support to this notion.
Previous work [10] indicated that maximal radiolabeling of KIAA1363 in mouse brain membranes by [3H]-CPO (1 nM) occurred at 5 min; 48 mol % of the added CPO was covalently bound to KIAA1363 [4 mol % was present as the monoethylphosphoryl adduct (“aged” adduct), while 44 mol % was present as the diethylphosphoryl adduct (non-aged adduct)]. By 60 min, the amount of covalent phosphoryl adduct on KIAA1363 was reduced to 25 mol % of the added CPO [12 mol % was present as the monoethylphosphoryl adduct, while 13 mol % was present as the diethylphosphoryl adduct]. The remaining CPO (75 mol %) had been completely hydrolyzed by KIAA1363 to trichloropyridinol and diethylphosphate products. Because “aging” of the diethylphosphoryl adduct accounted for only a small fraction (12 mol %) of the overall metabolism of 1 nM CPO by KIAA1363 during the 60 min incubation period, this reaction was not incorporated into our kinetic analysis. This assumption considerably simplified the equations needed to model covalent inhibition and spontaneous reactivation of KIAA1363, as has been discussed previously for esterases in general [31]. Furthermore, it should be noted that in another study [9], “aging” of the CPO-modified KIAA1363 was not apparent, because CPO was hydrolyzed completely by KIAA1363 during the 60 min incubation. This provided further justification for not including the aging step in the kinetic equations.
KIAA1363 is an integral membrane protein in the endoplasmic reticulum [18]. It appears to be a lipase with acetyl monoacylglycerol ethers (AcMAGEs) and cholesteryl esters as putative substrates [12, 32]. Inhibition of KIAA1363 in macrophages has been shown to lower production of pro-inflammatory cytokines through changes in key ether lipid metabolism pathways [33]. It should also be noted that other studies did not observe a cholesteryl esterase activity associated with KIAA1363 [14]. We also tested the ability of KIAA1363 to hydrolyze both cholesteryl oleate and 4-methylumbelliferyl oleate, but could not detect any activity with either substrate (data not shown). More work is required to identify the endogenous substrates of KIAA1363.
The bimolecular rate constant, which is the best metric for quantifying the potency of inhibition of enzymes by covalent inhibitors, for the reaction of human KIAA1363-transfected cell lysate with CPO was 4.4 × 106 M−1s−1 (Table 1). This value is similar to the bimolecular rate constant for human CES1-transfected cell lysate and CPO (6.7 × 106 M−1s−1), and indicates that KIAA1363 is highly susceptible to covalent inactivation by oxons. Similar conclusions can be made for PO and KIAA1363. Therefore, KIAA1363 is very efficient at detoxifying oxons, and is likely an important enzyme that protects against OP poisoning in humans, particularly in the brain because of its high expression. Furthermore, our data indicated that the KIAA1363-diethylphosphate protein is reactivated much faster than the CES1-diethylphosphate protein (t1/2 = 0.5 h for KIAA1363 versus t1/2 = 31 h for CES1 [8]). Thus, although carboxylesterases can reactivate following inactivation by select OPs [34, 35], KIAA1363 appears to be a more efficient OP hydrolase.
Although liver is a notable exception, KIAA1363 is widely distributed in several tissues including the brain [9]. The developmental expression (or ontogeny) of KIAA1363 in tissues is unknown. Whether changes in sensitivity to OP insecticides during development correlates with the expression levels of KIAA1363 is an unanswered but intriguing question. To our knowledge PBPK/PD models of OP metabolism in rodents and humans do not incorporate the metabolism of OPs by KIAA1363, which could be an important pathway of detoxication in tissues such as the brain. Given the high sensitivity of KIAA1363 to oxons, incorporating this pathway into PBPK/PD models that evaluate the toxicokinetics of oxons seems warranted. Thus, characterizing the interactions between oxons and the enzymes, such as KIAA1363, that detoxify these bioactive molecules is a useful addition to the knowledgebase. The kinetic parameters that were determined here should help the development of more sophisticated PBPK models, which take into account detoxication reactions occurring in sensitive target tissues, such as brain. The bimolecular rate constants and IC50s that we determined for CPO and PO with human KIAA1363 should prove useful in these modeling efforts.
Highlights.
Half-maximal inhibitory concentrations (IC50s) and the apparent bimolecular rate constant (kinact/Ki) for reactions of chlorpyrifos oxon and paraoxon with the serine hydrolase KIAA1363 were determined.
Human KIAA1363 was potently inhibited by both chlorpyrifos oxon and paraoxon, with IC50s in the nM range.
The bimolecular rate constants for the reactions of CPO and PO with human KIAA1363 should prove useful in PBPK/PD modeling efforts.
Acknowledgments
Research support was provided by NIH 1R15ES015348-01A1, NIH 1R15ES015348-02, 3R15ES015348-01A1S1, and 3R15ES015348-01A1S2. Kim Pluta was supported by NIH T35RR007071 (Summer Research Experience program for veterinary students). We also gratefully acknowledge the assistance of Shuqi Xie.
List of Abbreviations
- AcMAGE
acetyl monoacylglycerol ether
- CES1
carboxylesterase 1
- CES2
carboxylesterase 2
- CPO
chlorpyrifos oxon
- MGL
monoglyceride lipase
- MPO
methyl paraoxon
- 4-MUBA
4-methylumbelliferyl acetate
- pNPV
para-nitrophenyl valerate
- OPs
organophosphorus insecticides
- PO
paraoxon
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kiely T, Donaldson D, Grube A. Pesticide industry sales and usage - 2000 and 2001 market estimates. U.S. Environmental Protection Agency; Washington D.C: 2004. [Google Scholar]
- 2.Aldridge WN. Mechanisms and Concepts in Toxicology. Taylor and Francis; London: 1996. pp. 86–90. [Google Scholar]
- 3.Ebichon DJ. In: Casarett and Doull’s Toxicology. Klaassen CD, editor. McGraw-Hill; New York: 1996. pp. 643–698. [Google Scholar]
- 4.Mileson BE, Chambers JE, Chen WL, Dettbarn W, Ehrich M, Eldefrawi AT, Gaylor DW, Hamernik K, Hodgson E, Karczmar AG, Padilla S, Pope CN, Richardson RJ, Saunders DR, Sheets LP, Sultatos LG, Wallace KB. Toxicol Sci. 1998;41:8–20. doi: 10.1006/toxs.1997.2431. [DOI] [PubMed] [Google Scholar]
- 5.Aldridge WN. Biochem J. 1953;53:110–117. doi: 10.1042/bj0530110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxwell DM. Organophosphates. 1992:183–199. [Google Scholar]
- 7.Crow JA, Bittles V, Herring KL, Borazjani A, Potter PM, Ross MK. Toxicol Appl Pharmacol. 2012;258:145–150. doi: 10.1016/j.taap.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crow JA, Bittles V, Borazjani A, Potter PM, Ross MK. Biochemical pharmacology. 2012;84:1215–1222. doi: 10.1016/j.bcp.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nomura DK, Durkin KA, Chiang KP, Quistad GB, Cravatt BF, Casida JE. Chemical research in toxicology. 2006;19:1142–1150. doi: 10.1021/tx060117m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nomura DK, Leung D, Chiang KP, Quistad GB, Cravatt BF, Casida JE. Proc Natl Acad Sci U S A. 2005;102:6195–6200. doi: 10.1073/pnas.0501915102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nomura DK, Dix MM, Cravatt BF. Nature reviews Cancer. 2010;10:630–638. doi: 10.1038/nrc2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang KP, Niessen S, Saghatelian A, Cravatt BF. Chemistry & biology. 2006;13:1041–1050. doi: 10.1016/j.chembiol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Sekiya M, Osuga J, Igarashi M, Okazaki H, Ishibashi S. J Atheroscler Thromb. 2011;18:359–364. doi: 10.5551/jat.7013. [DOI] [PubMed] [Google Scholar]
- 14.Buchebner M, Pfeifer T, Rathke N, Chandak PG, Lass A, Schreiber R, Kratzer A, Zimmermann R, Sattler W, Koefeler H, Frohlich E, Kostner GM, Birner-Gruenberger R, Chiang KP, Haemmerle G, Zechner R, Levak-Frank S, Cravatt B, Kratky D. Journal of lipid research. 2010;51:2896–2908. doi: 10.1194/jlr.M004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh S. Circ Res. 2011;108:e6–7. doi: 10.1161/CIRCRESAHA.110.239137. author reply e8–e9. [DOI] [PubMed] [Google Scholar]
- 16.Nomura DK, Fujioka K, Issa RS, Ward AM, Cravatt BF, Casida JE. Toxicol Appl Pharmacol. 2008;228:42–48. doi: 10.1016/j.taap.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timchalk C, Nolan RJ, Mendrala AL, Dittenber DA, Brzak KA, Mattsson JL. Toxicological sciences : an official journal of the Society of Toxicology. 2002;66:34–53. doi: 10.1093/toxsci/66.1.34. [DOI] [PubMed] [Google Scholar]
- 18.Chang JW, Moellering RE, Cravatt BF. Angewandte Chemie. 2012;51:966–970. doi: 10.1002/anie.201107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prescher JA, Bertozzi CR. Nature chemical biology. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 20.Lee I, Berdis AJ. Biochim Biophys Acta. 2015 [Google Scholar]
- 21.Chambers H, Brown B, Chambers JE. Pesticide Biochemistry and Physiology. 1990;36:308–315. [Google Scholar]
- 22.Feaster SR, Quinn DM. Methods in enzymology. 1997;286:231–252. doi: 10.1016/s0076-6879(97)86013-2. [DOI] [PubMed] [Google Scholar]
- 23.Main AR, Dauterman WC. Nature. 1963;198:551–553. [Google Scholar]
- 24.Streit TM, Borazjani A, Lentz SE, Wierdl M, Potter PM, Gwaltney SR, Ross MK. Biological chemistry. 2008;389:149–162. doi: 10.1515/BC.2008.017. [DOI] [PubMed] [Google Scholar]
- 25.Feaster SR, Lee K, Baker N, Hui DY, Quinn DM. Biochemistry. 1996;35:16723–16734. doi: 10.1021/bi961677v. [DOI] [PubMed] [Google Scholar]
- 26.Crow JA, Middleton BL, Borazjani A, Hatfield MJ, Potter PM, Ross MK. Biochim Biophys Acta. 2008;1781:643–654. doi: 10.1016/j.bbalip.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estevez J, Mangas I, Sogorb MA, Vilanova E. Chemico-biological interactions. 2013;203:245–250. doi: 10.1016/j.cbi.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Quistad GB, Liang SN, Fisher KJ, Nomura DK, Casida JE. Toxicol Sci. 2006;91:166–172. doi: 10.1093/toxsci/kfj124. [DOI] [PubMed] [Google Scholar]
- 29.Costa LG, Giordano G, Cole TB, Marsillach J, Furlong CE. Toxicology. 2013;307:115–122. doi: 10.1016/j.tox.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li WF, Costa LG, Richter RJ, Hagen T, Shih DM, Tward A, Lusis AJ, Furlong CE. Pharmacogenetics. 2000;10:767–779. doi: 10.1097/00008571-200012000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Estevez J, Vilanova E. Critical reviews in toxicology. 2009;39:427–448. doi: 10.1080/10408440802412309. [DOI] [PubMed] [Google Scholar]
- 32.Okazaki H, Igarashi M, Nishi M, Sekiya M, Tajima M, Takase S, Takanashi M, Ohta K, Tamura Y, Okazaki S, Yahagi N, Ohashi K, Amemiya-Kudo M, Nakagawa Y, Nagai R, Kadowaki T, Osuga J, Ishibashi S. J Biol Chem. 2008;283:33357–33364. doi: 10.1074/jbc.M802686200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunerdosse DM, Morris PJ, Miyamoto DK, Fisher KJ, Bateman LA, Ghazaleh JR, Zhong S, Nomura DK. ACS chemical biology. 2014;9:2905–2913. doi: 10.1021/cb500717g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hemmert AC, Otto TC, Wierdl M, Edwards CC, Fleming CD, MacDonald M, Cashman JR, Potter PM, Cerasoli DM, Redinbo MR. Molecular pharmacology. 2010;77:508–516. doi: 10.1124/mol.109.062356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maxwell DM, Brecht KM. Journal of applied toxicology : JAT. 2001;21(Suppl 1):S103–107. doi: 10.1002/jat.833. [DOI] [PubMed] [Google Scholar]