

Abstract
The vital importance of the Leloir pathway of galactose metabolism has been repeatedly demonstrated by various uni-/multicellular model organisms, as well human patients who have inherited deficiencies of the key GAL enzymes. Yet, other than the obvious links to the glycolytic pathway and glycan biosynthetic pathways, little is known about how this metabolic pathway interacts with the rest of the metabolic and signaling networks. In this study, we compared the growth and the expression levels of the key components of the PI3K/Akt growth signaling pathway in primary fibroblasts derived from normal and galactose-1 phosphate uridylyltransferase (GalT)-deficient mice, the latter exhibited a subfertility phenotype in adult females and growth restriction in both sexes. The growth potential and the protein levels of the pAkt(Thr308), pAkt(Ser473), pan-Akt, pPdk1, and Hsp90 proteins were significantly reduced by 62.5%, 60.3%, 66%, 66%, and 50%, respectively in the GalT-deficient cells. Reduced expression of phosphorylated Akt proteins in the mutant cells led to diminished phosphorylation of Gsk-3β (−74%). Protein expression of BiP and pPten were 276% and 176% higher respectively in cells with GalT-deficiency. Of the 24 genes interrogated using QIAGEN RT2 Profiler PCR Custom Arrays, the mRNA abundance of Akt1, Pdpk1, Hsp90aa1 and Pi3kca genes were significantly reduced at least 2.03-, 1.37-, 2.45-, and 1.78-fold respectively in mutant fibroblasts. Both serum-fasted normal and GalT-deficient cells responded to Igf-1-induced activation of Akt phosphorylation at +15 minutes, but the mutant cells have lower phosphorylation levels. The steady-state protein abundance of Igf-1 receptor was also significantly reduced in mutant cells. Our results thus demonstrated that GalT deficiency can effect down-regulation of the PI3K/Akt growth signaling pathway in mouse fibroblasts through distinct mechanisms targeting both gene and protein expression levels.
Keywords: Galactose-1 phosphate uridylyltransferase (GalT), Endoplasmic reticulum (ER) stress, Protein kinase B (Akt), 3-Phosphoinositide-dependent kinase-1 (Pdk1), Heat shock protein 90 (Hsp90), Galactosemia
Introduction
The phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/ protein kinase B (Akt) signaling pathway has been intensively studied because of its prominent role in regulating cell growth and metabolism (see [1] for review). Since many human cancers involve amorphic mutations in the phosphatase and tensin homolog (PTEN) gene, a key negative regulator of the PI3K/Akt pathway [2, 3], many studies focused on the aberrant up-regulation of the pathway and its positive regulators. Indeed, a master positive regulator of the pathway, 3-Phosphoinositide-dependent kinase-1 (PDK1), has been hailed as a promising anti-neoplastic target for human cancers [4, 5]. By contrast, little is known about conditions in which the PI3K/Akt pathway is down-regulated in the absence of anti-cancer therapy or genetic modulation of pro-apoptotic signaling pathways. Brace and coworkers were the first to show that galactokinase (GALK1) deficiency induced by GALK1-specific siRNA leads to reduced Akt phosphorylation in human cancer cells in culture [6].
Earlier, we constructed a new mouse model of galactose-1 phosphate uridylyltransferase (GalT) deficiency [7]. Our characterization of the GalT-deficient mice revealed subfertility in adult females and growth restriction in both sexes [7]. To study the mechanisms behind the growth impairment caused by GalT deficiency, we examined the expression level of the canonical PI3K/Akt growth signaling pathway in mouse fibroblasts derived from the GalT-deficient mice. We found that GalT deficiency induces pleiotropic down-regulation of the PI3K/Akt pathway at both mRNA and protein abundance levels. Our findings not only reaffirm the link between galactose metabolism and the PI3K/Akt growth signaling pathway at the molecular level, but also offer novel insights into the pathophysiological mechanisms of Classic Galactosemia [8] – a rare disorder where human patients suffer from the inherited deficiency of the human GALT enzyme.
Materials and Methods
Cells and Culture Conditions
Primary skin fibroblasts were isolated from young adult female GalT-deficient mice [7] and their sex-matched normal littermates using the method described by Seluanov and coworkers [9]. After confirmation of cell type identity by a Board-Certified pathologist, the isolated fibroblast strains were propagated in regular Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 15% fetal bovine serum (FBS), 2.5% penicillin and streptomycin at 37°C with 5% CO2. Collagenase and cell culture media were purchased from Invitrogen (Waltham, MA).
Antibodies
Antibodies against selected components of the PI3K/Akt signaling pathway were included in the Phospho-Akt Pathway Antibody Sampler Kit (Cell Signaling Technology, Danvers, MA). Anti-mouse BiP/GRP78 were purchased from BD Bioscience (San Jose, CA). Anti-mouse Hsp90 was obtained from Santa Cruz Biotechnology Inc. (Dallas, TX). Anti-mouse Gapdh was purchased from Sigma Inc. (St. Louis, MO). Anti-mouse Igf-1 receptor (Igf-1r) β was purchased from Cell signaling Technology (Danvers, MA).
Cell growth assessment
20 × 104 fibroblast cells derived from each of the three normal and three GalT-deficient mice were seeded in each of the 16 wells of a 24-well cell culture plate at day 0. Cell growth was monitored daily by counting the cells from four aliquots (two aliquots from each of the duplicate wells) for seven days consecutively.
Igf-1 activation of Akt phosphorylation
To study the kinetics of mouse Igf-1 activation of Akt phosphorylation, fibroblast cells derived from the three normal and three GalT-deficient mice were cultured in replicates overnight in DMEM supplemented with 2% FBS and 2.5% penicillin/streptomycin. On the day of the experiment, the cells were serum-starved for two hours before Igf-1 stimulation (100ng/ml in serum- and antibiotics-free DMEM). Cells were stimulated for four hours and samples of the stimulated cells were collected at +15 minutes, +30 minutes, and +4 hours for Akt phosphorylation assessment. At the end of Igf-1 treatment, the culture media was replaced with serum-free DMEM. Another sample was collected at +24 hours for Akt phosphorylation assessment. Recombinant mouse Igf-1 was obtained from Invitrogen Inc. (Frederick, MD).
Quantitative Real Time PCR
Steady-state mRNA expression levels of 24 selected components of the PI3K/Akt signaling in three normal and three GalT-deficient cell strains were assessed by QIAGEN Custom RT2 Profiler PCR Arrays on a contractual fee-for-service basis.
Immunoblot Analysis
Protein lysates from the fibroblasts were prepared by mechanical disruption of the cells in hypotonic buffer [25mM NaCl, 0.5mM EDTA, 25mM Tris HCl (pH 7.2)] with protease and phosphatase inhibitors at 4°C. The cell debris were removed by centrifugation at 13,000rpm for 15 minutes at 4°C. Pierce BCA protein estimation kit (Thermo Scientific, Rockford, IL) was used to determine the total protein content. 40μg of the total protein was resolved by 12% SDS-PAGE before being transferred to a nitrocellulose membrane. The immunoblotting was carried out by the procedure described by Lai and coworkers [10]. Manufacturers’ recommended titers for the respective primary/secondary antibodies were followed in all experiments. Primary antibodies were detected with IR dye-conjugated secondary antibodies and visualized by Odyssey Image Analyzer (Li-Cor Biotechnology, Lincoln, NE). Quantitative analysis of the fluorescence signals was performed by Image Studio™ Lite software (Li-Cor Biotechnology, Lincoln, NE), and the results were normalized to the corresponding Gapdh abundance detected from the same blot. Student’s t-test was used to determine statistical significance of the results.
Galactose-1 phosphate Assay
Intracellular galactose 1-phosphate level was assayed by the enzyme-coupled assay previously described [11].
Results
Accumulation of galactose-1 phosphate in GalT-deficient mouse fibroblasts
Previously, we constructed a new mouse model of GalT deficiency [7]. In this study, we isolated skin fibroblasts from six age-, and sex-matched normal (N/N) and GalT-deficient (G/G) mice (three for each group). We found that when the GalT-deficient cells were exposed to high level of galactose (0.1% ≈ 5.5mM) in the growth medium, the amount of gal-1P accumulated rose up to 12.59 ± 2.84 ng gal-1P/ μg cell protein (Fig. 1a). Even when the mutant cells were grown in the absence of exogenous galactose, they accumulated 3.12 ± 1.79 ng gal-1P/ μg cell protein (Fig. 1a).
Fig. 1. Comparative biochemical, growth and molecular studies of normal (N/N) and GalT-deficient (G/G) mouse fibroblasts under different culture conditions.
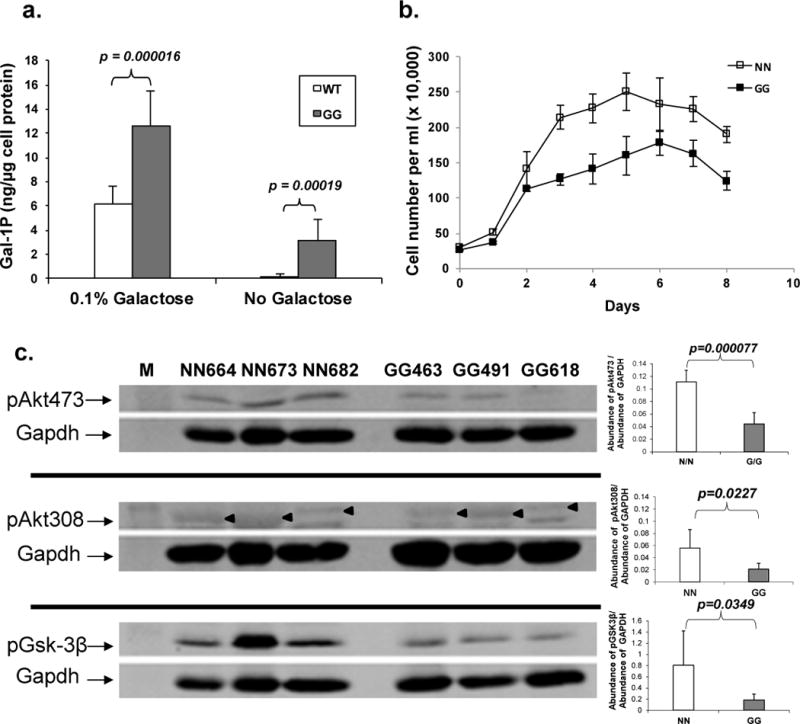
a. Galactose-1 phosphate contents were determined from normal and GalT-deficient cells (n = 6 for each group) grown in regular DMEM with and without the addition of 0.1% galactose. In the former case, the cells were challenged with 0.1% galactose for three hours before being harvested for analysis.
b. Normal and GalT-deficient fibroblasts were seeded in equal numbers at day 0 and growth of cells in regular DMEM medium was monitored for the next seven days. Each data point represents the mean value of four cell counts.
c. Levels of pAkt and pGsk3β in three normal and three GalT-deficient cell strains cultured in regular DMEM were compared by Western Blot Analysis. Exemplary Western Blots were shown on the left, and the quantified results for the abundances of pAkt’s and pGsk-3β from the two groups (n = 6) were presented on the right. “◄” denotes the signal from the anti-pAkt(Thr308) antibody, which is quite close to a non-specific signal below.
Growth restriction in GalT-deficient mouse fibroblasts
We followed the growth of three normal and three GalT-deficient mouse fibroblast strains in regular culture medium for consecutive seven days. We found that the mutant fibroblasts reached 177±27 × 104 cells/ml at the peak of the growth curve while the normal cells could reach up to 250±26 × 104 cells cells/ml (Fig. 1b). These results agree with the growth restriction phenotype we reported in our mouse model with GalT deficiency [7].
Altered expression levels of phosphorylated Akt proteins and their downstream target in GalT-deficient cells
To investigate the molecular basis behind the reduced growth of the mutant mouse fibroblasts, which could shed clues to underlying mechanisms for the growth restriction phenotype of the mouse model [7] and human patients with GALT-deficiency Galactosemia [12], we examined for signs of attenuation of the PI3K/Akt growth signaling pathway in these cells. We performed immunoblot analysis using monoclonal antibodies against Akt proteins phosphorylated at threonine-308 and serine-473, respectively, in normal and GalT-deficient fibroblasts cultured in regular DMEM supplemented with 15% FBS. Our results revealed significantly reduced steady-state abundance of pAkt(Ser473) (−60.3%, p<0.05) and pAkt(Thr308) (−62.5%, p<0.05) in the GalT-deficient cells (Fig. 1c). The diminished level of pAkt also resulted in a significant decrease in pGsk-3β (−74%), which is a downstream target of the phosphorylated Akt [13] (Fig. 1c). This confirmed a functional consequence of reduced pAkt.
Altered protein expression levels of the key regulators of Akt phosphorylation in GalT-deficient cells in regular DMEM medium
To determine the cause(s) for the decreased steady-state abundance of pAkt(Thr308) and pAkt(Ser473) in the GalT-deficient cells, we evaluated the key regulators of Akt phosphorylation (Pten, Pdk1, pan-Akt) and other known factors such as endoplasmic reticulum (ER) stress, which has been shown to occur in human GALT-deficient fibroblasts [14]. We found that GalT-deficient mouse fibroblasts exhibited approximately 66% decrease in pPdk1 (active phosphorylated form of Pdk1) and pan-Akt (total Akt), as well as over 50% reduction in Hsp90 (Fig. 2a). By contrast, we saw 176% and 276% increases in pPten and BiP, respectively, in the mutant cells.
Fig. 2. Relative protein and mRNA abundance of key regulators of Akt phosphorylation in normal and GalT- deficient mouse fibroblasts cultured in regular DMEM.
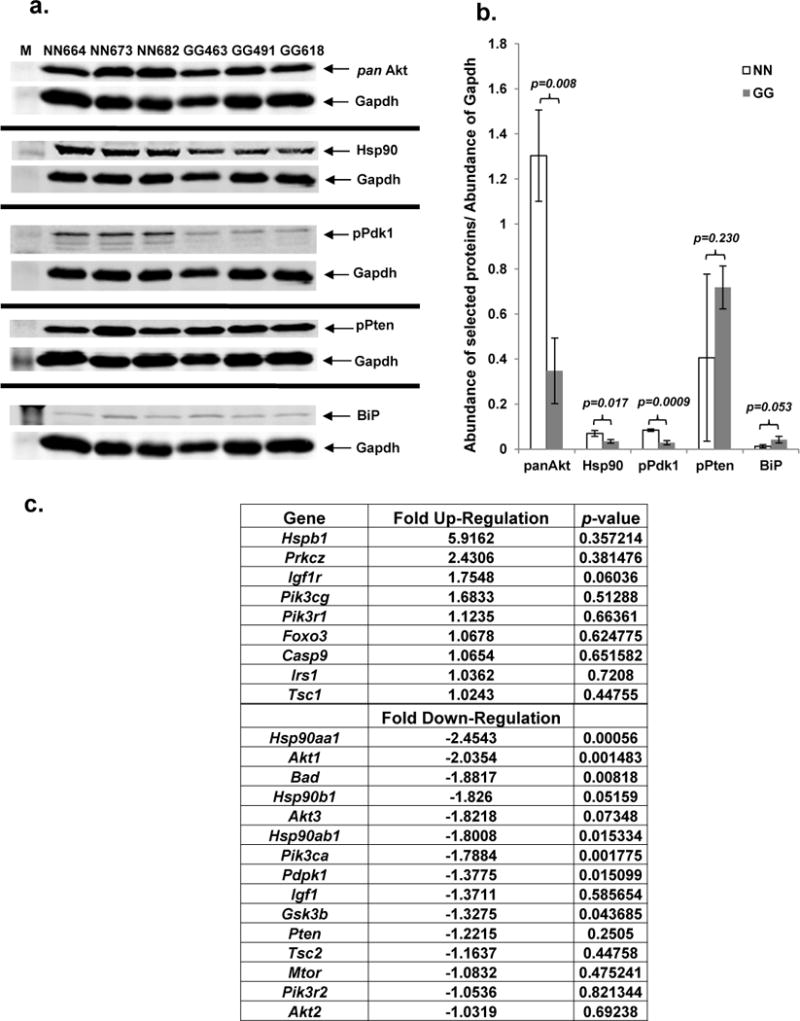
a. Protein expression levels of pPdk1, pPten, pan Akt, BiP, Hsp90 and Gapdh in respective mouse fibroblasts (n=3 in each group) were compared by immunoblotting.
b. Normalized protein levels of key regulators of Akt phosphorylation.
c. Relative fold change of mRNA expression of the selected components of PI3K/Akt signaling pathway in GalT deficient mouse fibroblasts as determined by RT-PCR.
mRNA expression levels of PI3K/Akt signaling pathway components
To determine if the changes in the abundances of the PI3K/Akt signaling pathway components in the mutant fibroblasts resulted from the corresponding changes in the mRNA expression of the respective genes, we quantified the steady-state mRNA abundances of 24 selected components of the signaling pathway using QIAGEN RT2 Profiler PCR Custom Arrays. The top three up-regulated genes in the mutant cells were Hspb1 (5.9-fold), Prkcz (2.4-fold), and Igf-1r (1.75-fold) (Fig. 2c), but their associated p values were greater than 0.05. On the other hand, the mRNA abundance of Akt1, Pdpk1, Hsp90aa1, Hsp90b1, Hsp90ab1 and Pi3kca genes were significantly reduced at least 2.03-, 1.37-, 2.45-, 1.83-, 1.80- and 1.78-fold respectively in mutant fibroblasts, with the associated p values less than 0.05 (Fig. 2c). No significant changes were seen for Irs1, Igf1, Mtor, and Pten.
Igf-1 activation of Akt phosphorylation
The attenuated steady-state expression of the PI3K/Akt signaling pathway in GalT-deficient mouse fibroblasts prompted us to examine the proximal events that lead to Akt phosphorylation in the cells. As shown in Fig. 3a, Akt phosphorylation was rapidly induced within 15 minutes in both the normal and GalT-deficient cells. However, it is apparent that the levels of phosphorylation were lower in the mutant cells, similar to what we saw at steady-state levels (Fig. 1c). We also compared the steady-state protein abundance of Igf-1 receptor (Igf-1r) in both normal and GalT-deficient cells. We found that the expression of Igf-1r was significantly down-regulated (−78%) in the mutant cells (Fig. 3b).
Fig. 3. Igf-1 induced Akt phosphorylation and steady-state of Igf-1 receptor in normal and GalT deficient mouse fibroblasts.
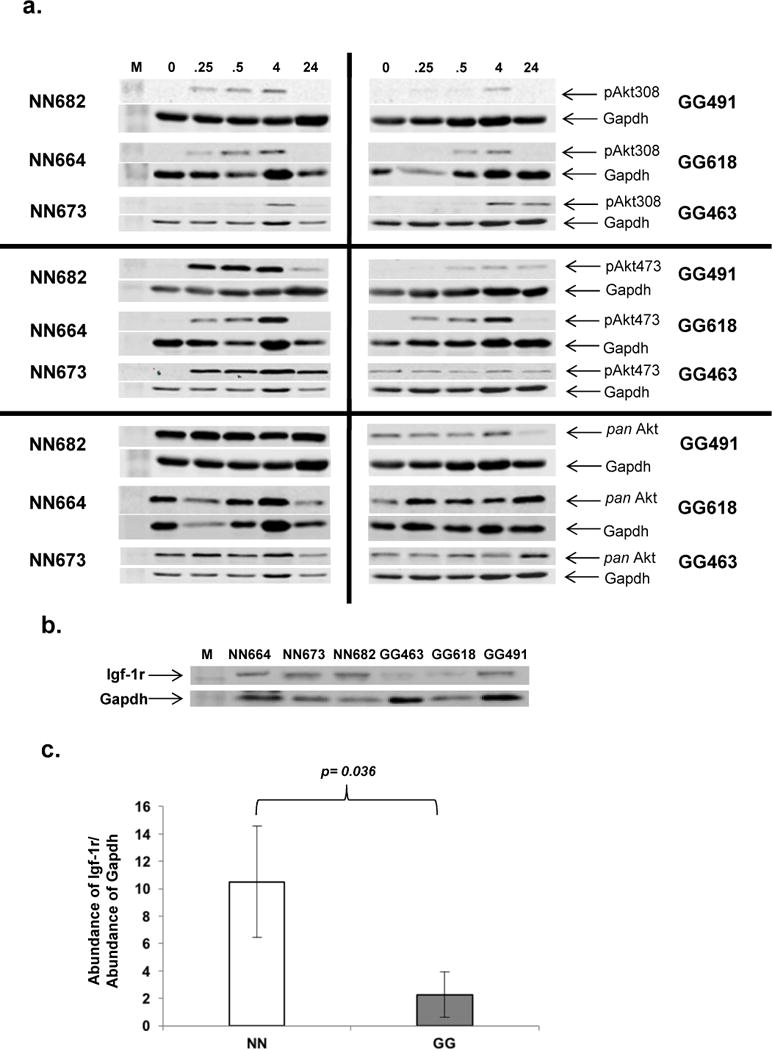
a. Three normal and three GalT-deficient mouse fibroblasts were cultured and stimulated with Igf-1 (100 ng/ml) at time =0 as described in Materials and Methods. Levels of Akt phosphorylation at the indicated time intervals (0.25 hour, 0.5 hour, 4 hours, 24 hours) was analyzed by Immunoblot analysis as described above.
b. Immunoblot analysis of steady-state abundance of Igf-1 receptor (Igf-1r) in normal and GalT-deficient cells cultured in DMEM.
c. Normalized protein level of steady state Igf-1r in mouse fibroblasts.
Discussion
Earlier, Brace and colleagues found that silencing of the human galactokinase (GALK1) gene led to the decreased Akt phosphorylation in cultured cancer cells and tumors [6]. While their findings were novel, the authors did not disclose whether the down-regulation occurs solely at the post-translational level. In addition, it is unclear the degree of the GALK1 knockdown in their experiments, which could have significant implications on the extent of the down-regulation. In our studies described here, we examined the link between galactose metabolism and the PI3K/Akt signaling pathway in fibroblasts derived from mice that are completely devoid of a different key enzyme of the galactose metabolic pathway – galactose-1 phosphate uridylyltransferase (GalT). Additionally, our results indicated that the down-regulation of the PI3K/Akt pathway occurs at both mRNA and protein levels.
But how can perturbation of the galactose metabolic pathway caused by GalT deficiency lead to pleiotropic down-regulation of the PI3K/Akt growth signaling pathway? In Fig. 4, we proposed a model based upon the current and past experimental data gathered by our laboratory and those of other investigators. For quite some time, others and we have shown that GALT deficiency in human patient cells could lead to ER stress [15, 16], which could be the result of metabolic dysregulation and/or the aberrant glycosylation widely reported in patients [17, 18]. In fact, the increase in BiP protein abundance (Fig. 2a, b) and the up-regulation of the Hspb1 (Heat shock 27kD protein 1) gene (Fig. 2c) in the mutant cells indicated that they were experiencing ER stress. It has been documented, and confirmed by us here, that ER stress could result in reduced mRNA level of pan-Akt and its abundance [19] (Fig. 2), which could lead to decreased Akt available for phosphorylation.
Fig. 4. Proposed molecular mechanisms that account for the changes in the PI3K/Akt signaling pathway revealed in the GalT-deficient mouse fibroblasts.
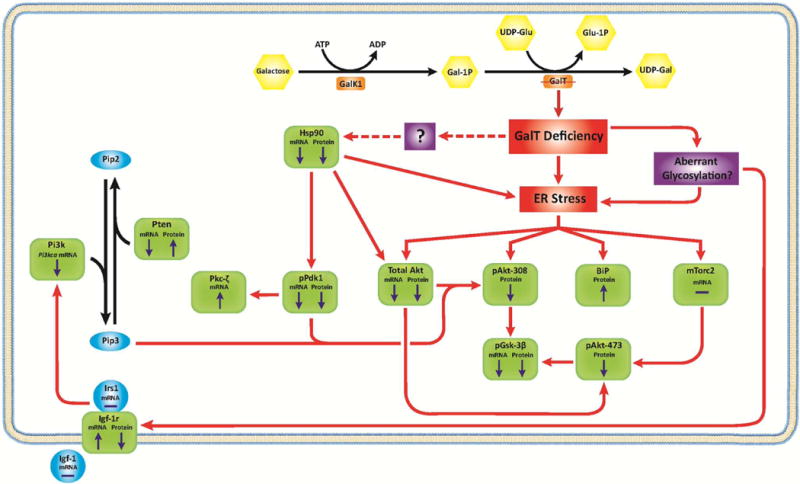
Key components of the PI3K/Akt signaling pathway examined in the GalT-deficient mice are depicted in green squares. “⬆” and “⬇” indicates the direction of change in mRNA and protein levels of each components.
The 66% decrease in pan-Akt level could, in theory, explain the diminished pAkt(Thr308) and pAkt(Ser473) phosphorylation. Yet, we were surprised to find reduced amounts of pPdk1 (active form of Pdk1) (−66%) and Hsp90 (−50%) in this case. Since Pdk1 is the major kinase that phosphorylates Akt at threonine-308 (see review [1]), our data suggested that decreased pPdk1 could have negatively affected the abundance of pAkt(Thr308). But what could have caused the 66% reduction of pPdk1 protein abundance in the GalT-deficient cells at the first place; especially we only saw a 1.37-fold (28%) decrease in the Pdpk1 mRNA (Fig. 2c)? Fujita et al. were among the few to show that Hsp90 is critical for the stability of Pdk1 [20]. In Fig. 2, we showed that Hsp90 is diminished at both mRNA and protein levels in the GalT-deficient cells, which may partly explain the 66% reduction in pPdk1 protein abundance. We are not certain about the cause for the diminished Hsp90 mRNA and protein levels in the GalT-deficient cells, although inhibition of Hsp90 activity elicits ER stress and apoptosis [21]. The phosphorylation of Pdk1 to pPdk1 is dependent by PI3K activity (see review [1]). Therefore, reduction in pPdk1 can also be a result of diminished PI3K activity. Interestingly, we detected a 1.78-fold (45%) decrease (p < 0.05) in Pi3kca gene mRNA (Fig. 2c). Since pPdk1 is capable of activating protein kinase C zeta subunit (Prkcz) [22], it is possible that the canonical protein kinase C signaling pathway is affected in GalT deficiency as well. Just as the lower expression levels of pPdk1 and Hsp90 in GalT-deficient cells preclude reduced pan-Akt level as the sole mechanism for the diminished pAkt(Thr308) abundance, ER stress might also contribute to the negative change in pAkt(Ser473) abundance through reduced mTORC2 enzymatic activity [19], which is required to phosphorylate pAkt at serine-473 [23].
The down-regulation of the PI3K/Akt signaling pathway at steady-state in the GalT-deficient cells prompted us to examine the proximal events leading to Akt phosphorylation in these cells. Despite lower level of Akt phosphorylation was observed in the GalT-deficient cells, induction of Akt phosphorylation occurred within 15 minutes after addition of mouse Igf-1 in both normal and mutant cells (Fig. 3a). This suggested that kinetics of the Igf-1 induced Akt phosphorylation is not impaired in GalT deficiency. However, we found decreased steady-state level of Igf-1r in the mutant cells (Fig. 3b), possibly the result of GalT-induced aberrant glycosylation and the subsequent destabilization of the growth receptor [24]. Although such decrease in Igf-1r abundance could explain the lower level of Igf-1-induced Akt phosphorylation, it is important to point out that the reduced Igf-1r alone failed to account for the simultaneous diminished steady-state levels of pPdk1, Hsp90 and pan-Akt in the mutant cells. All these indicate that distinct mechanisms are employed in the down-regulation of the PI3K/Akt signaling pathway in the mutant cells. While ER stress and aberrant glycosylation remain the best candidate factors that connect GalT deficiency and the signaling pathway in our model (Fig. 4), it remains to be elucidated how GalT deficiency causes ER stress and aberrant glycosylation at the first place. Our future studies will thus focus on the identification of the signaling molecules that trigger the down-regulation of PI3K/Akt in GalT-deficient cells and delineation of the relationships, if any, of these signaling molecules to ER stress and glycan biosynthesis.
In conclusion, our studies of GalT-deficient mouse fibroblasts not only reaffirmed the role played by normal galactose metabolism in PI3K/Akt signaling, but also yielded novel insights into the regulatory mechanisms that were not documented in the studies conducted by Brace and coworkers [6]. From the perspective of Classic Galactosemia, it makes perfect sense to learn that the PI3K/Akt signaling pathway is affected in the GalT-deficient mouse fibroblasts. After all, Pdk1 and Prkcz play a crucial role in mammalian oocytes development and neuronal survival [25–27], both of which are believed to be affected in patients with the disease [12].
Supplementary Material
Highlights.
GalT deficiency induces Endoplasmic Reticulum (ER) stress in mouse fibroblasts.
Down-regulation of PI3K/Akt signaling occurs in GalT-deficient mouse fibroblasts.
GalT activity controls mRNA levels of positive regulators of the PI3K/Akt pathway.
GalT activity affects protein levels of selected regulators of the PI3K/Akt pathway.
Acknowledgments
We would like to thank Dr. Clinton C. Mason (Department of Pediatrics, U. of Utah) for his advice on statistical analysis of the data. Grant support to KL include 1R01HD074844 (NIH/NICHD), a Research Grant from the Galactosemia Foundation (USA), a generous gift from the Dershem Family (Warrior Benefits), the K2R2R award from the Primary Children’s Hospital Foundation (Intermountain Healthcare).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Annals of medicine. 2014;46:372–383. doi: 10.3109/07853890.2014.912836. [DOI] [PubMed] [Google Scholar]
- 2.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J, Pandolfi PP. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta A, Dey CS. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Molecular biology of the cell. 2012;23:3882–3898. doi: 10.1091/mbc.E12-05-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erlanson DA, Arndt JW, Cancilla MT, Cao K, Elling RA, English N, Friedman J, Hansen SK, Hession C, Joseph I, Kumaravel G, Lee WC, Lind KE, McDowell RS, Miatkowski K, Nguyen C, Nguyen TB, Park S, Pathan N, Penny DM, Romanowski MJ, Scott D, Silvian L, Simmons RL, Tangonan BT, Yang W, Sun L. Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorganic & medicinal chemistry letters. 2011;21:3078–3083. doi: 10.1016/j.bmcl.2011.03.032. [DOI] [PubMed] [Google Scholar]
- 5.Nagashima K, Shumway SD, Sathyanarayanan S, Chen AH, Dolinski B, Xu Y, Keilhack H, Nguyen T, Wiznerowicz M, Li L, Lutterbach BA, Chi A, Paweletz C, Allison T, Yan Y, Munshi SK, Klippel A, Kraus M, Bobkova EV, Deshmukh S, Xu Z, Mueller U, Szewczak AA, Pan BS, Richon V, Pollock R, Blume-Jensen P, Northrup A, Andersen JN. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric kinase inhibitor. J Biol Chem. 2011;286:6433–6448. doi: 10.1074/jbc.M110.156463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brace A, Bjorke ML, Nieto-Bergman S, Friedman LS, Ward K, Blake RA. GALK1s as modifiers of the PTEN/AKT Pathway and methods of use. Application#11/993,191. USA: US Patent Application Publication. 2010 Jul 22;
- 7.Tang M, Siddiqi A, Witt B, Yuzyuk T, Johnson B, Fraser N, Chen W, Rascon R, Yin X, Goli H, Bodamer OA, Lai K. Subfertility and growth restriction in a new galactose-1 phosphate uridylyltransferase (GALT) – deficient mouse model. Eur J Hum Genet. 2014;22:1172–1179. doi: 10.1038/ejhg.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science. 1956;123:635–636. doi: 10.1126/science.123.3198.635. [DOI] [PubMed] [Google Scholar]
- 9.Seluanov A, Vaidya A, Gorbunova V. Establishing primary adult fibroblast cultures from rodents. Journal of visualized experiments: JoVE. 2010 doi: 10.3791/2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai K, Bolognese CP, Swift S, McGraw P. Regulation of inositol transport in Saccharomyces cerevisiae involves inositol-induced changes in permease stability and endocytic degradation in the vacuole. J Biol Chem. 1995;270:2525–2534. doi: 10.1074/jbc.270.6.2525. [DOI] [PubMed] [Google Scholar]
- 11.Heinrich MR, Howard SM. Galactokinase, Methods Enzymol. 1966;9:407–412. [Google Scholar]
- 12.Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, Rubio-Gozalbo E, Schomer D, Welt C, Anastasoaie V, D’Anna K, Gentile J, Guo CY, Hecht L, Jackson R, Jansma BM, Li Y, Lip V, Miller DT, Murray M, Power L, Quinn N, Rohr F, Shen Y, Skinder-Meredith A, Timmers I, Tunick R, Wessel A, Wu BL, Levy H, Elsas L, Berry GT. The adult galactosemic phenotyp. J Inherit Metab Dis. 2012;35:279–286. doi: 10.1007/s10545-011-9372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.M GA, Uddin S, Mahmud D, Damacela I, Lavelle D, Ahmed M, van Besien K, Wickrema A. Regulation of myeloma cell growth through Akt/Gsk3/forkhead signaling pathway. Biochem Biophys Res Commun. 2002;297:760–764. doi: 10.1016/s0006-291x(02)02278-7. [DOI] [PubMed] [Google Scholar]
- 14.Gallerne C, Prola A, Lemaire C. Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochim Biophys Acta. 2013;1833:1356–1366. doi: 10.1016/j.bbamcr.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 15.Slepak T, Tang M, Addo F, Lai K. Intracellular galactose-1-phosphate accumulation leads to environmental stress response in yeast model. Mol Genet Metab. 2005;86:360–371. doi: 10.1016/j.ymgme.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Slepak TI, Tang M, Slepak VZ, Lai K. Involvement of endoplasmic reticulum stress in a novel Classic Galactosemia model. Mol Genet Metab. 2007;92:78–87. doi: 10.1016/j.ymgme.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coss KP, Hawkes CP, Adamczyk B, Stockmann H, Crushell E, Saldova R, Knerr I, Rubio-Gozalbo ME, Monavari AA, Rudd PM, Treacy EP. N-glycan abnormalities in children with galactosemia. Journal of proteome research. 2014;13:385–394. doi: 10.1021/pr4008305. [DOI] [PubMed] [Google Scholar]
- 18.Sturiale L, Barone R, Fiumara A, Perez M, Zaffanello M, Sorge G, Pavone L, Tortorelli S, O’Brien JF, Jaeken J, Garozzo D. Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology. 2005;15:1268–1276. doi: 10.1093/glycob/cwj021. [DOI] [PubMed] [Google Scholar]
- 19.Yung HW, Charnock-Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PloS one. 2011;6:e17894. doi: 10.1371/journal.pone.0017894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita N, Sato S, Ishida A, Tsuruo T. Involvement of Hsp90 in signaling and stability of 3-phosphoinositide-dependent kinase-1. J Biol Chem. 2002;277:10346–10353. doi: 10.1074/jbc.M106736200. [DOI] [PubMed] [Google Scholar]
- 21.Taiyab A, Sreedhar AS, Rao Ch M. Hsp90 inhibitors, GA and 17AAG, lead to ER stress-induced apoptosis in rat histiocytoma. Biochemical pharmacology. 2009;78:142–152. doi: 10.1016/j.bcp.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 23.Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, Wu J, Lin HK, Sarbassov dos D. ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Science signaling. 2011;4:ra10. doi: 10.1126/scisignal.2001731. [DOI] [PubMed] [Google Scholar]
- 24.Siddals KW, Allen J, Sinha S, Canfield AE, Kalra PA, Gibson JM. Apposite insulin-like growth factor (IGF) receptor glycosylation is critical to the maintenance of vascular smooth muscle phenotype in the presence of factors promoting osteogenic differentiation and mineralization. J Biol Chem. 2011;286:16623–16630. doi: 10.1074/jbc.M110.202929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kharebava G, Makonchuk D, Kalita KB, Zheng JJ, Hetman M. Requirement of 3-phosphoinositide-dependent protein kinase-1 for BDNF-mediated neuronal survival. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:11409–11420. doi: 10.1523/JNEUROSCI.2135-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy P, Adhikari D, Zheng W, Liang S, Hamalainen T, Tohonen V, Ogawa W, Noda T, Volarevic S, Huhtaniemi I, Liu K. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum Mol Genet. 2009;18:2813–2824. doi: 10.1093/hmg/ddp217. [DOI] [PubMed] [Google Scholar]
- 27.Lie S, Morrison JL, Williams-Wyss O, Suter CM, Humphreys DT, Ozanne SE, Zhang S, Maclaughlin SM, Kleemann DO, Walker SK, Roberts CT, McMillen IC. Periconceptional undernutrition programs changes in insulin-signaling molecules and microRNAs in skeletal muscle in singleton and twin fetal sheep. Biology of reproduction. 2014;90:5. doi: 10.1095/biolreprod.113.109751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.