

Abstract
A lack of intracellular delivery systems has limited the use of biologics such as monoclonal antibodies (mAb) that abrogate molecular signaling pathways activated to promote escape from cancer treatment. We hypothesized that intracellular co-delivery of the photocytotoxic chromophore benzoporphyrin derivative monoacid A (BPD) and the anti-VEGF mAb bevacizumab in a nanophotoactivatable liposome (nanoPAL) might enhance the efficacy of photodynamic therapy (PDT) combined with suppression of VEGF-mediated signaling pathways. As a proof-of-concept we found that nanoPAL-PDT induced enhanced extra- and intracellular bevacizumab delivery and enhanced acute cytotoxicity in vitro. In an in vivo subcutaneous mouse model of pancreatic ductal adenocarcinoma, nanoPAL-PDT achieved significantly enhanced tumor reduction. We attribute this to the optimal incorporation of insoluble BPD into the lipid bilayer, enhancing photocytotoxicity, and the simultaneous spatiotemporal delivery of bevacizumab, ensuring efficient neutralization of the rapid but transient burst of VEGF following PDT.
Keywords: Pancreatic cancer, Liposomes, Combinatorial chemotherapy, Bevacizumab, Benzoporphyrin derivative, Photodynamic therapy, Nanomedicine, Avastin, Nanoparticle
Graphical Abstract
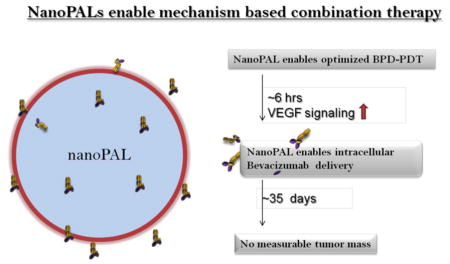
NanoPALs depicted as ‘smart’ nanovectors optimized to achieve mechanism based therapeutic intervention in subcutaneous models of pancreatic ductal adenocarcinomas. It has been established that there is a transient increase in VEGF in the tumor microenvironment as well as intracellular VEGF levels after ~ 6 hrs after PDT. Hence, nanoPALs have been designed to co-deliver the benzoporphyrin derivative (BPD) - the photosensitizer for photodynamic therapy (PDT) - and intracellular delivery of bevacizumab - a therapeutic antibody to abrogate the increased VEGF signaling after PDT.
Introduction
The fourth leading cause of cancer death in the US1, 2, pancreatic ductal adenocarcinoma (PDAC) accounts for greater than 85% of pancreatic cancer cases3. PDAC is characterized by its tenacious resistance to gemcitabine-based regimens (the standard chemotherapy for this condition) and radiotherapy, making surgical resection the best option when the disease is localized, but most patients harbor advanced-stage disease at the time of diagnosis3. Only 15–20% of patients present localized, operable tumors and are candidates for surgery. The 5-year survival rate for those patients who do qualify for surgery is still less than 20%, up from a dismal overall 5-year survival rate of 6.7%1. A key component of this poor outcome is the co-opting of molecular signaling pathways by the tumor to escape chemotherapy, radiotherapy and molecular-targeted agents4. The existing standard of care to perform sequential drug delivery has a poor efficiency and ultimately induces dose-limiting toxicities, acquired drug-resistance and poor response rates. Therefore, nanoscale drug delivery systems capable of multidrug delivery are being engineered to reduce systemic exposure to toxic drugs while safely enabling deposition of multiple agents simultaneously within the tumor compartment.
Photodynamic therapy (PDT) is an effective modality against chemo- and radioresistant cancers due to its unique mechanisms of action, causing direct lipid and protein damage leading to mitochondrial-induced apoptosis that bypasses many mechanisms of classic multidrug-resistance5–9. This light-activated strategy makes use of a photosensitizing compound that is nontoxic in the dark but generates cytotoxic species when exposed to light of the appropriate wavelength. PDT has shown promising results in both preclinical10–12 and clinical PDAC studies13–15. PDT is particularly promising due to its efficacy against PDAC cells refractory to extreme gemcitabine dosages16. In a pilot clinical study, the median survival of a small patient cohort with inoperable, locally advanced PDAC was 12.5 months for patients that received PDT whereas 6–10 months is the median survival for current therapies of non-metastatic disease13. Recently, a phase I/II clinical dose escalation trial of BPD-PDT demonstrated feasibility, safety and efficacy in patients with locally advanced PDAC17. BPD-PDT had a 100% tumor necrosis response rate with no adverse events. In fact, PDT led to shrinkage of an initially inoperable tumor involving a major artery such that one of the patients then qualified for and underwent surgical tumor resection17.
Despite these promising results, the incidence of tumor recurrence is high for many advanced stage cancers even after a complete clinical response to frontline therapy. An important recurrence mechanism is the high adaptability of cancer cells to any therapeutic modality, specifically by utilizing inter- and intracellular signaling pathways to promote tumor cell survival, proliferation and metastasis. Up-regulation of VEGF signaling, a key mediator of tumor angiogenesis, has been observed in response to a number of treatment modalities, including: radiotherapy18, 19; chemotherapy20–22; and PDT23, 24. PDT is known to induce tumor hypoxia via oxygen consumption and microvascular shutdown resulting from PDT25, 26. PDT-induced tumor hypoxia is known to induce VEGF expression via induction of hypoxia inducible factor-1α (HIF-1α) stabilization and promotion of vegf transcription27, 28. This stress response to PDT corroborates prior reports of up-regulated VEGF signaling in response to a variety of therapies, including chemotherapy and radiotherapy18, 20, 22. In regards to PDT, Gomer and colleagues previously demonstrated that Photofin-PDT induces increased tumor VEGF expression via HIF-1α-induced vegf gene transcription27, 28. Our group has shown that increased tumor secreted VEGF levels in response to subcurative BPD-PDT occur via p38 mitogen-activated protein kinases (MAPK) and stress-activated protein kinase (c-Jun NH2-terminal kinase, JNK)29.
Thus, targeting the VEGF pathway in combination with cytotoxic modalities is a rational approach to help circumvent escape from the primary therapy. Multiple pathways ultimately need to be addressed simultaneously, possibly using cocktails of biologics and small molecular inhibitors. Selective tumor delivery and drug release will be key to limit additive systemic toxicities for such approaches. This study addresses the challenge of PDT and biologic agent co-delivery using nanoliposomes based on the hypothesis that an anti-VEGF mAb therapy combined with a photosensitizer-loaded nanoliposome can impede tumor recurrence and regrowth—using a single administration rather than chronic dosing such that the biologic therapy is spatiotemporally synchronized with the molecular response to the photocytotoxic arm.
Here, we report the development of nanoPALs that successfully enable the co-packaging of PDT (BPD) and anti-VEGF monoclonal antibody (bevacizumab) agents, and that the optimized nanoPAL formulation is significantly more effective than the administration of the individual, unpackaged drugs both in vitro and in a subcutaneous mouse model of PDAC. The nanoPAL builds on advances in chemical synthesis that offer exquisite control over the physicochemical properties of liposomes-enabling novel strategies for co-delivery and providing an ideal route for enhancing photosensitizer delivery while also neutralizing the tumor-localized burst in secreted VEGF immediately following PDT24, 28, 30–34. We hypothesized that a rationally-designed unilamellar liposome optimized for BPD packaging—can create a robust BPD microenvironment ideal for PDT. In this work, the following properties were considered and optimized: size; surface charge; drug-to-lipid ratio; lipid membrane packing; and, steric stabilization. While liposomal vectors are well characterized for tuning the loading of lipophilic therapeutic agents, they are relatively unexplored for formulating biologic agents, such as proteins, which require additional considerations to preserve biomolecular functionality both during synthesis as well as in the nanoliposomal environment35. In fact, for these reasons there are few reports of successful intracellular protein delivery using nanomaterials. Liposomes are an attractive technology but concerns remain about their compatibility with biomolecules due to the standard use of freeze-thaw cycling35.
Methods
Visudyne® (liposomal Verteporfin, BPD-MA) was a kind gift from QLT Inc. (Vancouver, BC, Canada). BPD-MA (Verteporfin) was purchased from VWR. Bevacizumab (Avastin®) was purchased from Genentech (San Francisco, CA). AlexaFlour488 or AlexaFlour680 were used to label bevacizumab and Slow Fade® Gold Antifade Reagent with DAPI was purchased from Molecular Probes (Invitrogen Life Technologies, Carlsbad, California). 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), Cholesterol and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000(DSPE-mPEG-2000), 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) were purchased from Avanti Polar Lipids (Alabaster, AL). Dialysis was carried out in phosphate-buffered saline (PBS) without calcium or magnesium at pH 7.4 using Float-A-Lyzer® G2, MWCO=300 kD (Spectrum Laboratories, Inc., Rancho Dominguez, CA). Human pancreatic ductal adenocarcinoma cell line AsPC-1 was purchased from ATCC (American Type Culture Collection, Manassas, VA) and maintenance media, RPMI 1640 (Roswell Park Memorial Institute), fetal bovine serum (FBS), trypsin with 0.5% EDTA and PBS were purchased from Mediatech, Inc (Manassas, VA). For in vivo studies, nude mice, 8 wks old weighing ~20g, were purchased from Charles River Laboratories Inc (Wilmington, MA).
Determination of release profile of NanoPAL
The release studies were carried out based on dialysis36, modified for nanoPALs (Supplementary Information).
Mouse model of subcutaneous pancreatic tumor
All animal studies were approved by the Subcommittee on Research Animal Care at the Massachusetts General Hospital, and conformed to the guidelines established by the NIH. All animal studies were conducted with appropriate humane care.
Results
Stability and efficacy of bevacizumab
To ensure the stability of the bevacizumab payload, we first carefully optimized the temperature at which the full affinity of bevacizumab is retained, in order to identify the lipids that can be used, which usually have differing acyl chain lengths and hence different transition membrane temperatures (Tm). In order to preserve the specificity and the therapeutic efficacy of bevacizumab during the formulation process, we first investigated the stability of the antibody at three different temperatures that were relevant for the eventual formulation of nanoPALs. We observed that bevacizumab incubated at 65°C for 1 h formed precipitants in a dose-dependent manner (Figures 1A, 1B). The mAb incubated at 65°C was also not recognized by a secondary antibody during western blotting whereas the secondary antibody recognized the mAbs incubated at both of the lower temperatures (Figure 1C).
Figure 1.

Bevacizumab incubated at 4, 45 and 65°C for 1 h at 1 mg/mL (A) and 2 mg/mL (B); (C) Western blots of bevacizumab treated at varying temperatures. An anti-rabbit IgG antibody was used to detect bevacizumab on the membrane; (D) Western blots showing the bevazicumab binding capacity to hVEGF-A (45kDa) at 1:80,000 at different exposure times from top to bottom. The samples in A were used as primary antibody to detect hVEGF-A protein on the gel. Each lane with sample is followed by pre-stained protein ladder to help identify the molecular weight of VEGF on the gel; (E) The bar graphs show the average signal intensities of the bevacizumab bands in figure D, normalized to the non-treated bevacizumab. The results are mean +/− s.d.’s (n =5). Asterisks denote statistically significant differences, *p<0.05 (t-test).
Next we directly investigated the potential loss of mAb affinity during the synthesis process at various temperatures. We observed that bevacizumab was still able to bind hVEGF after incubation at 45°C on the blots when the hVEGF was on the gel (1:80,000) (Figure 1D). We observed a significant decrease in the sensitivity of bevacizumab treated at 65°C (Figure 1E). Therefore, the optimal lipid mixture for nanoPAL should have a Tm ≤ 45°C. To meet this requirement, DPPC (Tm = 45°C) was used as the neutral lipid in the nanoPAL formulation.
Optimization of synthesis steps for nanoPAL
Using the ≤ 45°C temperature window defined above, we next investigated two potential synthesis procedures for the formulation of the nanoPAL. We generated two populations of nanoparticles—freeze-thawed vesicles and dehydrated-rehydrated vesicles (Figure 2A, 2B, 2C). Using transmission electron microscopy (TEM), we observed that freeze-thawed vesicles (Figure 2B) were more regular in shape and robust compared to the dehydrated-rehydrated vesicles (Figure 2C; Figure S1). Thus, the freeze thaw vesicles are more likely to retain and elicit nanoparticlulate properties and we utilized freeze-thawing nanoPAL synthesis for this study.
Figure 2.
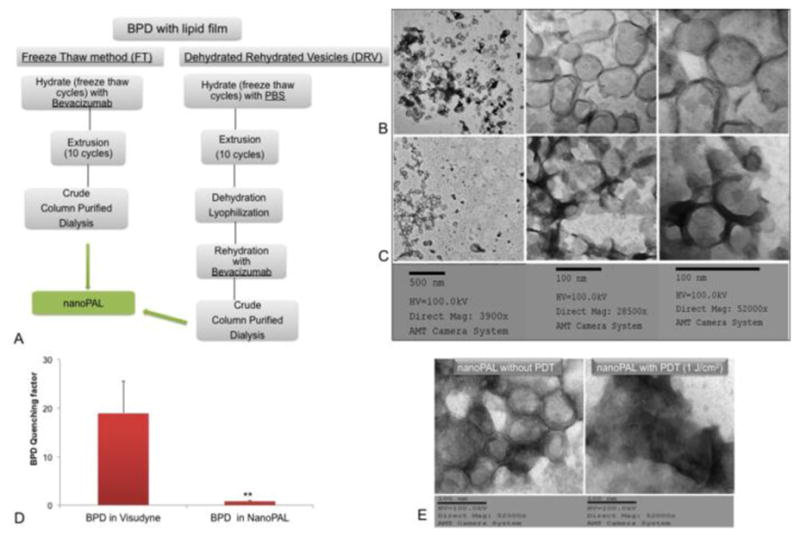
(A) Flow chart summarizing the synthesis steps to prepare freeze thawed or dehydrated and rehydrated vesicles; TEM microgrpahs of (B) freeze-thawed vesicles, (C)dehydrated-rehydrated vesicles; (D) The quenching factor (ΔRFU/Initial RFU) of BPD for Visudyne or nanoPALs. Results are mean +/− s.d.’s (n =4). **p=0.0016 (t-test); (E) TEM micrograph is a representative image of intact nanoPALs without any PDT exposure and nanoPALs exposed to a 1 J·cm−2 light dose, over 20 s, typically employed in the cytotoxicity studies.
NanoPALs maintain monomeric BPD in the lipid bilayer
Aggregation of BPD, which is hydrophobic, decreases the concentration of monomeric BPD and compromises photophysical activity due to quenched fluorescence, intersystem crossing and population of the triplet state, which impedes the generation of photo-generated cytotoxic species. At a given concentration of BPD, the nanoPAL formulation displayed significantly less quenching than Visudyne, suggesting the nanoPAL retains BPD in a monomeric form with a concomitant enhancement of PDT efficacy (Figure 2D). In order to explore potential physical effects of laser administration on the nanoPAL, we compared TEM images of nanoPAL without PDT and the nanoPAL subjected to a low-level light dose of 1J/cm2. We observed that nanoPALs exposed to PDT showed significant loss of nanoparticle structure compared to the control TEM, indicating potential for photo-triggered release of the nanoPAL payloads (Figure 2E).
PDAC cells uptake cationic nanoPALs
We tailored the nanoPAL charge to operate near the edge of neutrality but with sufficient cationic charge to target the vasculature and to enhance cellular-uptake32, 37, 38. Stronger cationic charges enhance tumor and cellular uptake but decrease selectivity and elicit unacceptable toxicities. An elegant study demonstrated that a minimal cationic lipid component is sufficient to achieve a favorable benefit-to-risk ratio32. With this in mind, DOTAP, a second generation synthetic cationic lipid, was incorporated to balance toxicity versus tumor uptake38. We performed a pilot screening study of a small set of preliminary nanoPALs with varying zetapotential (−9 mV, +1mV and +15 mV) and found that the minimal cationic charge (+15 mV) had a favorable uptake profile (Figure S2). We further tested the slightly cationic nanoPALs by imaging their in vivo tumor uptake in an orthotopic pancreatic cancer model (Figure S3). These pilot in vivo imaging experiments showed promising bulk tumor uptake of both BPD and bevacizumab labeled with a fluorescent dye. The BPD uptake and PDT efficacy of the nanoPAL precursors was determined at both 1 and 24 hours post injection. Although the imaging experiments show a higher uptake of BPD at 1 hour (Figure S3–E), we did not find any significant difference in efficacy between PDT at 1 hour versus 24 hours (Figure S3–F). The significant decrease in the BPD fluorescence at 24 hours may be due to intracellular aggregation of free BPD or uptake by serum proteins, which decreases the bioavailability after nanoPAL degradation in the tumor. Hence, we chose 1 h as the time point for PDT in further studies. After these screening and optimization steps, the final cationic nanoPAL (+15 mV) formulation was used for the remaining studies (henceforth termed nanoPAL and extensively characterized in the following sections).
Physicochemical properties of nanoPAL
The reproducibility of the nanoPAL was determined in terms of particle size (Table 1), 120 nm (standard deviation = 8.67%), and mean zeta potential +15mV (standard deviation = 0.33%) of several batches of formulation. The incorporation efficiency of BPD into the nanoPAL was around 80% of the initial concentration and the loading efficiency of bevacizumab ranged from 60–80% over individual batches of nanoPAL (Table 1).
Table 1.
Physicochemical Characteristics of nanoPALs
| NanoPAL | Particle size (nm) | PDI | Zetapotential (mV) |
|---|---|---|---|
| Batch-1 | 122 | 0.1 | 14.2 |
| Batch-2 | 100.9 | 0.1 | 14.9 |
| Batch-3 | 111.8 | 0.04 | 14.2 |
| Batch-4 | 110.7 | 0.05 | 14.4 |
Shelf life stability of nanoPAL
The shelf life stability of the nanoPAL was determined at days 1, 30 and 60 following synthesis. NanoPALs were stored at 4°C after nitrogen purging and the following properties were determined at each time point: particle size (Figure 3A); zetapotential (Figure 3B); polydispersity index (PDI) (Figure 3C); and, the percentage of BPD and bevacizumab retained (Figure 3D). The particle size, PDI and zetapotential were all stable and did not undergo any statistically significant change even after 60 days of storage. Although the nanoPALs retained a significant amount of BPD over time, loss of the initial BPD payload was significant within 30 days of storage. After 60 days, a nearly complete leakage of bevacizumab is observed. This indicates that a potential pharmaceutical strategy for clinical use would be freeze-drying nanoPAL and reconstituting it at the time of injection. For all the treatment studies reported here, the nanoPAL formulations were administered within 2 days of synthesis with storage at 4°C as described above.
Figure 3.
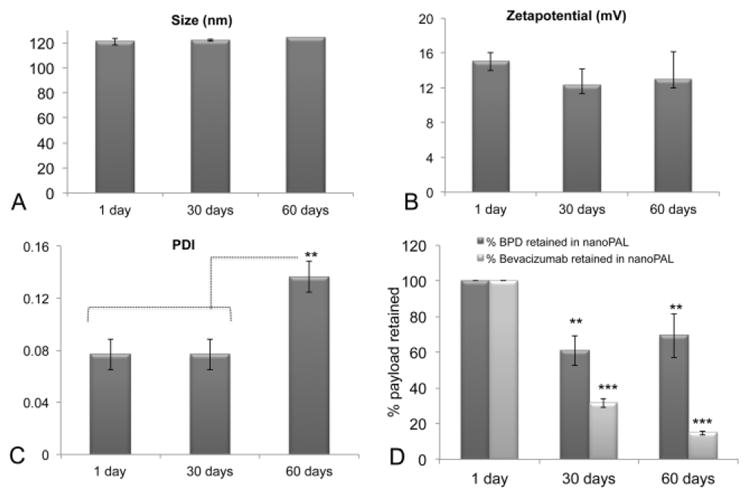
Shelf life stability and release profile of nanoPAL
NanoPAL stability over a period of 60 days in terms of particle size (A), zetapotential (B) and PDI (C) is shown; (D) Stability of nanoPALs in terms of association efficiency is plotted as the percent of payload leaked out during the storage period at 4°C under nitrogen atmosphere. The results are mean +/− s.d.’s, (n=3). Asterisks denote **P<0.01, ***P<0.0001 (Bonferroni post hoc- one-way ANOVA test).
NanoPAL therapeutic cargo release profiles
Cargo release rates have important implications on the bioavailability of the therapeutic payloads and the encapsulated agents are often not deemed fully bioavailable until release at the tumor site. We evaluated the release profiles of BPD and bevacizumab from nanoPAL in human serum at 37°C under dark conditions to simulate the in vivo microenvironment encountered by nanoPALs injected into the blood circulation. By controlling the lipid membrane packing, the nanoPAL was tuned to retain BPD for a residence time in blood stream for up to 72 hours to facilitate maximal payload delivery to the tumor. This was accomplished by designing nanoPAL to have a Tm slightly above 37°C via the inclusion of DPPC39 and by the relatively conservative use of PEG for steric stabilization. The release profile was interrogated using a modified dialysis bag method36, where the release media was periodically sampled. The release profiles show a 15% leakage of BPD and 85% of bevacizumab from nanoPAL when normalized to the leakage of Visudyne and free bevacizumab from the dialysis sample chamber into the reservoir over 72 hours, suggesting that the nanoPAL is a favorable controlled delivery system (Figure S4). We note that the nanoPAL payload stability enables spatiotemporal synchronized tumor delivery of BPD and bevacizumab prior to photoirradiation, and PDT, 1 hour after administration.
NanoPALs enhance the internalization of biologics into PDAC cells
NanoPALs were incubated with human pancreatic ductal adenocarcinoma (PDAC) AsPC1 cells at 37°C. Confocal microscopy visualized the intracellular localization of BPD and fluorescently labeled bevacizumab. Fluorescence-activated cell sorting (FACS) was used to observe a significant increase in bevacizumab uptake using nanoPALs (78% of cell population) as compared to the free drug cocktail group (0.5% of cell population), characterized as a shift in the FACS histogram heat map into the double positive quadrant, indicating that both BPD and bevacizumab were taken up by the cells. Similarly, at 24 h the dual agent uptake increased to 92% of the cell population in the nanoPAL group versus 5% of the cell population in the conventional agent formulation control group (Figure 4A). We observed an increased association of bevacizumab with the cells in the nanoPAL-treated group compared to free drug control at both 1 h and 24 h, which was subsequently internalized into the cytosol within 24 h (Figure 4B)
Figure 4.

(A) The heat maps show the quantitative uptake pattern of BPD and bevacizumab-AF488 by PDAC cells. The top panel shows gated quadrants generated by using an unstained PDAC population. (B) Confocal images represent the qualitative PDAC cellular uptake pattern of BPD (red) and bevacizumab-AF488 (green). Each image is a mosaic stitch of (3×3) ×3 = 27 random fields on each coverslip (n=3). Uptake of Visudyne + bevacizumab control (a, d) and nanoPAL-treated cells (b, e, c, f) 1 and 24 h post-administration. Scale bars are 50 μm (b,e), or 10 μm (c, f). (C) Percent cell viability of PDAC cells after the combination drug treatment with nanoPALs. Results are mean +/− s.d.’s, n ≥ 7 replicates per group. Asterisks denote statistically significant differences between nanoPAL treated groups, #p<0.05 compared to Visudyne+Bev, ***p<0.001 compared to NT (Bonferroni post hoc, one-way ANOVA test).
NanoPALs enhance treatment efficacy in vitro
The above results suggest that PDT of cells incubated with nanoPAL for 1 hour can lead to significant payload delivery to cells. To evaluate cytotoxicity after light application, the MTT assay was employed. We observed that the cell killing efficacy (Figure 4C) with nanoPAL was significantly higher (82% cell death) than with the conventional control (54% cell death).
NanoPAL tumor uptake in vivo
To ensure that BPD reaches the tumor and that bevacizumab delivery was within the therapeutically relevant time window of 6 hours post-PDT, we also performed drug delivery imaging studies (Table 2). Whole tumors were excised and imaged using a small animal hyperspectral fluorescence imaging system. Spectral unmixing analysis was applied to quantify bulk BPD and fluorescently labeled bevacizumab (Alexa Fluor 488) delivery. The bulk tumor accumulation was found to be similar for both nanoPAL and the Visudyne + bevacizumab controls (Figure 5). The whole tumors from the same experiment were then sectioned and stained for microvasculature (PECAM antibody), nuclei (DAPI) and human cancer cells (cytokeratin 8 antibody) to further analyze the bevacizumab localization amongst the various cell populations via confocal microscopy (Figure 5A–5E). Due to the potential for mechanical perturbation of the tissue sections and the tight packing of cells within the tumor, unambiguous resolution and quantification of enhanced intracellular mAb delivery was not possible. However, we observed that irrespective of the mode of delivery (i.e., nanoPAL or conventional formulations), there was a significant uptake and intratumoral accumulation of bevacizumab-AF488 within 2 h of injection (~70% tumor permeation), and no loss of signal was seen at 24 h in both groups (Figure 5C–5F), possibly due to weak lymphatic tumor flow and continued delivery via the circulation. Applying PDT 1 hour post nanoPAL injection was found to have no statistically significant effect on the bevacizumab AF488 distribution 2 hours after injection.
Table 2.
Tumor Distribution study design
| Group | Injection | N | Bev-AF488 Dose (mg/kg) |
|---|---|---|---|
| 1 | NanoPAL (BPD + BevAF488) | 4 | 15 |
| 2 | NanoPAL (BPD) + BevAF488 | 4 | ~15 |
| 3 | Visudyne + BevAF488 | 4 | ~15 |
Figure 5.

Confocal imaging of fluorescently labeled bevacizumab delivery in subcutaneous PDAC tumors. (A) A single tumor cross-section collected 2 h post-nanoPAL administration, imaged with 1.6 μm x-y sampling, illustrates bevacizumab (green) permeation of the tumor space (outlined in white based on a selective immunostain of human epithelial cancer cells). Scale bar, 1 mm. (B) Enlarged view of the region marked in A, demonstrating nanoPAL(bev) extravasation from microvessels labeled with a selective immunostain for mouse endothelial cells (magenta). Scale bar, 250 μm. (C) Fluorescence colocalization of bevacizumab or nanoPAL (bev) with microvessels, and (D) with the extravascular tumor space at various time-points. Regardless of the formulation, bevacizumab enters the tumor interstitium within 2 h. (E) Tumoral bevacizumab payload, and percent tumor permeation. The results are mean +/− s.e.m, n ≥ 3 mice per group. Asterisks denote statistically significant differences compared to the corresponding treatment at 1 h, *p<0.05 **p<0.01 ***p<0.001 (Kruskal-Wallis one-way ANOVA test).
NanoPALs significantly enhance PDT in vivo
We next tested nanoPAL tumor destruction in a xenograft, subcutaneous model of pancreatic cancer using AsPC1 cells. Non-invasive tumor measurement with calipers permitted longitudinal studies of treatment efficacy. Therapeutic agents were injected via the tail vein and light for PDT was administered transcutaneously 1 h after injection. To compare the efficacy of the nanoPAL packaging on PDT with Visudyne, studies were performed using nanoPAL containing only BPD and no bevacizumab—nanoPAL(BPD). The tumor volume curves show a significant enhancement of PDT-related tumor destruction with the nanoPAL(BPD) versus Visudyne (Figure 6A). Similarly, to determine the efficacy of bevacizumab monotherapy versus nanoPAL-delivered bevacizumab monotherapy, studies were performed using nanoPAL containing only bevacizumab and no BPD—nanoPAL(Bev) (Figure 6B). We did not observe any significant differences between these treatment groups, potentially attributing to the similar overall amount of mAb accumulation as seen in the tumor distribution studies (Figure 5), and the lack of increased intracellular production of VEGF in the absence of PDT. In addition, bevacizumab monotherapy normally requires chronic treatment cycles over a period of several weeks to achieve significant tumor reduction. It is also possible that nanoPAL-associated bevacizumab was not fully liberated to interact with VEGF until light irradiation disrupted the liposome structure (e.g., via lipid peroxidation) to release the antibody.
Figure 6.

Absolute tumor volumes recorded after single i.v. bolus administration of (A) Visudyne or nanoPAL(BPD) on day 1 as indicated on the timeline(bottom). *p = 0.0176 indicates statistical significance between untreated (NT) and nanoPAL(BPD)-treated mice. (B) Bevacizumab versus nanoPAL(bev) monotherapy; (C) Visudyne+bevacizumab versus nanoPAL(BPD+bevacizumab) combination therapy. **p=0.0051 ***p=0.000016, indicate statistical significance compared with NT; #p=0.00016 denotes significant difference between Visudyne+bevacizumab and nanoPAL(BPD+bevacizumab), number of mice allocated to each group is indicated in the parenthesis. Results are mean +/− s.e.m. (D) Percent body weight changes normalized to initial weight; (E) Vertical scatterplot depicts the distribution of tumor weights of mice from 3 different experiments. Each data point represents a single mouse. Statistics were performed using Mann Whitney U analysis.
NanoPALs enhance the combinatorial regimen efficacy in vivo
We next explored the therapeutic efficacy of the combinatorial regimen of PDT with bevacizumab. In the nanoPAL treatment group, 24 h post PDT, we observed a gradual incidence of temporary edema at the tumor site followed by erythema, which both gradually resolved after the treatment. On days 3 to 4 (Figure 6C), all mice treated with nanoPAL showed extensive tumor necrosis (n=8), and there was almost no tumor regrowth for 35 days after the treatment (Figure 6C) in all mice. At the end of the experiment, 33% of the nanoPAL-treated mice appeared to have a complete response and residual tumors exhibited minimal regrowth by day 35. In contrast, at nanoPAL-matched PDT and mAb doses, the Visudyne and bevacizumab combination therapy was able to stabilize the tumor but did not lead to measurable tumor destruction (Figure 6C). These observations were confirmed at the end of the study when tumors from each group were harvested and weighed (Figure 6E). Treatment toxicity was also monitored via body weight loss. All treatment groups were within standard limits of toxicity (Figure 6D).
NanoPAL-treated tumors show extensive necrosis
To further analyze the extent of tumor necrosis, haematoxylin and eosin staining was used to study tumors that received nanoPAL-PDT. Tumor sections from mice without light exposure revealed that the cells were aggressively dividing (Figure 7A). Abundant and irregular nuclei were also seen with a clear and continuous peripheral layer surrounding the tumor boundary, indicating an actively growing tumor. In contrast, tumors exposed to nanoPAL-PDT (72 h post PDT) exhibited significantly fewer cancer cells and mitotic nuclei than untreated controls. Morphological inspection revealed distorted plasma membranes indicating extensive necrosis (Figure 7B).
Figure 7.

The figure shows a representative H&E-stained, 5-μm-thick tumor section at 5× and 20× magnification pre-PDT (A) and 72 hrs post-PDT (B).
Discussion
NanoPALs enable concomitant, spatiotemporally synchronized photodamage with release of biologic agents to hinder molecular signaling pathways associated with PDAC treatment escape. Here we demonstrate the co-delivery of bevacizumab with BPD to pancreatic tumor cells. The nanoPALs developed here ensure that the biologic released at the tumor site retains its specificity to abrogate its molecular target and the PDT agent remains monomeric to elicit its photophysical reactions. The surface charge of nanoPAL was also optimized as negatively charged molecules like GAG and heparin sulfate are frequently over-expressed by cancer cells and tumor-associated vasculature, which render charge-dependent specificity to cationic carrier systems40. Cationic charge also affects toxicity via enhanced uptake by a variety of normal cell types, especially within the reticuloendothelial system (including the liver, kidneys and spleen)41, 42. Thus, cationic lipids were conservatively incorporated into the design to balance the tradeoff in tumor delivery versus systemic toxicity.
PDAC remains a lethal cancer with a dismal prognosis. Existing clinical strategies, although valuable for palliation, fail to significantly impact patient survival. There is a critical need for the development of rationally designed combinatorial therapies where multiple, mechanistically distinct and complimentary anticancer agents are delivered simultaneously to leverage existing FDA approved treatments such as Folfirinox43, 44, gemcitabine-based combinations45–47 and chemo-biologic drug based combinations48, 49. In order to achieve a higher therapeutic index, cytotoxic agents may be combined with complimentary agents that target key pathways promoting survival and regrowth keeping in mind that chronic administration of toxic agents should be avoided. A more efficient approach delivers molecular-targeted agents at time points critical for suppressing transient signaling bursts elicited by finite cytotoxic therapies such as PDT, radiotherapy and potentially burst-release chemotherapy with the advent of optically activated nanodelivery systems. In this way, cancer cells are forced to face a far more formidable barrier for re-establishing a thriving cancer microenvironment.
The utility and feasibility of PDT continues to improve16, 50–52 and despite a proven clinical record as an adjuvant therapy after the surgical resection of tumors it is often used after all other options have failed50. Its unique mechanisms induce cell death pathways that bypass classical drug resistance and BPD-PDT is under development as a novel therapy for PDAC based on promising phase I/II trials17. Visudyne, as used in the PDAC clinical trials, is a FDA-approved photosensitizer for treating age-related macular degeneration and is a liposomal formulation of mainly aggregated BPD, although, upon intravenous injection the BPD partitions from the lipid to blood plasma proteins where it regains some monomeric photochemical activity53, 54. Our strategy to enhance PDT uses a rationally-designed nanoparticle that protects BPD from the blood plasma while simultaneously delivering active, monomeric BPD to the tumor, which enhances PDT potency. The nanoPAL was designed to be stable in plasma for a few hours, long enough to deliver a therapeutic payload to the tumor. Differing mechanisms of cellular uptake between nanoPAL and BPD-bound serum proteins is a potential explanation for the enhanced potency of the nanoPAL versus Visudyne. Diminished reactive oxygen species generation from plasma bound BPD is a possible but unlikely explanation as efficient triplet state formation of BPD-serum albumin complexes has been observed54. Both the endocytosis (lysosomal) pathway and membrane localization pathways (e.g. through liposome-plasma membrane fusion) can be effective forms of BPD delivery. Once released, BPD tends to preferentially accumulate within the mitochondria and facilitates mitochondrial PDT. In contrast, lysosomal BPD-PDT using activatable immunoconjugates, for example, requires 24 h post-delivery to reach maximum efficacy but the molecular-targeted, activation feature increases photodamage selectivity for wide-field irradiation of disseminated microscopic tumors55. Overall, this suggests that the nanoPAL enables efficient delivery of monomeric BPD to the microvasculature and parenchyma of macroscopic tumors.
Based on its clinical promise, PDT is also being developed as a combinatorial treatment modality56. One of the salient features of PDT is its ability to counteract resistance mechanisms exhibited by cancer cells and the tumor microenvironment against traditional modalities. For instance, PDT is observed to reverse chemoresistance, re-sensitizing drug-resistant cancer cells to chemotherapy19, but as for all cytotoxic treatments, the surviving cancer cells activate a multitude of pro-survival signaling pathways to promote recurrence. VEGF is a key cytokine that promotes tumor regrowth, and it is up-regulated and secreted by cancer cells following therapy18, 20–22, 29. PDT, and potentially other finite treatments, stimulate transient increases in VEGF production24. Therefore, there exists a window of opportunity to mop up this transient signaling cascade.
We reasoned that inhibiting VEGF within the tumor cells—where it is produced—may be optimal, as opposed to the conventional use of bevacizumab that primarily targets circulating VEGF. Neutralizing tumor-derived VEGF prior to extracellular release would obviate the signal cascade prior to activation of the tumor microenvironment and recruitment of bone marrow-derived progenitors and other systemic effects. The in vitro studies indicate nanoPAL-mediated intracellular delivery of bevacizumab. In vivo combinatorial studies indicate that nanoPAL dramatically enhanced efficacy compared to the combination of conventional treatments, strongly suggesting that advances in liposomal co-packaging can add potency to combinations of leading therapeutic agents.
As breakthroughs in the molecular etiology of pancreatic cancer and molecular mechanisms of treatment escape continue, investigating multimodal treatment approaches that leverage these new discoveries seems germane. With this mindset, the nanoPAL is a robust liposome-based nanodelivery system capable of incorporating cytotoxic and biologic agents. Here, nanoPAL co-packaging enhanced the combination of BPD-PDT with anti-VEGF therapy. NanoPAL derivatives can be formulated to co-deliver a variety of combinatorial payloads targeted to additional mechanistic pathways by incorporating other biologics or small molecule inhibitors (e.g., co-inhibition of VEGF and c-MET57), which is likely key for addressing the multiple, crosstalking mechanisms of treatment escape.
Supplementary Material
Table 3.
Therapeutic response study design
| Group # | Treatment | N | Bev Dose (mg/kg) | BPD Dose (mg/kg) |
|---|---|---|---|---|
| 1 | Nontreated | 10 | N/A | N/A |
| 2 | Visudyne (PDT) | 8 | N/A | 0.5 |
| 3 | Bevacizumab Only | 5 | 15 | N/A |
| 4 | Visudyne + Bevacizumab (PDT) | 7 | 15 | 0.5 |
| 5 | NanoPAL (Bevacizumab) | 8 | 15 | N/A |
| 6 | NanoPAL (BPD) | 9 | N/A | 0.5 |
| 7 | NanoPAL (BPD + Bevacizumab) | 7 | 15 | 0.5 |
Acknowledgments
Funding: This study was supported by the National Institutes of Health (NIH) – National Cancer Institute (NCI) Grants F32CA144210, RO1CA160998 and PO1CA084203.
We thank Dr. R. Rox Anderson for interim support of S.T.; Dr. Jie Zhao, Margaret Sherwood and Danny Cao (Wellman Center Photopathology Core) for expert technical assistance with TEM and FACS; Dr. Esther Oliva (Dept. of Pathology, Massachusetts General Hospital) for providing expert histopathology; and Dmitriy Timerman for valuable discussions.
Abbreviations
- mAb
Monoclonal Antibody
- BPD
Benzoporphyrin Derivative
- PDT
Photodynamic Therapy
- nanoPAL
nanophotoactivatable liposomes
- PDAC
Pancreatic Ductal Adenocarcinoma
- VEGF
Vascular Endothelial Growth Factor
- MAPK
Mitogen Activated Protein Kinases
- HIF
Hypoxia Inducible Factor
- JNK
c-Jun NH2-terminal kinase
- MET
hepatocyte growth factor receptor
- PDI
Poly Dispersity Index
- DAPI
4′,6-diamidino-2-phenylindole
- DPPC
2-dipalmitoyl-sn-glycero-3-phosphocholine
- DOTAP
1,2-dioleoyl-3-trimethylammonium-propane
- DSPE-PEG2000
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000
Footnotes
Financial disclosure: The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Horner MJ, Ries L, Krapcho M, Neyman N, Aminou R, Howlader N, et al. Seer cancer statistics review, 1975–2006; J Natl Cancer Inst Bethesda, MD. 2009 [Google Scholar]
- 3.Bardeesy N, DePinho R. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 4.Bardeesy N. Molecular Signaling Pathways in Pancreatic Cancer. In: Von Hoff DD, Evans DB, Hruban RH, editors. Pancreatic Cancer. 1. Boston: Jones and Bartlett Publishers; 2005. pp. 43–70. [Google Scholar]
- 5.Kessel D, Erickson C. Porphyrin photosensitization of multiresistant cell types. Photochem Photobiol. 1992;55:397–9. doi: 10.1111/j.1751-1097.1992.tb04253.x. [DOI] [PubMed] [Google Scholar]
- 6.Canti G, Lattuada D, Morelli S, Nicolin A, Cubeddu R, Taroni P, et al. Efficacy of photodynamic therapy against doxorubicin-resistant murine tumors. Cancer Lett. 1995;93:255–9. doi: 10.1016/0304-3835(95)03818-h. [DOI] [PubMed] [Google Scholar]
- 7.Duska LR, Hamblin MR, Miller JL, Hasan T. Combination Photoimmunotherapy and Cisplatin: Effects on Human Ovarian Cancer Ex Vivo. J Natl Cancer Inst. 1999;91:1557–63. doi: 10.1093/jnci/91.18.1557. [DOI] [PubMed] [Google Scholar]
- 8.Preise D, Mazor O, Koudinova N, Liscovitch M, Scherz A, Salomon Y. Bypass of Tumor Drug Resistance by Antivascular Therapy. Neoplasia. 2003;5:475–80. doi: 10.1016/s1476-5586(03)80031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holt JJ, Gannon MK, Tombline G, McCarty TA, Page PM, Bright FV, et al. A cationic chalcogenoxanthylium photosensitizer effective in vitro in chemosensitive and multidrug-resistant cells. Bioorg Med Chem. 2006;14:8635–43. doi: 10.1016/j.bmc.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 10.Xie Q, Jia L, Liu YH, Wei CG. Synergetic anticancer effect of combined gemcitabine and photodynamic therapy on pancreatic cancer in vivo. World J Gastroentero. 2009;15:737–41. doi: 10.3748/wjg.15.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mikvy P, Messman H, MacRobert AJ, Pauer M, Sams VR, Davies CL, et al. Photodynamic therapy of a transplanted pancreatic cancer model using meta-tetrahydroxyphenylchlorin (mTHPC) Br J Cancer. 1997;96:713–8. doi: 10.1038/bjc.1997.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajri A, Coffy S, Vallat F, Evrard S, Marescaux J, Aprahamian M. Human pancreatic carcinoma cells are sensitive to photodynamic therapy in vitro and in vivo. Br J Surg. 1999;86:899–906. doi: 10.1046/j.1365-2168.1999.01132.x. [DOI] [PubMed] [Google Scholar]
- 13.Bown SG, Rogowska AZ, Whitelaw DE, Lees WR, Lovat LB, Ripley P, et al. Photodynamic therapy for cancer of the pancreas. Gut. 2002;50:549–57. doi: 10.1136/gut.50.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayaru L, Bown SG, Pereira SP. Photodynamic therapy for pancreatic and biliary tract carcinoma. Int J Gastrointest Cancer. 2005;35:1–13. doi: 10.1385/IJGC:35:1:001. [DOI] [PubMed] [Google Scholar]
- 15.Fan BG, Andren-Sandberg A. Photodynamic therapy for pancreatic cancer. Pancreas. 2007;34:385–9. doi: 10.1097/mpa.0b013e3180439c50. [DOI] [PubMed] [Google Scholar]
- 16.Celli JP, Solban N, Liang A, Pereira SP, Hasan T. Verteporfin-based photodynamic therapy overcomes gemcitabine insensitivity in a panel of pancreatic cancer cell lines. Lasers Surg Med. 2011;43:565–74. doi: 10.1002/lsm.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer. 2014;110:1698–704. doi: 10.1038/bjc.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorski DH, Beckett MA, Jaskowiak NT, Calvin DP, Mauceri HJ, Salloum RM, et al. Blockade of the Vascular Endothelial Growth Factor Stress Response Increases the Antitumor Effects of Ionizing Radiation. Cancer Res. 1999;59:3374–8. [PubMed] [Google Scholar]
- 19.Abdollahi A, Lipson KE, Han X, Krempien R, Trinh T, Weber KJ, et al. SU5416 and SU6668 attenuate the angiogenic effects of radiation-induced tumor cell growth factor production and amplify the direct anti-endothelial action of radiation in vitro. Cancer Res. 2003;63:3755–63. [PubMed] [Google Scholar]
- 20.Tran J, Master Z, Yu JL, Rak J, Dumont DJ, Kerbel RS. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc Natl Acad Sci U S A. 2002;99:4349–54. doi: 10.1073/pnas.072586399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wild R, Dings RP, Subramanian I, Ramakrishnan S. Carboplatin selectively induces the VEGF stress response in endothelial cells: Potentiation of antitumor activity by combination treatment with antibody to VEGF. Int J Cancer. 2004;110:343–51. doi: 10.1002/ijc.20100. [DOI] [PubMed] [Google Scholar]
- 22.Justinger C, Schluter C, Oliviera-Frick V, Kopp B, Rubie C, Schilling MK. Increased growth factor expression after hepatic and pancreatic resection. Oncol Rep. 2008;20:1527–31. [PubMed] [Google Scholar]
- 23.Kosharskyy B, Solban N, Chang SK, Rizvi I, Chang Y, Hasan T. A Mechanism-Based Combination Therapy Reduces Local Tumor Growth and Metastasis in an Orthotopic Model of Prostate Cancer. Cancer Res. 2006;66:10953–8. doi: 10.1158/0008-5472.CAN-06-1793. [DOI] [PubMed] [Google Scholar]
- 24.Chang SK, Rizvi I, Solban N, Hasan T. In vivo Optical Molecular Imaging of Vascular Endothelial Growth Factor for Monitoring Cancer Treatment. Clin Cancer Res. 2008;14:4146–53. doi: 10.1158/1078-0432.CCR-07-4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B, Pogue BW, Luna JM, Hardman RL, Hoopes PJ, Hasan T. Tumor vascular permeabilization by vascular-targeting photosensitization: effects, mechanism, and therapeutic implications. Clin Cancer Res. 2006;12:917–23. doi: 10.1158/1078-0432.CCR-05-1673. [DOI] [PubMed] [Google Scholar]
- 26.Mallidi S, Watanabe K, Timerman D, Schoenfeld D, Hasan T. Prediction of tumor recurrence and therapy monitoring using ultrasound-guided photoacoustic imaging. Theranostics. 2015;5:289–301. doi: 10.7150/thno.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrario A, von Tiehl KF, Rucker N, Schwarz MA, Gill PS, Gomer CJ. Antiangiogenic treatment enhances photodynamic therapy responsiveness in a mouse mammary carcinoma. Cancer Res. 2000;60:4066–9. [PubMed] [Google Scholar]
- 28.Gomer CJ, Ferrario A, Luna M, Rucker N, Wong S. Photodynamic therapy: Combined modality approaches targeting the tumor microenvironment. Lasers Surg Med. 2006;38:516–21. doi: 10.1002/lsm.20339. [DOI] [PubMed] [Google Scholar]
- 29.Solban N, Pål SK, Alok SK, Sung CK, Hasan T. Mechanistic Investigation and Implications of Photodynamic Therapy Induction of Vascular Endothelial Growth Factor in Prostate Cancer. Cancer Res. 2006;66:5633–40. doi: 10.1158/0008-5472.CAN-06-0604. [DOI] [PubMed] [Google Scholar]
- 30.Jiang F, Zhang X, Kalkanis SN, Zhang Z, Yang H, Katakowski M, et al. Combination Therapy with Antiangiogenic Treatment and Photodynamic Therapy for the Nude Mouse Bearing U87 Glioblastoma. Photochem Photobiol. 2008;84:128–37. doi: 10.1111/j.1751-1097.2007.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen TM. Liposomes. Opportunities in drug delivery. Drugs. 1997;54(Suppl 4):8–14. doi: 10.2165/00003495-199700544-00004. [DOI] [PubMed] [Google Scholar]
- 32.Campbell RB, Fukumura D, Brown EB, Mazzola LM, Izumi Y, Jain RK, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62:6831–6. [PubMed] [Google Scholar]
- 33.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–743. [PubMed] [Google Scholar]
- 34.Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, et al. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568–72. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 35.Fu A, Tang R, Hardie J, Farkas ME, Rotello VM. Promises and pitfalls of intracellular delivery of proteins. Bioconjugate Chem. 2014;25:1602–8. doi: 10.1021/bc500320j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crielaard BJ, van der Wal S, Le HT, Bode AT, Lammers T, Hennink WE, et al. Liposomes as carriers for colchicine-derived prodrugs: Vascular disrupting nanomedicines with tailorable drug release kinetics. Eur J Pharm Sci. 2012;45:429–35. doi: 10.1016/j.ejps.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 37.Campbell RB, Balasubramanian SV, Straubinger RM. Influence of cationic lipids on the stability and membrane properties of paclitaxel-containing liposomes. J Pharm Sci. 2001;90:1091–105. doi: 10.1002/jps.1063. [DOI] [PubMed] [Google Scholar]
- 38.Tangutoori S, Ohta A, Gatley S, Campbell RB. Repurposing an Erstwhile Cancer Drug: A Quantitative and Therapeutic Evaluation of Alternative Nanosystems for the Delivery of Colchicine to Solid Tumors. J Cancer Sci Ther. 2014;6:236–46. [Google Scholar]
- 39.Loura LM, Fernandes F, Fernandes AC, Ramalho JP. Effects of fluorescent probe NBD-PC on the structure, dynamics and phase transition of DPPC. A molecular dynamics and differential scanning calorimetry study. Biochim Biophys Acta. 2008;1778:491–501. doi: 10.1016/j.bbamem.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 40.Denekamp J. The tumour microcirculation as a target in cancer therapy: a clearer perspective. Eur J Clin Invest. 1999;29:733–6. doi: 10.1046/j.1365-2362.1999.00558.x. [DOI] [PubMed] [Google Scholar]
- 41.Dokka S, Toledo D, Shi X, Castranova V, Rojanasakul Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm Res. 2000;17:521–5. doi: 10.1023/a:1007504613351. [DOI] [PubMed] [Google Scholar]
- 42.Filion MC, Phillips NC. Major limitations in the use of cationic liposomes for DNA delivery. Int J Pharm. 1998;120:159–70. [Google Scholar]
- 43.Lorgis V, Chauffert B, Gentil J, Ghiringhelli F. Influcence of localization of primary tumor on effectiveness of 5-fluorouracil/leucovorin combined with irinotecan and oxaliplatin (FOLFIRINOX) in patients with metastatic pancreatic adenocarcinoma: a retrospective study. Anticancer Res. 2012;32:4125–30. [PubMed] [Google Scholar]
- 44.Assaf E, Verlinde-Carvalho M, Delbaldo C, Grenier J, Sellam Z, Pouessel D, et al. 5-fluorouracil/leucovorin combined with irinotecan and oxaliplatin (FOLFIRINOX) as second-line chemotherapy in patients with metastatic pancreatic adenocarcinoma. Oncology (Willston Park) 2011;80:301–6. doi: 10.1159/000329803. [DOI] [PubMed] [Google Scholar]
- 45.Francois E, Bennouna J, Chamorey E, Etienne-Grimaldi MC, Renée N, Senellart H, et al. Phase I Trial of Gemcitabine Combined with Capecitabine and Erlotinib in Advanced Pancreatic Cancer: A Clinical and Pharmacological Study. Chemotherapy. 2012;58:371–80. doi: 10.1159/000343969. [DOI] [PubMed] [Google Scholar]
- 46.Mattiucci GC, Ippolito E, D’Agostino GR, Alfieri S, Antinori A, Crucitti A, et al. Long-term Analysis of Gemcitabine-based Chemoradiation after Surgical Resection for Pancreatic Adenocarcinoma. Ann Surg Oncol. 2013;20:423–9. doi: 10.1245/s10434-012-2767-7. [DOI] [PubMed] [Google Scholar]
- 47.Hong DS, Choe JH, Naing A, Wheler JJ, Falchook GS, Piha-Paul S, et al. A phase 1 study of gemcitabine combined with dasatinib in patients with advanced solid tumors. Invest New Drugs. 2013;31:918–26. doi: 10.1007/s10637-012-9898-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brell JM, Krishnamurthi SS, Rath L, Bokar JA, Savvides P, Gibbons J, et al. Phase I trial of sunitinib and gemcitabine in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;70:547–53. doi: 10.1007/s00280-012-1936-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sweeney CJ, Chiorean EG, Verschraegen CF, Lee FC, Jones S, Royce M, et al. A phase I study of sunitinib plus capecitabine in patients with advanced solid tumors. J Clin Oncol. 2010;28:4513–20. doi: 10.1200/JCO.2009.26.9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, et al. Imaging and photodynamic therapy: mechanisms, monitoring, and optimization. Chem Rev. 2010;110:2795–838. doi: 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindenmann J, Matzi V, Neuboeck N, Anegg U, Baumgartner E, Maier A, et al. Individualized, multimodal palliative treatment of inoperable esophageal cancer: Clinical impact of photodynamic therapy resulting in prolonged survival. Lasers Surg Med. 2012;44:189–98. doi: 10.1002/lsm.22006. [DOI] [PubMed] [Google Scholar]
- 52.Lee Y, Baron ED. Photodynamic Therapy: Current Evidence and Applications in Dermatology. Semin Cutan Med Surg. 2011;30:199–209. doi: 10.1016/j.sder.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 53.Chowdhary RK, Shariff I, Dolphin D. Drug release characteristics of lipid based benzoporphyrin derivative. J Pharm Sci. 2003;6:13–9. [PubMed] [Google Scholar]
- 54.Aveline BM, Hasan T, Redmond RW. The effects of aggregation, protein binding and cellular incorporation on the photophysical properties of benzoporphyrin derivative monoacid ring A (BPDMA) J Photochem Photobiol B. 1995;30:161–9. doi: 10.1016/1011-1344(95)07174-z. [DOI] [PubMed] [Google Scholar]
- 55.Spring BQ, Abu-Yousif AO, Palanisami A, Rizvi I, Zheng X, Mai Z, et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc Natl Acad Sci U S A. 2014;111:E933–42. doi: 10.1073/pnas.1319493111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rizvi I, Celli JP, Evans CL, Abu-Yousif AO, Muzikansky A, Pogue BW, et al. Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer. Cancer Res. 2010;70:9319–28. doi: 10.1158/0008-5472.CAN-10-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, et al. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71:4758–68. doi: 10.1158/0008-5472.CAN-10-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.