

Abstract
The mechanical properties of the tumor microenvironment have been increasingly recognized as potent modulators of cell behavior and function. In particular, tissue rigidity is functionally important during tumor progression. In this review, we survey recent advances in our understanding of the role of tissue rigidity in tumor progression and metastasis, the mechanisms by which mechanical cues integrate with biochemical signals from the microenvironment, and the underlying mechanotransduction pathways involved in tumor progression. These findings highlight the importance of understanding and defining cellular mechanotransduction pathways and the breadth of signals derived from the tumor microenvironment that influences tumor progression.
Keywords: Metastasis, EMT, tissue rigidity, matrix stiffness, extracellular matrix
Introduction
The tumor microenvironment presents cancer cells with a diverse set of extracellular cues to potently influence tumor cell behavior and function. Past decades of cancer research have mainly focused on the role of various extracellular and intracellular biochemical signals on cancer cell proliferation, invasion, and metastasis. Increasingly, mechanical properties of the extracellular matrix (ECM) have been recognized as also being functionally important during tumor progression [1, 2]. Recently, tumor mechanics, and tissue rigidity in particular, has emerged as an important factor during tumor progression and metastasis [3]. In the patient setting, increasing tissue rigidity of primary breast tumors correlates with metastatic recurrence and poor patient survival [4–6]. Studies in 3D culture systems and mouse tumor models showed that increased tissue rigidity plays a functional role in driving tumor invasion and malignancy [2, 7, 8]. Understanding the function and mechanisms of mechanical cues in this diverse set of contexts will be critical and may elucidate essential signaling pathways regulating tumor progression. Many critical aspects of tumor mechanosensing remain unanswered. For example, how do disseminated tumor cells react to the diverse biochemical and mechanical properties of the environments they invade? Do the same rules apply in metastatic sites versus the primary tumor?
Regulation of Tumor Dissemination by Matrix Stiffness
Tumor dissemination is a critical step in the initiation of tumor metastasis. Several lines of evidence suggest that increasing matrix stiffness functionally contributes to this process. The presence of a fibrotic focus, a dense cluster of collagen fibers and fibroblasts, in breast tumors is a prognostic marker of distant metastasis and poor survival [9–11]. Besides the biochemical factors from a fibrotic stroma, fibrotic tumor lesions are associated with a 20–50 fold increase in tissue rigidity. In mouse breast tumor models, tumor progression is also characterized by an incremental stiffening of the tissue [7, 12]. One key question in tumor mechanobiology is how mechanical forces generated by the rigid tumor stromal matrix impact tumor progression and metastasis. Attempts to study this issue have often been hampered by the technical difficulty in distinguishing the effect of biochemical and mechanical signals in vivo. Surrogate markers, such as the presence of fibrotic foci or organized collagen fibers, have thus been often utilized. Another significant obstacle is that matrix stiffness is in large part defined by deposition and modification of ECM proteins, further convoluting interpretation of mechanical signals from biochemical ones. In vitro systems utilizing hydrogels and other synthetic matrices to modulate matrix stiffness independently of biochemical cues have yielded new insights into mechanobiology and mechanotransduction.
Landmark studies showed that human mammary epithelial cells form normal ductal acini with intact adherens junctions and underlying basement membrane in the compliant matrix stiffness that mimics normal mammary glands, whereas they present a malignant phenotype characterized by weaker junctions and invasion through basement membrane in the rigid matrix stiffness similar to breast tumors [7, 8]. These cell morphological changes observed in response to increasing mechanical forces resemble a highly conserved cellular program, termed Epithelial-Mesenchymal Transition (EMT). During EMT, cells lose their epithelial characteristics and acquire mesenchymal characteristics, such as adherens junctions and apical-basal polarity, and the ability to migrate [13]. EMT may be defined at the morphological, biochemical, and transcriptional levels, largely due to contextual differences during development and tumor metastasis [14]. In this review, we define EMT as the partial or complete loss of epithelial gate keepers, such as E-cadherin, and the up-regulation of one or more canonical mesenchymal markers, including vimentin, N-cadherin, and fibronectin. During EMT, one or more of the key EMT-inducing transcription factors, including the Twist, Snail, and Zeb families, are induced and/or activated to promote these molecular changes. A number of studies demonstrate that aberrant reactivation of EMT can potently drive tumor cell local invasion and systemic dissemination [15–18]. Increasing matrix stiffness is also shown to regulate TGF-β signaling, a potent inducer of EMT in tumor cells [19]. This suggests two important concepts - first, that matrix stiffness could potentially regulate EMT to facilitate tumor dissemination; and second, that mechanical cues could signal in concert with biochemical signals to regulate EMT.
A recent study provided the first molecular pathway directly linking matrix stiffness to EMT. Twist1, a key EMT-inducing transcription factor, was found to be essential for high matrix stiffness-induced EMT and tumor invasion [20]. Furthermore, matrix stiffness-driven Twist1 mechanotransduction signals, together with TGF-β, can cooperatively induce EMT and tumor invasion. In response to increasing matrix stiffness, Twist1 protein is released from its cytoplasmic anchor G3BP2 to translocate into the nucleus, thus driving the transcriptional program of EMT and tumor invasion. Depletion of G3BP2 promoted breast cancer invasion and metastasis in mice, and reduced expression of G3BP2 in combination with increased matrix stiffness predicts poor survival in breast cancer patients.
Other mechanical cues, including tissue geometry and intracellular forces have also been shown to regulate EMT. TGF- β differentially induces EMT in epithelial cell sheets, with cells at edges of sheets responding more than those in the center. This response is mediated by changes in mechanical stress within the epithelial cell sheet, suggestive of a convergence of mechanical and biochemical signals that regulate this key process [21]. YAP/TAZ, the key transcription co-activator in the Hippo pathway, is regulated by various mechanical cues, including matrix stiffness and cell shape [22]. While not a key regulator of EMT, YAP is implicated in the induction of EMT in several studies [23–25]. These results suggest that multiple distinct mechanotransduction signaling pathways could impinge on the EMT program to drive tumor cell invasion. Indeed, epithelial polarization can also facilitate changes in cell shape through regulation of adherens junctions positioning to define epithelial sheet folding [26]. Adherens junctions can act as cellular mechanosensors, using vinculin as a molecular bridge to impinge on actinomyosin cytoskeleton dynamics [27]. During the EMT event of neural crest formation, E- to N-cadherin switch leads to redistribution of intracellular forces from intercellular junctions to cell-matrix adhesions, thus promoting adherens junction disassembly [28]. Consistent with this notion, tissue tension can drive tumor progression through destabilization of adherens junctions and nuclear translocation of β-catenin [29]. These recent discoveries raise the possibility that tumor cells that have undergone EMT lose adherens junction-mediated mechanotransduction. Thus, disseminated tumor cells may respond to mechanical cues differently until they re-epithelialize. A more complete understanding of adherens junction-mediated mechanotransduction may also shed light into the differences between EMT-dependent and EMT-independent modes of tumor metastasis.
In addition to EMT, collective cell migration is another important cellular machinery driving primary tumor invasion. In contrast to EMT in which tumor cells migrate as single cells, during collective cell migration carcinoma cells can invade as a group of cells while maintaining cell-cell junctions. Leader cells at the forefront of the invasive edge mediate migration through the surrounding ECM and tissue architecture. Recently, mechanical cues have been reported to regulate collective cell migration. In contrast to mechanoregulation of EMT, intercellular forces from leader cells induce actinomyosin contractility within the epithelial sheet to facilitate invasion. [30]. This report corroborates previous findings that ECM molecules including laminin 111 modulate collective cell migration in both normal and malignant breast tissues. In fact, loss of basement membrane integrity and subsequent activation of collagen I-dependent signaling was shown to be sufficient to promote tumor cell migration and dissemination in 3D culture models [31]. Mechanoregulation of collective cell migration likely represents a distinct mechanotransduction pathway, prompting the question of how many tumor mechanosensitive pathways exist. Further investigation into these molecular mechanisms of tumor mechanotransduction will clarify their individual roles and potential interactions between these pathways. Another key question is whether and how collective cell migration and EMT may cooperatively drive tumor dissemination. Both collective cell migration and EMT are critical processes during embryogenesis and germ layer formation. Tensile forces regulate both germ band extension prior to gastrulation and large scale cell movement later during Drosophila embryogenesis [32]. How these pathways interact and how a multitude of mechanical and biochemical signals are parsed to form a coherent cellular response to regulate tumor dissemination clearly requires further investigation.
Detailed characterization of tumor mechanical properties also helps to provide the physiological relevance for a functional role of tissue rigidity in the regulation of tumor dissemination and EMT. Force mapping of human and mouse breast tumors indicates that while benign lesions have markedly increased rigidities compared to normal mammary tissue, invasive tumors have heterogeneous mechanical properties [33]. This may suggest active regulation of ECM modifications and regulation of tissue mechanics. Interestingly, compared to the tumor interior core, the tumor periphery is relatively stiffer [33, 34], consistent with the observation that cells from the invasive front of tumors are prone to undergo EMT prior to dissemination [35]. This raises the enticing possibility that individual tumor cell movement is controlled locally by the mechanical properties of its surrounding niche. Direct testing of the functional roles of mechanical properties of the tumor periphery and inner core in vivo is still required. This notion, however, meshes well with the recent observation that durotaxis, directional cellular migration towards a higher stiffness substrate, is mediated by differential focal adhesion signaling at the single cell scale [36]. Focal adhesions are able to sense minute changes in matrix rigidity, which direct focal adhesion polarization and cell movement. This provides a potential mechanism by which tumor cells may migrate preferentially to the tumor periphery, hence facilitating dissemination. Together, these observations highlight the range of scales that mechanosensing acts at, spanning from the subcellular to tissue level.
ECM Remodeling Mechanisms
Given that increased matrix stiffness can induce tumor cell invasion and dissemination, it is critical to understand when the underlying modifications of tumor ECM occur and by what mechanism. Recent observations support a model in which peripheral tumor stiffening can be mediated by tumor cells themselves. First, it is worth noting that the stiffness of isolated tumor cells may vary during tumor progression. Transformed mammary epithelial cells in PyMT mice are stiffer than nearby normal cells [12]. However, analyses of breast and ovarian cell lines demonstrated that cell with increased metastatic potential were softer and more deformable than less invasive cells [37, 38]. Physical properties of the nucleus also change throughout tumor progression and nuclear rigidity may restrict tumor dissemination [39, 40]. How physical properties are modulated at the cellular and subcellular level and the degree to which these attributes correlate with mechanical properties of the tumor microenvironment remains unclear. These findings raise the possibility that there are distinct mechanical requirements, both inside and outside of tumor cells, during tumor progression.
Second, several studies have suggested that tumor cells that have undergone EMT can in turn modulate mechanical properties of the ECM. One such mechanism is that invasive cells can produce increased contractile forces. Twist1 induces tumor cell invasion through Rac1, which can increase cell tension and tissue rigidity [41]. These findings are consistent with the functional requirement of the Drosophila Twist homolog to generate contractile forces during embryonic development [42]. Analysis of lymph-node negative breast tumors reveals that expression of Twist1 correlates significantly with the expression of many ECM remodeling molecules including collagens, matrix metalloproteinases, and lysyl oxidases. This finding suggests that disseminating tumor cells that have undergone EMT can further remodel the tumor microenvironment [43]. Generation and modification of the ECM by tumor cells that have undergone EMT is consistent with the normal function of mesenchymal cells in connective tissues. Accordingly, highly metastatic breast cancer cells have high expression of lysyl oxidase (LOX), which can be further induced by stromal cells [44, 45]. Fibronectin and other ECM molecules are also upregulated following induction of EMT [46, 47]. EMT can also induce the expression and deposition of fibrillin, the main component of the elastic fiber microfibrils. Loss of syndecan-4 results in reduced microfibrils, but upon TGF-β treatment fibrillin-1 expression is partially rescued, bypassing the requirement for syndecan-4 [48]. Ras-transformed mouse mammary epithelial cells express tenascin-C but only secrete it to the ECM upon undergoing TGF-β-induced EMT [49]. These results suggest that induction of EMT can induce extensive ECM deposition and remodeling processes.
Tumor cells can also hijack normal functions of stromal cells to facilitate ECM remodeling. LOX secreted by cancer-associated fibroblasts (CAFs) can drive matrix stiffening through collagen crosslinking to facilitate tumor dissemination and metastasis [7, 50]. Matrix stiffening can also be mediated by CAFs through caveolin-1 (Cav1) to promote tumor invasion and metastasis [51]. Loss of Cav1 leads to disorganized stromal architecture while expression of Cav1 in CAFs promotes directional migration of tumor cells. Mechanical remodeling of the tumor microenvironment also in turn regulates CAFs, thus feeding forward to maintain CAF abundance and function. Furthermore, conditioned media from tumor cells induced CAF-mediated ECM remodeling, suggesting that tumor cells can activate CAFs via paracrine signals to further stiffen matrix [52]. Consistent with this notion, myofibroblast contraction can induce the release and activation of latent TGF-β from the ECM. This response could generate a positive feedback loop between matrix stiffness and growth factors. Fibroblast polarization in response to increased matrix stiffness activates a network of protein tyrosine kinases to promote fibroblast cell contractility and focal adhesion formation [53]. Consistent with this notion, clinical observation shows that the presence of fibrotic foci and atypical stromal fibroblasts are associated with poor outcome in breast tumors [54]. These studies suggest that increasing matrix stiffness and activation of EMT feed forward to each other to further enhance tumor cell invasion.
Mechanosensing Feedback Mechanisms
While matrix stiffness can drive EMT, a few recent reports suggest that induction of EMT can influence how tumor cells interpret and respond to changes in matrix stiffness. Altered expression of cell surface receptors of various ECM components could be a potential mechanism to regulate the cellular response to matrix stiffness. Integrin expression and dimerization is regulated during tumor cell transformation and EMT, which in turn can modulate tumor cell adhesion and ECM recognition [55]. Integrin signaling feedback mechanisms also play a role in this process. On compliant matrices, integrin attachments are not stabilized and thus focal adhesions are not allowed to mature [56]. Consequently, in low stiffness conditions focal adhesions become destabilized and are disassembled, preventing effective cell spreading and movement. Extracellular mechanical properties similarly affect intracellular strain, providing a distinct regulatory mechanism. Recent work demonstrated that integrins bind directly to the actin binding protein, filamin A (FLNA), and changes in cell strain result in modulation of the FLNA network and release of the FLNA-binding GTPase activating protein (FilGAP) [57, 58]. Regulation of RAC by FilGAP provides a direct mechanism by which cell strain can modulate cell spreading and motility in an integrin-dependent manner. These findings link intracellular strain from the actinomyosin cytoskeletal network to inside-out integrin signaling.
Interestingly, these results raise the possibility that changes in cell shape during EMT may affect the cellular responses to mechanical cues. Supportive of this hypothesis, Twist mediates the generation of cell contractility during embryogenesis, which is required for proper development [42]. Both cellular tension and extrinsic mechanical forces can activate molecular mechanotransduction switches such as integrins [59]. In mesenchymal stem cells, cell adherence and spreading induces a change in vimentin protein folding as shown by a change in the surface availability of cysteines [60]. This suggests that changes in cell shape can induce changes in the molecular fold of intracellular proteins, thus regulating their functions through altering accessibility to their binding sites and/or post-translational modification sites. Mechanical strain can also directly induce integrin conformational changes [61, 62]. Modulation of integrin affinity and avidity by mechanical forces thus is a major venue through which cell shape changes during EMT could directly impact their responses to matrix stiffness.
In addition to integrins, other receptors such as discoidin domain receptors (DDR) play a critical role in sensing changes in the ECM. DDR1 and DDR2 are expressed exclusively in epithelial and mesenchymal tissues, respectively and are regulated during EMT [63]. This dichotomy allows for cell type-specific ECM recognition and interaction. Indeed, DDR2, which facilitates tumor metastasis by promoting Snail1 protein stability is induced by TGF-β [64, 65]. This raises an interesting possible mode of regulation by DDR proteins given their cell-type specific expression and being the only receptor tyrosine kinases that specifically recognize ECM ligands. Crosstalk between integrin and DDR signaling opens the possibility that tumor cells may utilize both collagen receptors in concert to modulate cell-matrix interactions and invasive responses [66]. How extracellular signals are interpreted by DDR and integrin proteins in concert and how downstream signals are later integrated requires further investigation.
Mechanoregulation of Tumor Cell Plasticity
Metastatic colonization is the rate-limiting step during metastasis [67, 68]. Modification of the pre-metastatic niche to alter its mechanical properties contributes significantly to this process [69, 70]. Force mapping of matched mouse primary tumors and metastatic breast lesions in the lung indicate that metastases largely present low rigidity, while primary tumors show heterogeneity in rigidities ranging from hard to soft [33]. These results mirror the phenomenon that while primary tumors show heterogeneous cell morphologies ranging from epithelial to mesenchymal types within the same tumor, the corresponding metastatic lesions present largely an epithelial morphology. Further studies demonstrate that tumor cells activate the EMT program to disseminate and subsequently undergo Mesenchymal-to-Epithelial Transition (MET) to regain proliferation and form secondary lesions at distant sites [15, 18]. Several lines of evidence strongly support the notion that regulation of the mechanical properties of the premetastatic niche contributes to the dynamic regulation of EMT and MET cellular programs (Figure 1).
Figure 1. Model of the potential dynamic regulation of epithelial cell plasticity during tumor metastasis.
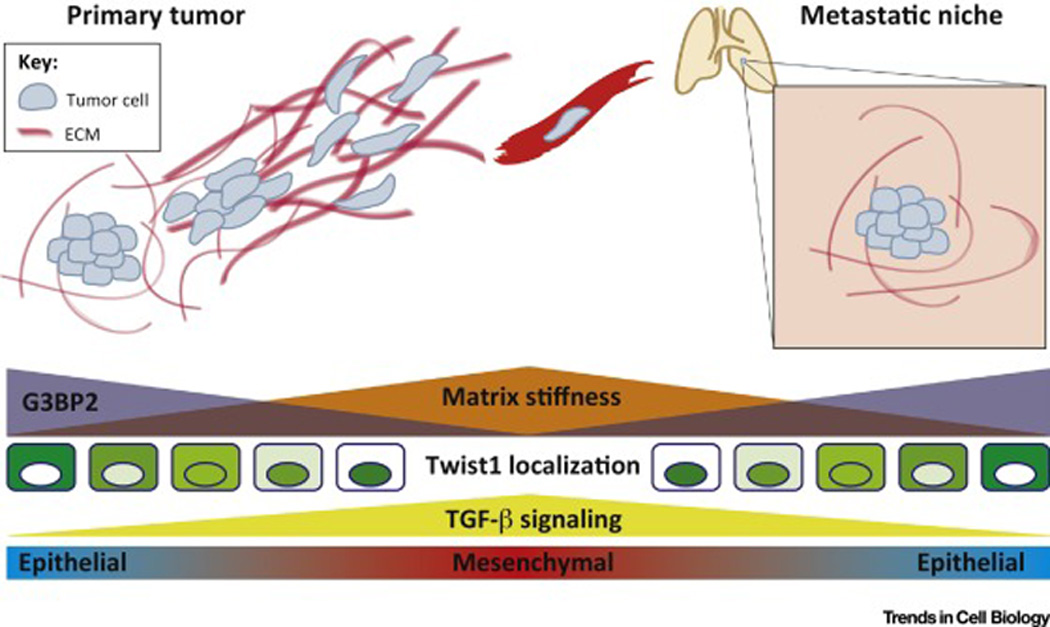
Matrix stiffness is modulated in combination with G3BP2 expression and TGF-β signaling during the metastatic cascade. Increasing tissue rigidity, TGF-β signaling, and loss of G3BP2 in the primary tumor induces EMT and tumor invasion. A compliant environment in the pre-metastatic niche in concert with reacquisition of G3BP2 expression and loss of TGF-β signals may allow for tumor cell re-epithelialization through MET, facilitating metastatic colonization and outgrowth.
Breast tumors are shown to remodel the lung microenvironment prior to arriving in the distant site, a process that is functionally critical to metastatic colonization [69, 71]. Tumor-derived LOX facilitates recruitment of CD11b+ stromal cells to enable metastatic outgrowth of disseminated tumor cells. Moreover, inhibition of the LOX family member, LOXL2, attenuates development of the metastatic niche as demonstrated by reduced stromal fibroblast activation, ECM deposition and modification, and growth factor availability [72]. Recently it was revealed that hypoxia induces secretion of LOX to permit bone metastasis through modulation of bone homeostasis [73]. Hypoxia has also been shown to induce collagen deposition and modification of ECM proteins through induction of an array of enzymes including LOX family members [74, 75]. Collectively, these findings suggest that a key effect for hypoxia in tumor metastasis is ECM remodeling, and potentially matrix stiffening. Furthermore, these functions suggest that in addition to directly regulating EMT through induction of core transcription factors [76–78], hypoxia also indirectly regulates EMT through matrix remodeling processes.
When does remodeling of the primary and secondary tumor microenvironment occur, and what cell type mediates remodeling in vivo are key outstanding questions. Remodeling of the metastatic niche may occur prior to metastatic colonization but the kinetics and mechanism of this process remain unclear [69, 73]. Fibronectin is deposited in the lung premetastatic niche prior to recruitment of stromal bone marrow derived cells and tumor cells [79]. The tumor secretome can be regulated by miR200s to promote metastasis via Sec23a secretion into the pre-metastatic niche, providing an additional mechanism of metastatic niche remodeling [80]. Sec23a is responsible for secretion of suppressive ECM molecules such as TINAGL1 as well as collagens, which could potentially regulate ECM rigidity in the metastatic niche [80, 81]. It remains to be determined what effect down regulation of miR200s and Sec23a or other elements of the secretory pathway has on the rigidity of the tumor microenvironment and metastatic niche. Another hypothesis is that direct effects of mechanical remodeling on stromal cells influence the formation of a metastatic niche. For example, the generation of heterotypic adherens junctions between tumor cells and stromal cells mediates metastatic bone colonization [82]. How these cellular interactions are regulated and whether these heterotypic interactions influence responses to mechanical cues is of interest, particularly given that adherens junctions can mediate mechanotransduction [27]. Further investigation into the function and interplay between biochemical and mechanical properties of the metastatic niche is clearly warranted.
A related but distinct topic regarding tumor cell plasticity is the relationship between tumor mechanical properties and the generation of tumor initiating cells (TICs). Matrix rigidity presents a potential stimulus to regulate EMT and MET processes during metastasis. EMT is shown to promote TIC properties, thus raising the possibility that matrix stiffness could regulate the generation and maintenance of TICs [83, 84]. Recent identification of Twist1 as a critical element in tumor mechanosensing supports further investigation into this possibility [20]. ECM remodeling by both tumor and stromal cells has previously been shown to regulate stemness. Tenascin-C secreted by disseminated cancer cells has also been identified as a critical molecule in the formation of the breast cancer metastatic niche, enhancing stem cell signaling [85]. Stromal-derived periostin (POSTN) can induce Wnt signaling in cancer stem cells to promote maintenance of stemness [86]. POSTN is an ECM protein and has been recently implicated in modifying mechanical properties of connective tissues [87]. Mechanistically, POSTN may contribute to matrix stiffening by acting as a scaffolding protein, bridging interactions between other ECM molecules including collagen I, tenascin-C, and fibronectin through multiple interaction domains [88]. How various biochemical and mechanical cues from the tumor microenvironment regulate TIC formation and maintenance, and furthermore how these mechanisms vary in different tumor types and tissue contexts remains unclear.
Concluding Remarks
In summary, mechanical signals from the tumor microenvironment can impinge on individual steps of the metastatic cascade. We pose a model in which initial genetic and epigenetic aberrations leading to the formation of a non-invasive primary tumor also generate a desmoplastic stromal response (Figure 2). This response is mediated through a variety of factors including stromal-derived LOX and up-regulation of Cav1 in fibroblasts. This desmoplastic response results in increased matrix stiffness and release of growth factors such as TGF-β, both of which promote EMT to allow tumor cell local invasion and dissemination. Tumor cells that have undergone EMT feed forward to enhance matrix remodeling through deposition of additional ECM molecules, modification of the ECM, and increases in the pool of available growth factors. Together, this positive feedback loop facilitates tumor dissemination to distant sites. Once at the secondary site, the local microenvironment attenuates this mechano-signaling cascade, allowing for the re-epithelialization of tumor cells and colonization of the premetastatic niche (Figure 1). This model suggests that mechanical cues from the tumor microenvironment play a critical role in modulating tumor cell plasticity during metastasis. This model will be revised and improved as we learn more about the contributions of the mechanical and biochemical cues of the primary and metastatic tumor microenvironment to tumor progression.
Figure 2. Model of ECM remodeling mechanisms underlying tumor cell invasion and dissemination.
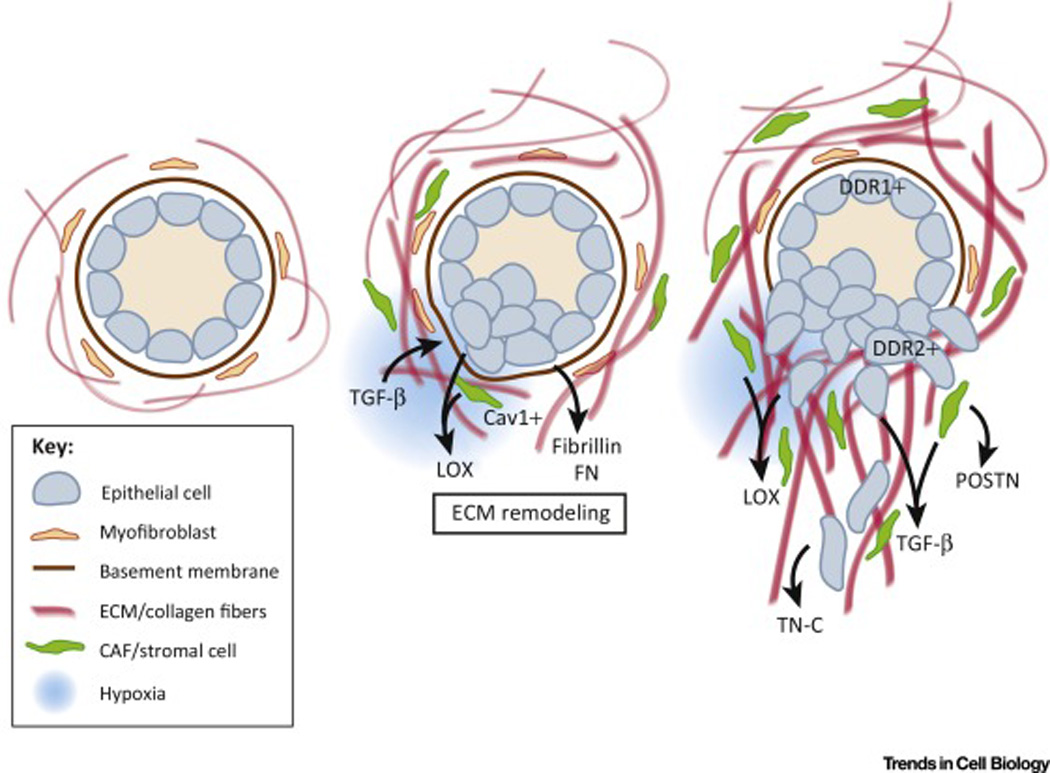
Initial remodeling of the tumor microenvironment is mediated by tumor cells, Cav1-positive CAFs and other stromal cells such as bone marrow-derived cells. Deposition and modification of ECM by these cells in combination with other environmental cues including hypoxia and TGF-β induces EMT, facilitating tumor invasion. As EMT is induced in tumor cells, they form feed forward signaling loops by further remodeling the tumor microenvironment by secreting a variety of factors including LOX, TGF-β, Tenascin-C, and POSTN. Furthermore, EMT results in the modulation of the expression of discoidin domain receptors DDR1 and DDR2, affecting tumor cell responses to the ECM.
In particular, many unanswered questions remain surrounding the role of matrix stiffness and other mechanical cues during tumor progression (Outstanding questions). When do critical ECM modifications occur during tumor progression and predominantly by what cell types in the tumor? What signaling pathways in cancer are mechanosensitive and how do these cues impact tumor progression and metastasis? It is clear that tissues utilize distinct mechanotransduction pathways to recognize and respond to unique physical signals [89]. Could different mechanical cues contribute differentially to different tumor types? Systematic and comprehensive analysis of the physical properties of multiple tumor and tissue types, combined with improved understanding of the cellular mechanotransduction pathways may help to answer these questions [38]. Physical aspects of the cellular microenvironment play critical roles during diverse processes including embryogenesis, tissue homeostasis, and disease progression [90] – are the mechanosensing mechanisms conserved in these varied contexts? In summary, mechanical properties play an important role during tumor progression and additional insights into mechanotransduction pathways in cancer will have significant implications for our fundamental understanding of tumor initiation, dissemination, and metastasis.
Outstanding questions.
At what point(s) during tumor progression do ECM remodeling and increases in matrix rigidity occur? Does matrix stiffening play a role in tumor initiation as well as dissemination?
What is the temporal scale in which ECM remodeling occurs in human tumors and metastatic niches?
What is the main cell type that remodels ECM in the tumor microenvironment? Does this change as tumors progress and/or vary by tissue type?
Do different tumor types respond differentially to mechanical signals (e.g. increasing matrix stiffness)? If so, what is the molecular basis for cell-type specific sensitivities to diverse matrix rigidities?
In addition to directly signaling through actinomyosin contractility, integrins also couple to other signal transduction pathways. What are the signaling pathways that cooperate with integrin engagement to induce EMT in response to increasing matrix stiffness?
Does matrix stiffness or other mechanical cues play a role in inducing MET of disseminated tumor cells at secondary sites?
Can the physical properties of tumors be exploited for clinical benefit? Can tumor stiffening be specifically and therapeutically targeted, given that the rigidities of normal tissues range by orders of magnitude?
Trends.
Physical properties of the tumor microenvironment and of tumor cells play functional roles in driving tumor metastasis.
Matrix stiffness regulates tumor dissemination and metastasis through induction of epithelial-mesenchymal transition.
Extracellular matrix remodeling of the primary and secondary tumor microenvironment represents a key step of tumor metastasis.
Mechanical cues are sensed by a repertoire of cellular mechanosensors. Integrins are key cellular mechanosensors of matrix stiffness.
Extracellular matrix deposition and remodeling is mediated by a variety of cell types, including both tumor and stromal cells.
Acknowledgements
We apologize to the many researchers in this field whose work we were unable to cite due to space restrictions. We thank Laurent Fattet for helpful discussions. Our research on tumor metastasis is supported by grants from National Cancer Institute 1RO1CA168689, 1R01CA174869, and 1R21CA191442, American Cancer Society grant RSG-09-282-01-CSM, and DOD Breast Cancer Program W81XWH-13-1-0132 to J.Y. and a NIH Cancer Cell Biology Training grant (2T32CA067754), NIH Molecular Pathology of Cancer Training grant (5T32CA077109), and ARCS Foundation Scholarship to S.C.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10(1):63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9(2):108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conklin MW, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178(3):1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Provenzano PP, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu H, Mouw JK, Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol. 2011;21(1):47–56. doi: 10.1016/j.tcb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer cell. 2005;8(3):241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Van den Eynden GG, et al. A fibrotic focus is a prognostic factor and a surrogate marker for hypoxia and (lymph) angiogenesis in breast cancer: review of the literature and proposal on the criteria of evaluation. Histopathology. 2007;51(4):440–451. doi: 10.1111/j.1365-2559.2007.02761.x. [DOI] [PubMed] [Google Scholar]
- 10.Colpaert C, et al. The presence of a fibrotic focus is an independent predictor of early metastasis in lymph node-negative breast cancer patients. Am J Surg Pathol. 2001;25(12):1557–1558. doi: 10.1097/00000478-200112000-00016. [DOI] [PubMed] [Google Scholar]
- 11.Hasebe T, et al. Prognostic significance of fibrotic focus in invasive ductal carcinoma of the breast: a prospective observational study. Modern Pathology. 2002;15(5):502–516. doi: 10.1038/modpathol.3880555. [DOI] [PubMed] [Google Scholar]
- 12.Lopez JI, et al. In situ force mapping of mammary gland transformation. Integr Biol (Camb) 2011;3(9):910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154(1):8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 14.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai JH, et al. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–2206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ocana OH, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer prrx1. Cancer Cell. 2012;22(6):709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Leight JL, et al. Matrix rigidity regulates a switch between TGF-beta1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23(5):781–791. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei SC, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015;17(5):678–688. doi: 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez EW, et al. Tissue geometry patterns epithelial-mesenchymal transition via intercellular mechanotransduction. J Cell Biochem. 2010;110(1):44–51. doi: 10.1002/jcb.22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dupont S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 23.Shao DD, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158(1):171–184. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamar JM, et al. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109(37):E2441–E2450. doi: 10.1073/pnas.1212021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao B, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang YC, et al. Differential positioning of adherens junctions is associated with initiation of epithelial folding. Nature. 2012;484(7394):390–393. doi: 10.1038/nature10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.le Duc Q, et al. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol. 2010;189(7):1107–1115. doi: 10.1083/jcb.201001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scarpa E, et al. Cadherin Switch during EMT in Neural Crest Cells Leads to Contact Inhibition of Locomotion via Repolarization of Forces. Dev Cell. 2015;34(4):421–434. doi: 10.1016/j.devcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samuel MS, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19(6):776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das T, et al. A molecular mechanotransduction pathway regulates collective migration of epithelial cells. Nat Cell Biol. 2015;17(3):276–287. doi: 10.1038/ncb3115. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen-Ngoc KV, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A. 2012;109(39):E2595–E2604. doi: 10.1073/pnas.1212834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler LC, et al. Cell shape changes indicate a role for extrinsic tensile forces in Drosophila germ-band extension. Nat Cell Biol. 2009;11(7):859–864. doi: 10.1038/ncb1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plodinec M, et al. The nanomechanical signature of breast cancer. Nat Nanotechnol. 2012;7(11):757–765. doi: 10.1038/nnano.2012.167. [DOI] [PubMed] [Google Scholar]
- 34.Provenzano PP, et al. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4(1):38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brabletz T, et al. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005;179(1–2):56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- 36.Plotnikov SV, et al. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell. 2012;151(7):1513–1527. doi: 10.1016/j.cell.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu W, et al. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PLoS One. 2012;7(10):e46609. doi: 10.1371/journal.pone.0046609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Physical Sciences - Oncology Centers, N., et al. A physical sciences network characterization of non-tumorigenic and metastatic cells. Sci Rep. 2013;3:1449. doi: 10.1038/srep01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davidson PM, et al. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell Mol Bioeng. 2014;7(3):293–306. doi: 10.1007/s12195-014-0342-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denais C, Lammerding J. Nuclear mechanics in cancer. Adv Exp Med Biol. 2014;773:435–470. doi: 10.1007/978-1-4899-8032-8_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang WH, et al. RAC1 activation mediates Twist1-induced cancer cell migration. Nat Cell Biol. 2012;14(4):366–374. doi: 10.1038/ncb2455. [DOI] [PubMed] [Google Scholar]
- 42.Martin AC, et al. Integration of contractile forces during tissue invagination. J Cell Biol. 2010;188(5):735–749. doi: 10.1083/jcb.200910099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riaz M, et al. High TWIST1 mRNA expression is associated with poor prognosis in lymph node-negative and estrogen receptor-positive human breast cancer and is co-expressed with stromal as well as ECM related genes. Breast Cancer Res. 2012;14(5):R123. doi: 10.1186/bcr3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirschmann DA, et al. A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res. 2002;62(15):4478–4483. [PubMed] [Google Scholar]
- 45.El-Haibi CP, et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A. 2012;109(43):17460–17465. doi: 10.1073/pnas.1206653109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Weinberg RA, et al. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 48.Baldwin AK, et al. Epithelial-mesenchymal status influences how cells deposit fibrillin microfibrils. J Cell Sci. 2014;127(Pt 1):158–171. doi: 10.1242/jcs.134270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maschler S, et al. Enhanced tenascin-C expression and matrix deposition during Ras/TGF-beta-induced progression of mammary tumor cells. Oncogene. 2004;23(20):3622–3633. doi: 10.1038/sj.onc.1207403. [DOI] [PubMed] [Google Scholar]
- 50.Pickup MW, et al. Stromally Derived Lysyl Oxidase Promotes Metastasis of Transforming Growth Factor-beta-Deficient Mouse Mammary Carcinomas. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goetz JG, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146(1):148–163. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calvo F, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prager-Khoutorsky M, et al. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat Cell Biol. 2011;13(12):1457–1465. doi: 10.1038/ncb2370. [DOI] [PubMed] [Google Scholar]
- 54.Hasebe T, et al. Atypical tumor-stromal fibroblasts in invasive ductal carcinoma of the breast. Am J Surg Pathol. 2011;35(3):325–336. doi: 10.1097/PAS.0b013e31820afab9. [DOI] [PubMed] [Google Scholar]
- 55.Maschler S, et al. Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene. 2005;24(12):2032–2041. doi: 10.1038/sj.onc.1208423. [DOI] [PubMed] [Google Scholar]
- 56.Jiang G, et al. Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophys J. 2006;90(5):1804–1809. doi: 10.1529/biophysj.105.072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ehrlicher AJ, et al. Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature. 2011;478(7368):260–263. doi: 10.1038/nature10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang J, et al. Structurally Governed Cell Mechanotransduction through Multiscale Modeling. Sci Rep. 2015;5:8622. doi: 10.1038/srep08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geiger B, et al. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2(11):793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 60.Johnson CP, et al. Forced unfolding of proteins within cells. Science. 2007;317(5838):663–666. doi: 10.1126/science.1139857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323(5914):642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 62.Zhu J, et al. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Molecular cell. 2008;32(6):849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valiathan RR, et al. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31(1–2):295–321. doi: 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang K, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15(6):677–687. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walsh LA, Nawshad A, Medici D. Discoidin domain receptor 2 is a critical regulator of epithelial-mesenchymal transition. Matrix Biol. 2011;30(4):243–247. doi: 10.1016/j.matbio.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu H, et al. Discoidin domain receptors promote alpha1beta1- and alpha2beta1-integrin mediated cell adhesion to collagen by enhancing integrin activation. PLoS One. 2012;7(12):e52209. doi: 10.1371/journal.pone.0052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luzzi KJ, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153(3):865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 69.Erler JT, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer cell. 2009;15(1):35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 72.Barry-Hamilton V, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 73.Cox TR, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. 2015;522(7554):106–110. doi: 10.1038/nature14492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Wong CC, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci U S A. 2011;108(39):16369–16374. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014;14(6):430–439. doi: 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gort EH, et al. The TWIST1 oncogene is a direct target of hypoxia-inducible factor-2alpha. Oncogene. 2008;27(11):1501–1510. doi: 10.1038/sj.onc.1210795. [DOI] [PubMed] [Google Scholar]
- 77.Yang MH, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10(3):295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 78.Lester RD, et al. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178(3):425–436. doi: 10.1083/jcb.200701092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Korpal M, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17(9):1101–1108. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Townley AK, et al. Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J Cell Sci. 2008;121(Pt 18):3025–3034. doi: 10.1242/jcs.031070. [DOI] [PubMed] [Google Scholar]
- 82.Wang H, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27(2):193–210. doi: 10.1016/j.ccell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morel AP, et al. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3(8):e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oskarsson T, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17(7):867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Malanchi I, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481(7379):85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 87.Norris RA, et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. 2007;101(3):695–711. doi: 10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kii I, et al. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem. 2010;285(3):2028–2039. doi: 10.1074/jbc.M109.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475(7356):316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eyckmans J, et al. A hitchhiker's guide to mechanobiology. Dev Cell. 2011;21(1):35–47. doi: 10.1016/j.devcel.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]