

Abstract
Herein, we report the discovery of 2-amino-4-bis(aryloxybenzyl)aminobutanoic acids as novel inhibitors of ASCT2(SLC1A5)-mediated glutamine accumulation in mammalian cells. Focused library development led to two novel ASCT2 inhibitors that exhibit significantly improved potency compared with prior art in C6 (rat) and HEK293 (human) cells. The potency of leads reported here represents a 40-fold improvement over our most potent, previously reported inhibitor and represents, to our knowledge, the most potent pharmacological inhibitors of ASCT2-mediated glutamine accumulation in live cells. These and other compounds in this novel series exhibit tractable chemical properties for further development as potential therapeutic leads.
Keywords: ASCT2, SLC1A5, Glutamine, Cancer, Metabolism
Graphical Abstract
Cancer cells exhibit an altered metabolic profile compared to normal cells. In addition to increased utilization of glucose, they can become highly dependent on the amino acid glutamine. Mammalian cells can regulate glutamine import and export through an evolutionarily redundant series of cell surface transporters. A sodium-dependent transporter of glutamine, ASCT2 (gene symbol SLC1A5), stands out as a major transporter for glutamine uptake making it a promising target for probe development. In addition to being the primary glutamine transporter in cancer, SLC1A5 expression is associated with oncogenic MYC1,2 and KRAS,3,4 suggesting its relevance in many clinically important tumors, including those of the lung, colon, and pancreas.5–7 Fuchs and co-workers demonstrated that SLC1A5 antisense RNA triggered apoptosis in human hepatocellular carcinoma cells.8 Furthermore, Hassanein et al., more recently reported that SLC1A5 was expressed in 95% of squamous cell carcinomas (SCC), 74% of adenocarcinomas (ADC), and 50% of neuroendocrine tumors. In those studies, siRNA down-regulation of ASCT2 in lung cancer cells resulted in significant growth inhibition.9 Collectively, these studies suggest development of small molecules capable of inhibiting ASCT2 activity could be promising as precision cancer medicines.
To date, few compounds that target ASCT2 have been reported and most are derivatives of endogenous ASCT2 substrates. As an early entrant to the field, in 2004 Esslinger and co-workers described a series of glutamine analogs that explored pKa effects on the amide NH bond to probe the ASCT2 amino acid binding site through the addition of electron-donating and electron-withdrawing aryl groups to the terminal amide of glutamine. The best compound, L-γ-glutamyl-p-nitroanilide (GPNA, Compound 1), exhibited modest potency in the low millimolar range and no observations were made regarding steric requirements for binding to ASCT2.10 Our group was able to expand upon this class of inhibitors by exploring the steric requirements for binding to ASCT2 and found that while SAR was flat, 2-substituted glutamylanilides were preferred. We described N-(2-(morpholinomethyl)phenyl)-L-glutamine (Compound 2) as a novel glutamyl-anilide with three-fold improved activity against ASCT2 compared to GPNA.11 In 2011, Albers et al. described a series of ASCT2 inhibitors based on the ASCT2 substrate serine. Using an in silico approach along with experimental validation, they found that side chain aromaticity was required for high-affinity interaction. This lead to the discovery of O-(4-phenylbenzoyl)-L-serine (compound 3), an inhibitor of ASCT2 with an apparent affinity of 30 μM.12 In 2012, Oppedisano et al. identified the first small molecule lacking an amino acid that blocked glutamine uptake in ASCT2 reconstituted in proteoliposomes (compound 4). This series of 1,2,3-dithiazoles likely inhibited uptake of glutamine non-competitively through formation of mixed sulfides at Cys-207 or Cys-210 with the most potent compound exhibiting an IC50 of 3.7 μM (Figure 1).13
Figure 1.

Structures of previously reported ASCT2 inhibitors: GPNA (1) and an improved glutamylanilde (2), A potent inhibitor derived from the ASCT2 substrate serine (3), and a small molecule inhibitor of ASCT2 lacking an amino acid moiety (4).
To continue our efforts towards novel ASCT2 inhibitors, we focused our work around elaborating the glutamylanilide scaffold, particularly the amide linker. After screening multiple iterations of this scaffold, we arrived at a series of substituted 2,4-diaminobutanoic acids. While there was no SAR within a library of 4-N-mono-substituted derivatives, the resultant library, generated via reductive amination, afforded small amounts of 4-N-disubstituted products. We were able to cleanly isolate these byproducts and test their ability to block ASCT2-mediated 3H-glutamine uptake. Surprisingly, these compounds demonstrated up 10 fold better activity against ASCT2 compared with GPNA and had tractable structural elements for further development. While there were many areas on this new scaffold where SAR could be investigated, we chose to focus on the distal aromatic rings and the linker region connecting it (Figure 2).
Figure 2.

Chemical optimization plan for hit compound (S)-2-amino-4-(bis(2-(4-fluorophenoxy)benzyl)amino)butanoic acid via iterative parallel synthesis.
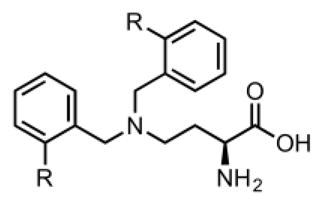
We developed a facile synthetic scheme to yield target 2-amino-4-bis(aryloxybenzyl)aminobutanoic acids. (Figure 3). This synthesis required just two steps starting from N-Boc-L-2,4-diaminobutyric acid tert-butyl ester hydrochloride and afforded products in overall yields ranging from 52–75%. Similar to the initial lead compound, libraries focused on 2-amino-4-bis(aryloxybenzyl)aminobutanoic acids with 2 identical functional groups on the 4-N position.
Figure 3.

Two-step synthesis of 2-amino-4-bis(aryloxybenzyl)-aminobutanoic acids. Reagents and conditions: a) RCHO, NaBH(OAc)3, CH2Cl2, RT, 12 h. b) HCl, dioxane, 40 °C, 5h.
Screening of the synthesized amino acids 5–11 (Table 1), in both C6 (rat) and HEK293 (human) cell lines revealed that compounds 6, 7, and 11 displayed roughly equal potencies in both cells lines.14 Interestingly, other compounds in this series, (Table 1, Compounds 8, 10) demonstrated some preference for blocking glutamine transport in the rat cell line. Our most potent compound in the rat cell line (Table 1, Compound 5) also exhibited this preference and blocked ASCT2-mediated glutamine uptake in C6 cells with an IC50 of 1.3 μM.
Table 1.
Structures and activities of phenoxybenzyl analogues. All IC50 values are reported as the mean of at least 3 biological replicates.
| |||||
|---|---|---|---|---|---|
| Compound | R= | IC50(rat) | SEM | IC50(human) | SEM |
| 5 |
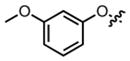
|
1.3 μM | ± 0.7 μM | 57.2 μM | ± 20.8 μM |
| 6 |
|
8.7 μM | ± 0.5 μM | 11.9 μM | ± 0.4 μM |
| 7 |
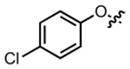
|
24.3 μM | ± 2.8 μM | 26.0 μM | ± 4.0 μM |
| 8 |
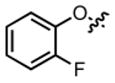
|
1.8 μM | ± 0.6 μM | 33.8 μM | ± 7.2 μM |
| 9 |
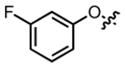
|
36.3 μM | ± 10.9 μM | 11.5 μM | ± 2.2 μM |
| 10 |
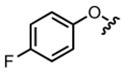
|
10.5 μM | ± 2.7 μM | 141.7 μM | ± 14.2 μM |
| 11 |
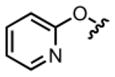
|
68.2 μM | ± 10.3 μM | 59.6 μM | ± 4.9 μM |
To further investigate the SAR of this scaffold, we prepared a library of 2-amino-4-bis(benzyloxybenzyl)aminobutanoic acids with various substitutions around the distal aromatic ring. Overall, we found that this addition of a rotatable bond between the two aromatic groups improved activity against glutamine uptake in both C6 and HEK293 cells. Our two most potent compounds (Table 2, Compounds 12, 19) blocked ASCT2- mediated glutamine uptake with potencies of 5.1 and 3.3 μM in C6 cells and 7.2 and 7.9 μM in HEK293 cells respectively. One compound in the benzyloxy series (Table 2, Compound 18) demonstrated significant preference for inhibiting glutamine uptake in the human form of ASCT2 (HEK293) over the rat form (C6). Full concentration response curves for the most potent compounds in each form of ASCT2 are shown in figure 4.
Table 2.
Structures and activities of benzyloxybenzyl analogues. All IC50 values are reported as the mean of at least 3 biological replicates.
|
| |||||
|---|---|---|---|---|---|
| Compound | R= | IC50(rat) | SEM | IC50(human) | SEM |
| 12 |
|
5.1 μM | ± 1.0 μM | 7.2 μM | ± 0.5 μM |
| 13 |
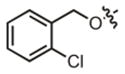
|
18.4 μM | ± 3.3 μM | 40.1 μM | ± 4.8 μM |
| 14 |
|
12.7 μM | ± 1.6 μM | 50.8 μM | ± 16.1 μM |
| 15 |
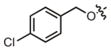
|
20.1 μM | ± 3.8 μM | 33.7 μM | ± 9.7 μM |
| 16 |
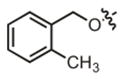
|
41.7 μM | ± 5.0 μM | 25.6 μM | ± 2.0 μM |
| 17 |
|
9.0 μM | ± 3.3 μM | 9.6 μM | ± 0.1 μM |
| 18 |
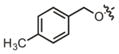
|
86.4 μM | ± 13.0 μM | 17.5 μM | ± 3.0 μM |
| 19 |
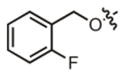
|
3.3 μM | ± 1.4 μM | 7.9 μM | ± 1.9 μM |
| 20 |
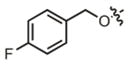
|
11.2 μM | ± 2.3 μM | 13.7 μM | ± 1.7 μM |
| 21 |
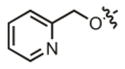
|
8.4 μM | ± 1.1 μM | 48.3 μM | ± 10.8 μM |
| 22 |
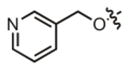
|
17.3 μM | ± 8.5 μM | 69.1 μM | ± 11.5 μM |
| 23 |
|
22.5 μM | ± 5.2 μM | 17.6 μM | ± 3.4 μM |
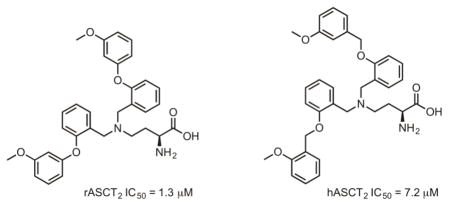
Figure 4.

Structures and activities of the most potent compounds in C6 cells (compound 5) and HEK293 cells (compound 12).
Biologically active compounds were also evaluated in silico in a human ASCT2 model. We employed computational approaches similar to the approach of Albers et al.12 and our previously published work11 to explore potential points of intermolecular interaction and binding pockets accessible to candidate probes. From a homology model based on the open structure of the bacterial aspartate transporter GltPh in complex with the inhibitor D,L-threo-benzyloxyaspartate (TBOA), PDB ID 2NWW, a number of targetable structural motifs were identified including a lipophilic pocket adjacent to the amino acid zwitterion binding site and potential hydrophilic points of contact within a loop region that was displaced by the inhibitor GPNA in the open form of the transporter. The best scoring poses for the most potent compounds identified demonstrated a compatible fit with the human ASCT2 model and interestingly, a tendency to exhibit points of interaction with both the amino acid zwitterion binding sites, particularly Ser353, Ser351, and Asp464, as well as the adjacent hydrophobic pocket, with possible pi-stacking and hydrogen bonding interactions with Trp461 (Figure 5).
Figure 5.

Docking of the most potent compound (Table 2, entry 1) into ASCT2 homology model. The compound 5 (colored sticks) fits the homology model (blue surface) generated for the inhibited form of human ASCT2 and is consistent with the displacement of a key loop region (right).
In summary, we report a novel series of 2-amino-4-bis(aryl-oxybenzyl)aminobutanoic acids as inhibitors of cellular glutamine uptake via ASCT2. Members of this series represent some of the most potent inhibitors of ASCT2-mediated glutamine uptake reported to date. Evaluation of the chemical series within the context of ligand docking to a homology model of human ASCT2 revealed reasonable compatibility with the ASCT2 binding site based on SurflexDock Total Scores. Based upon our data, we anticipate that compounds with the greatest potency may interact with multiple structural elements within the ASCT2 binding site, including the amino acid zwitterion binding site and the adjacent hydrophobic pocket. Efforts to further investigate SAR and optimize binding as well as selectivity within this scaffold are ongoing and advances from these studies will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (5P50-CA95103, 5P30-DK058404), the Kleburg Foundation, the Vanderbilt Center for Molecular Probes (VCMP), and the VICC Thoracic Oncology Program. The authors gratefully acknowledge Sam A. Saleh for assisting with the chemical synthesis. Ping Zhao and Allie Fu are acknowledged for assistance with compound screening.
Footnotes
Dedicated to the memory of Eric S. Dawson, Ph.D
Supplementary material (synthetic procedures, screening methods, and docking techniques) may be found online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Dang CV. Cancer Res. 2010;70:859. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dang CV, Le A, Gao P. Clin Cancer Res. 2009;15:6479. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Iinuma H, Ikeuchi H, Eshima K. Eur J Cancer. 2011;47:1946. doi: 10.1016/j.ejca.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 4.Gaglio D, Metallo CM, Gameiro PA, et al. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu K, Kaira K, Yomizawa Y, Sunaga N, Kawashima O, Oriuchi N, Tominaga H, Nagamori S, Kanai Y, Yamada M, Oyama T, Takeyoshi I. Br J Cancer. 2014;110:2030. doi: 10.1038/bjc.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witte D, Ali N, Carlson N, Younes M. Anticancer Res. 2002;22:2555. [PubMed] [Google Scholar]
- 7.Kaira K, Sunrose Y, Arakawa K, Sunaga N, Shimizu K, Tominaga H, Oriuchi N, Nagamori S, Kanai Y, Oyama T, Takeyoshi I. Histopathology. 2015;66:234. doi: 10.1111/his.12464. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs BC, Perez JC, Suetterlin JE, Chaudhry SB, Bode BP. Am J Physiol Gastrointest Liver Physiol. 2004;286:G467. doi: 10.1152/ajpgi.00344.2003. [DOI] [PubMed] [Google Scholar]
- 9.Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, Clark JE, Alborn WE, Eisenberg R, Massion PP. Clin Cancer Res. 2013;19:560. doi: 10.1158/1078-0432.CCR-12-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esslinger CS, Cybulski KA, Rhoderick JF. Bioorg Med Chem. 2005;13:1111. doi: 10.1016/j.bmc.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 11.Schulte ML, Dawson ES, Saleh SA, Cuthbertson ML, Manning HC. Bioorg Med Chem Lett. 2015;25:113. doi: 10.1016/j.bmcl.2014.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albers T, Marsiglia W, Thomas T, Gameiro A, Grewer C. Mol Pharmacol. 2012;81:356. doi: 10.1124/mol.111.075648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oppedisano F, Catto M, Koutentis PA, Nicolotti O, Pochini L, Koyioni M, Introcaso A, Michaelidou SS, Carotti A, Indiveri Toxicol Appl Pharmacol. 2012;265:93–102. doi: 10.1016/j.taap.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Brown JM, Hunihan L, Prack MM, Harden DG, Bronson J, Dzierba CD, Gentles RG, Hendricson A, Krause R, Macor JE, Westphal RS. J Neurochem. 2014;129:275. doi: 10.1111/jnc.12618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.