

Abstract
Mesenchymal stromal cells (MSCs) have shown promise as treatment for graft-versus-host disease (GvHD) following allogeneic bone marrow transplantation (alloBMT). Mechanisms mediating in vivo effects of MSCs remain largely unknown, including their biodistribution following infusion. To this end, human bone-marrow derived MSCs (hMSCs) were injected via carotid artery (IA) or tail vein (TV) into allogeneic and syngeneic BMT recipient mice. Following xenogeneic transplantation, MSC biodistribution was measured by bioluminescence imaging (BLI) using hMSCs transduced with a reporter gene system containing luciferase and by scintigraphic imaging using hMSCs labeled with [99mTc]-HMPAO. Although hMSCs initially accumulated in the lungs in both transplant groups, more cells migrated to organs in alloBMT recipient as measured by in vivo BLI and scintigraphy and confirmed by ex vivo BLI imaging, immunohistochemistry and quantitative RT-PCR. IA injection resulted in persistent whole–body hMSC distribution in alloBMT recipients, while hMSCs were rapidly cleared in the syngeneic animals within one week. In contrast, TV-injected hMSCs were mainly seen in the lungs with fewer cells traveling to other organs. Summarily, these results demonstrate the potential use of IA injection to alter hMSC biodistribution in order to more effectively deliver hMSCs to targeted tissues and microenvironments.
Keywords: Mesenchymal stromal cell, Graft-versus-host disease, Arterial injection, Biodistribution, Radionuclide imaging
Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) plays a significant role in the treatment of various malignant and nonmalignant diseases, including hematological disorders, metabolic storage disease, immune deficiencies and leukemia. Donor T-lymphocytes assist hematopoietic engraftment, restore adaptive immunity, mediate graft-versus-leukemia (GvL) activity, but also cause acute graft- versus-host disease (GvHD). GvHD is a leading cause of morbidity and mortality in allogeneic HSCT patients[1], resulting in mortality rates of around 20% despite the use of immunosuppressive drugs largely targeting T-cell activation and proliferation[2].
Mesenchymal stromal cells (MSCs) are multipotent adult progenitors that possess immunosuppressive properties, including the ability to attenuate lymphocyte activation, pro-inflammatory cytokine induction and proliferation[3]. Furthermore, MSCs from numerous sources, including bone marrow and umbilical cord blood, can be easily expanded and have minimal infusion-related toxicity and side effects[4]. Given their immunosuppressive and expansion capabilities, MSCs have been used as cellular therapy for GvHD[5]. In this regard, we have recently shown that human bone marrow-derived MSCs (hMSCs) injected via tail vein (TV) can attenuate acute GvHD severity while maintaining potent GvL activity following murine allogeneic bone marrow transplantation (BMT)[6].
We used bioluminescence imaging (BLI) to show that hMSCs migrated to GvHD target tissues including small and large bowel wherein they reduced GvHD-mediated cytotoxicity. Furthermore, novel cryoimaging demonstrated that MSCs decreased T-cell proliferation in secondary lymphoid organs like the spleen, providing mechanistic insight as to how hMSCs mediate in vivo effects[6].
Despite these observations, optimal delivery of hMSCs to attain greatest clinical effect and use of in vivo imaging relevant to the clinical setting remain undefined. For example, despite its convenience, simplicity and minimal invasiveness, intravenous injection results in significant hMSC deposition in the lung with markedly reduced localization to target tissues of interest[6–8], reflecting disparity of size between the smaller lung capillaries (10-15 μm) and larger MSCs (20-25 μm) as well as the adherent features of MSCs themselves. Therefore, homogenous or targeted distribution of MSCs to other target organs is suboptimal. Furthermore, rapid deterioration in hMSC signal intensity occurs within two weeks after TV injection[6]. Similar to hMSCs delivery, the techniques used to image hMSCs in animal models are also restricted; as they are not readily applicable to the clinical setting. Therefore, the purpose of the current study was to investigate if delivery of hMSCs via intra-arterial (IA) injection could enhance cell delivery to target organs and to validate radionuclide imaging as an imaging modality to measure hMSC biodistribution following xenogeneic transplantation.
Materials and Methods
Lentiviral vector construction
The construction of the second-generation, self-inactivating lentiviral vector pHR9-mnd-luc-mrfp-ttk was described previously[7].
Transient transfection and virus preparation
Viral particles were produced by triple transient co-transfection of 293T/17 cells (ATCC, Manassas, VA) with pHR'MND-LRT, pCMVΔR8.91 (packaging vector) and pMD.G (VSVG pseudotyping vector) as previously described[9]. Titer was determined by transduction of 293T cells (ATCC) followed by flow cytometry (iCyt Reflection, IL, USA) at the Cancer Center Core Facility of Case Western Reserve University.
Isolation and transduction of human bone marrow-derived mesenchymal stromal cells
Human mesenchymal stromal cells (hMSCs) were derived from bone marrow (BM) aspirates from healthy donors who had given consent in accordance with the Institutional Review Board of University Hospitals Case Medical Center. Specimens were collected and processed by the Hematopoietic Stem Cell Facility of the Case Comprehensive Cancer Center (P30 CA 43703), wherein hMSCs were expanded and characterized by surface protein expression (CD45-, CD73+, CD105+) and differentiated into adipocytes, chondrocytes and osteocytes as previously described[10]. Primary MSCs were seeded in 175 cm2 flasks to 30% confluence in hMSC growth medium supplemented with 1 g/L glucose, 10% hMSC-tested FBS (Gibco), 2 mM GlutaMAX, penicillin (100 U/mL) and streptomycin (100 μg/mL). The hMSCs were incubated in virus medium at a multiplicity of infection (MOI) of 4 in a total of 25 mL hMSC growth medium containing 8 μg/mL polybrene. The transfection protocol was repeated one time for all hMSC cultures. In each round, cells were incubated with viral supernatant for 8 hours, then in hMSC growth medium for 16 hours. After the second round, fresh medium was added to each flask and cells were incubated for an additional 24-48 hours. Cells were then passaged 1:4 and grown to 80-100% confluence. Three days post-transduction, cells were viewed on an Eclipse TE200 inverted microscope equipped with a fluorescent source and CCD camera. Transduction efficiency was determined by fluorescence-activated cell sorting (FACS) analysis of monomeric red fluorescent protein (mrfp) expression with the same settings as previously described[11].
Bone marrow transplantation and xenogeneic MSC transplantation
An established mouse model of allogeneic BMT was used[12]. Briefly, female C57BL/6J (H2b) and B6D2F1 (H2bxd) mice aged 8 to 12 weeks were purchased from Jackson Laboratory (Bar Harbor, ME). B6D2F1 (H2bxd) mice received 13 Gy (split dose) total body irradiation (TBI) prior to receiving BM and splenic T-cells from either naïve allogeneic B6 or syngeneic B6D2F1 donors. Five million (5M) bone marrow and two million (2M) T-cells were suspended in 200 μl Leibovitz L-15 media and injected intravenously into recipient mice on day 0 (D0). On Days 1 and 4 following BMT, 1M culture-expanded, BM-derived human MSCs were administered by either TV or IA injection. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Case Western Reserve University.
Modes of human bone marrow-derived mesenchymal stromal cell (hMSC) injection
The hMSCs were administered either by TV or IA injection. For IA injection, mice were anesthetized with isoflurane and shaved at the sites of incision. With the animal laying supine, an incision was made in the lateral neck. After locating the carotid artery via blunt dissection, surrounding tissue and nerves were carefully dissected away to completely expose the artery. A string was fed under and through the artery and cut at the folded end, making the sutures into two separate stitches. The cranial suture was tied tightly, while the caudal suture was tied loosely. A clamp was positioned over the artery as distal as possible from the caudal ligature, giving at least 0.5 to 0.8 cm of vessel between the clamp and the ligature. A small hole was cut on top of the vessel, one quarter the distance from the cranial ligature. The loose suture was gently pulled as close to the vessel clamp as possible. A cannula was placed into the artery and then the vessel clamp was carefully removed. The cannula was inserted to the point where the two parts of the arterial catheter meet and then tied up with the suture tightly. A 25G tuberculin syringe replaced the 0.02 mm metal wire. The needle holder was removed and the blood in the line was slowly drawn into the syringe until all bubbles were removed from the arterial line. Blood was pushed back into the line and then the syringe with 0.2 mL PBS containing hMSCs was inserted. Injection was done gradually to ensure a more homogenous distribution into the blood stream. After the injection, the line was flushed to ensure all the injectable had been taken up by the animal. The cannula was slightly pulled back, and the string was tied up tightly. After complete removal of the cannula, the back tie was tied two more times and the animal's neck was sutured.
Bioluminescence imaging (BLI)
Fresh and confluent hMSCs were detached from the cultured flasks, counted and resuspended in FBS-free fresh medium. One million hMSCs in 0.2 mL PBS were injected into the animal either via tail vein or artery on day 4 after BMT. Fresh medium without any cells was also injected into the mice as “vehicle” control. After injection, mice were imaged at a preset time sequence: 30 min, 24 h, 48 h, 96 h and once a week for up to two months under isoflurane anesthesia. Before each scan, 2 mg D-luciferin in 0.2 mL sterile PBS was injected into the peritoneum (IP). After 10 minutes injection, the animals were imaged by the PerkinElmer/Xenogen IVIS 200 System (Palo Alto, California) for 5 minutes. Data were analyzed with Living Image software (Xenogen).
After in vivo BLI at the last time point, mice were injected with an additional dose of 200 μL D-luciferin (150 μg/g body weight IP) and 10 min later, animals were sacrificed. Their chests were opened and imaged ex vivo for 5 minutes. Tissue processing itself occurred within the subsequent three minutes post-imaging. Selected tissues were placed in culture dishes and D-luciferin was added directly to the exposed organs. BLI was performed for 5 minutes. Ex vivo BLI resulted in high-resolution images displaying signal foci within organs of interest and showed the gene expression in a more direct way. Imaging data were analyzed and quantified with Living Image software (Xenogen).
Scintigraphic (radionuclide) imaging
On day 4 post-BMT, non-transduced hMSCs were detached from the culture flasks using 0.25% EDTA-trypsin (Invitrogen) and pelleted via centrifugation. Fifteen million resuspended cells were delivered to Tyco Health Company (Cleveland, OH) for radiolabeling with 10 mCi [99mTc]-HMPAO. Radiolabeled cells were counted, and cell death was calculated using the trypan blue exclusion dye. Radiolabeled hMSCs and reporter gene labeled hMSCs were mixed at a 1:1 ratio and TV or IA injected into transplant mice as described above. Mice underwent scintigraphic imaging and X-ray imaging at 5 h, 10 h and 1 d after injection. The X-ray anatomic structure was overlaid with the scintigraphic data by using Image J (NIH, Maryland). Pixels of some radiographic images were smoothed in the Matlab.
Histology and immunohistochemistry (IHC)
After sacrifice, harvested organs, including lungs, liver, spleen, small and large intestines were fixed immediately in 10% buffered formalin and then embedded in paraffin. Five micrometer frozen sections were cut from fixed tissues, placed on uncoated slides, and stained with hematoxyline-eosin (H&E) for histology. For IHC, sections were first treated with xylene twice for 5 minutes each, followed by serial rehydration with 100%, 95%, 70% and 50% alcohol aqueous solutions for 3 minutes in each solution. After rehydration in PBS for 30 minutes, slides were treated with 0.2% Triton 100X to permeabilize the cell membranes, followed by incubation with 0.5% trypsin for antigen retrieval at 37°C for 30 minutes. Treated slides were blocked with hydrogen peroxide block (Ultravision) for 20 minutes, washed, and then treated with V-block (Ultravision) for 8 minutes. After incubation with a goat blocking serum (1:10 dilution, Invitrogen) for 30 minutes, the slides were stained with a rabbit polyclonal anti-mrfp antibody (1:250, Clontech, Mountainview, CA). Primary antibodies were detected by goat anti-rabbit IgG poly-HRP amplification agent (Millipore, Temecula, CA). The color was developed with diaminobenzidine (DAB) after incubation for 5 min and then slides were counter stained with Mayer's hematoxyline solution (Sigma-Aldrich). Negative isotype control was used to verify the presence of positive stained cells. In order to further confirm mrfp staining, in situ nuclear hybridization staining of hMSCs using human Alu sequence was performed at the Pathology Department at Case Western Reserve University using established protocols to verify the IHC results obtained from mrfp staining (Alu Positive Control Probe II, Ventana Medical Systems).
Quantitative real time PCR
Real time quantitative PCR (RT qPCR) was used to detect the presence or engraftment of hMSCs in mouse recipients quantitatively. The harvested tissues were kept frozen at -80oC until total genomic DNA was isolated using Genomic DNA Mini purification kit (Invitrogen). The DNA sequence of the luciferase was obtained from the gene vector map provided by Sam Gambhir at Stanford University. TaqMan probes and forward and reverse primers were designed and synthesized by Applied Biosystems (Foster City, CA, USA). The sequences of primers and the probe include: forward primer: CATCACGGTTTTGGAATGTTTACTACA, reverse primer: GCTCCTCAGAAACAGCTCTTC, and the probe sequence ACGACTCGAAATCCAC. Briefly, extracted DNA served as a template for RT qPCR, in which a fluorescent reporter dye (6-carboxy-fluorescein; FAM) was released and quantified during each specific replication of the template. 9 μL DNA (7 ng/μL) was mixed with TaqMan Universal PCR Master Mix (Applied Biosystems) and TaqMan Gene Expression Assays (Applied Biosystems) for the detection of luc specific gene. Reactions were incubated in a 7500 Fast Real Time PCR Systems (Applied Biosystems) with the following parameters: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The threshold cycle (Ct) of the target gene was calculated by the Sequence Detection System software (Applied Biosystems). Each sample was quantified in triplicate.
To construct standard curves for the luc reporter in each specific tissue, hMSCs that were used to inject into the mice were processed for genomic DNA, and then serially diluted by the genomic DNA of the corresponding tissues of the mice that received no hMSCs injection, and subjected to quantitative real time PCR assay in triplicate using the same procedures. The generated threshold cycle (Ct) of the target gene from the real time PCR reaction was plotted against the hMSCs concentration in the logarithmic format. In TaqMan reactions, individual replicates of both DNA sample and the standards were always performed on the same plate in the same experimental run so as to reduce inter experimental noise. The rapsyn gene from the mouse sequence was used as an endogenous control to compare the difference from different tissues[13,14]. The value of DNA contained in each somatic cell (diploid) was 6.16 pg with two copies of non-repeated genes. The results were expressed as the percentage of human genes in the mouse tissue.
Statistical analysis
All values are expressed as the mean plus or minus standard deviation (±SD). Statistical comparisons between groups were completed using the Mann-Whitney test (non-parametric data) and unpaired t-test (parametric data).
Results
Patterns of hMSC biodistribution differ between types of bone marrow transplantation
To study their biodistribution in transplant recipient mice using BLI, hMSCs were lentiviral-transduced with a triple-fusion, firefly luciferase reporter gene. Specifically, one million hMSCs were tail-vein (TV) injected on days 1 and 4 following BMT and daily BLI was performed in animals for up to six weeks post-BMT. hMSCs initially migrated to the pulmonary circuit in both syngeneic and allogeneic BMT mice, but only in allogeneic BMT mice did hMSCs eventually redistribute throughout the animal with greatest BLI signal intensity noted in the gastrointestinal (GI) tract (Figure 1). Further, more hMSCs initially migrated to the abdomen within the first week post-BMT in allogeneic versus syngeneic mice, reflecting the early induction in gastrointestinal acute GvHD in the mouse model[6,12]. Only after several weeks did TV-injected hMSCs more widely biodistribute throughout alloBMT mice.
Figure 1.
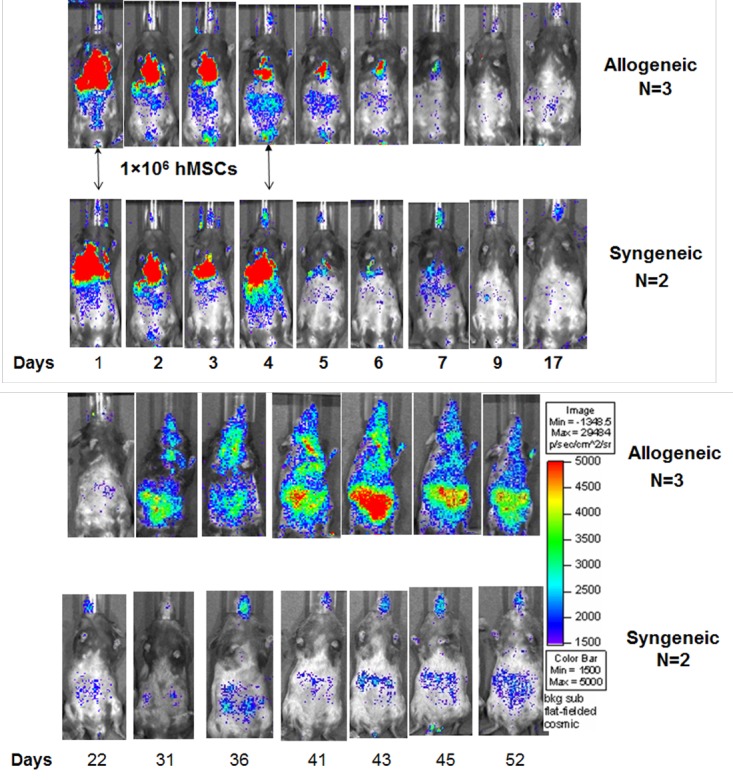
Bioluminescence imaging (BLI) of allogeneic (n=3) and syngeneic (n=2) transplant mice tail vein injected with one million hMSCs on days 1 and 4 after indicated transplant. Results from one of two representative experiments are shown.
Intra-arterial injection increases the biodistribution of human MSCs in transplant mice
Having observed the differences in hMSC biodistribution between syngeneic and allogeneic BMT mice, method of hMSC administration itself was next studied to determine its effect on hMSC biodistribution patterns in transplant recipients. To this end, hMSCs were labeled with either the luciferase reporter or 99mTC radionuclide for BLI and radionuclide imaging, respectively. Transplant recipients received one million hMSCs injected via TV or IA catheter and underwent whole-body imaging for 5 h, 10 h and one day following hMSC administration (Figure 2). At 5 h, hMSCs initially migrated to the lungs and abdomen in both transplant recipient groups irrespective of how they were injected. However, both TV and IA injection led to longer cell retention in the GI tract in allogeneic animals versus less retention in syngeneic animals, an observation noted at early (Figure 2) as well as later transplant times (Figure 3A). Furthermore, sustained, whole-body hMSCs signal intensity as measured by photon flux was only noted in IA-injected allogeneic animals (Figure 3B), while hMSC signal decreased in the GI tract of both TV-injected allogeneic and syngeneic animals over time (Figure 3C). Taken together, these results show that IA administration increases early hMSC biodistribution in both syngeneic and allogeneic animals, but causes sustained hMSC signal intensity in only allogeneic BMT mice. In addition, radionuclide imaging, similar to what is currently being used in the clinical setting, can be used as an imaging modality for in vivo hMSC biodistribution.
Figure 2.
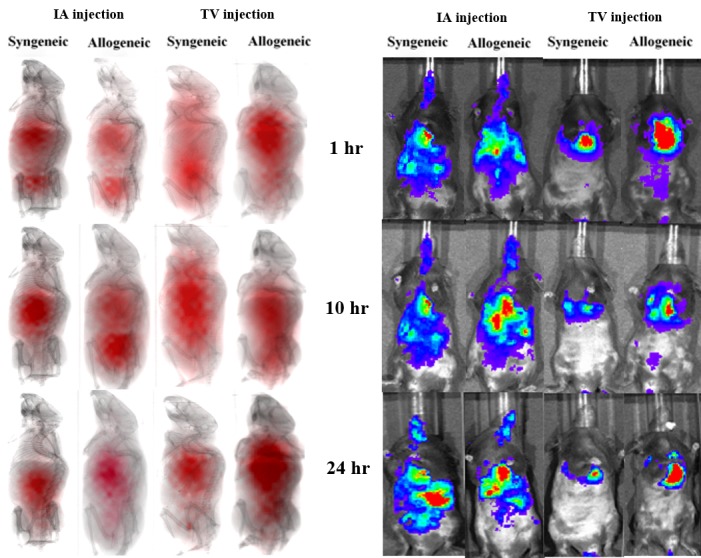
Radionuclide images (left) and BLI (right) for syngeneic and allogeneic mice receiving IA or TV injected hMSCs. Mice were imaged four days after BMT.
Figure 3.

(A) BLI of syngeneic and allogeneic transplant mice receiving one million hMSCs via IA and TV injection on days 1 and 4 after BMT. Indicated days refer to days after BMT. Results from one of two representative experiments are shown. (B) Quantitative analysis of the photon flux by drawing a region of interest (ROI) in the whole body. p < 0.05 (*) for mean photon flux between the IA allogeneic and the TV allogeneic group before day 17, and p < 0.05 (**) for mean photon flux between the IA allogeneic and IA syngeneic groups after day 7. (C) Quantitative analysis of the photon flux by drawing a region of interest (ROI) in the abdominal area. p < 0.05 (*) for mean photon flux between the IA allogeneic group and the TV allogeneic group before day 7; and p < 0.05 (**) for mean photon flux between IA allogeneic and IA syngeneic groups on days 7 and 10.
Human MSCs migrate to and are present in graft-versus-host disease target tissues
In general, BLI signal intensity was noted to be greater in the abdomens of allogeneic versus syngeneic animals following either TV or IA injection at later time points following BMT. Yet IA injection of hMSCs resulted in similar whole-body distribution patterns in both transplant recipient mice within the first day (Figure 2) and week (Figure 3) following administration. Ex vivo BLI on intestines from animals sacrificed 7 days after BMT was next performed to determine if hMSCs tracked to GvHD target tissues in alloBMT mice at earlier post-transplant times to support that microenvironmental differences in the GI tract existed between transplant groups. Allogeneic mice had strong BLI signal intensity noted in the abdomen upon necropsy (Figure 4). As previously seen, TV-injected hMSCs were localized primarily in the lungs, while IA-injected hMSCs were more holistically distributed in both allogeneic and syngeneic BMT mice at this early post-transplant time (Figure 4). Yet when these same animals were sacrificed, only the IA allogeneic mice had strong homogenous signal intensity throughout the intestines, whereas the IA syngeneic mice had heterogeneous signal intensity in their intestines (Figure 4). These results suggest that hMSCs indeed track to sites of GI inflammation and damage (i.e., radiation-induced in syngeneic mice), but that the alloreactive microenvironment in allogeneic mice provides an additional tracking signal resulting in more robust migration of hMSCs into the GI tract of these animals.
Figure 4.
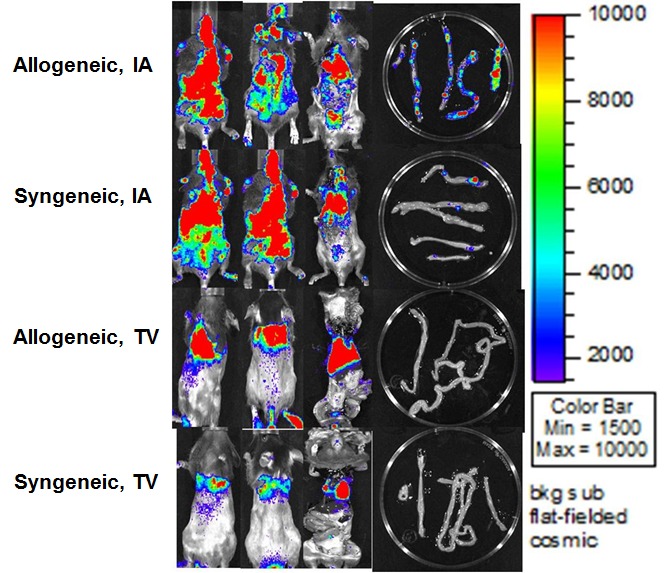
In vivo and ex-vivo BLI in allogeneic and syngeneic transplant groups receiving IA or TV injected hMSCs and sacrificed on D7 after indicated transplant. Results from one of two representative experiments are shown.
Confirmation of hMSCs in tissues
Immunohistochemistry (IHC) was performed on tissue samples of animals sacrificed on Day 7 post-BMT to confirm the presence of hMSCs in GvHD target tissues (Figure 5). Specifically, hMSCs labeled with the mrfp reporter gene were administered via IA injection and large intestines from both allogeneic and syngeneic animals were processed for IHC (Figure 5, left column). First, intestinal architecture in allogeneic mice (Figure 5, panels A and B) was distorted relative to syngeneic mice (Figure 5, panels E and F) consistent with GvHD-associated changes. Secondly, large conglomerates of brown-colored, mrfp-stained cells were seen in the intestines in transplant animals (Figure 5, panels A and E). At higher magnification, nuclei were clearly seen in these mrfp-positive cells (Figure 5, panels C and G). To verify that these cells were human in origin, mouse tissues were also stained against human-specific Alu repeat DNA sequence (Figure 5, right column). Blue-stained cellular aggregates were noted at low magnification (Figure 5, panels B and F), which were noted to contain nuclei at higher power (Figure 5, panels D and H). Positive human-Alu specific cells were also seen in other tissues including lung (Figure 6, panels A and B) and spleen (Figure 6, panels C and D) of allogeneic and syngeneic mice following IA administration of hMSCs.
Figure 5.
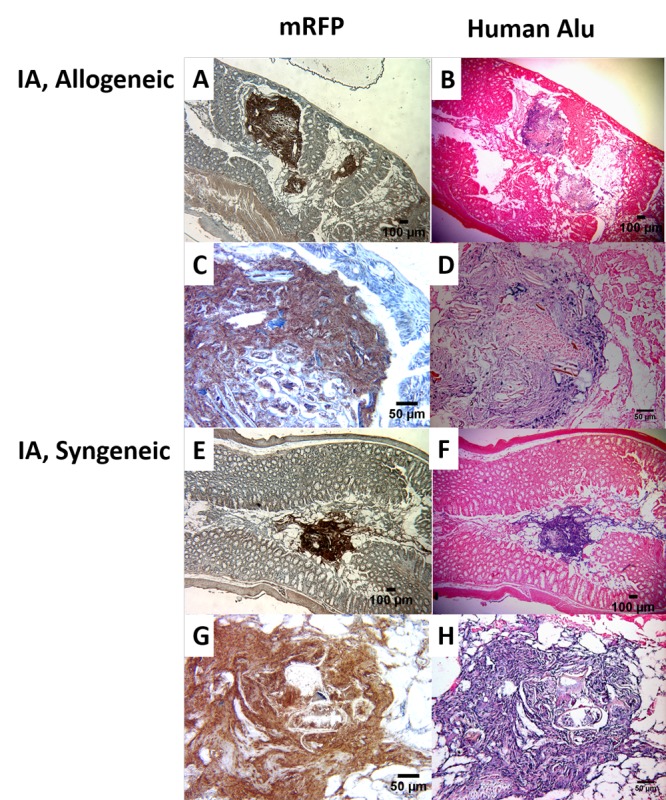
Immunohistochemistry of the large intestines of the allogeneic and syngeneic mice with intra-arterial injected hMSCs. The slides on the left panel were stained with mrfp, while the slides on the right were stained against Alu human repeat sequence. Mice were sacrificed on D7 after indicated BMT. Negative isotype control was used to verify the presence of positive stained cells (data not shown).
Figure 6.
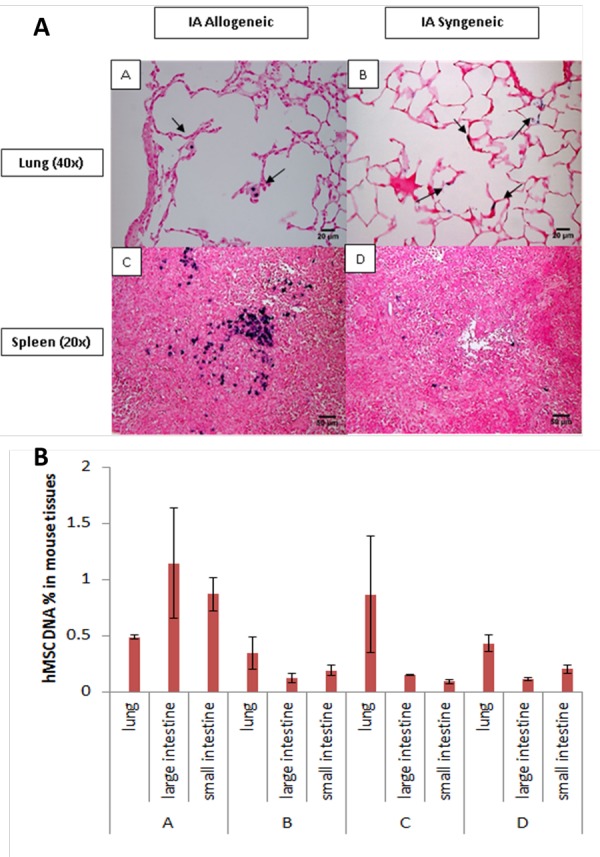
(A) Immunohistochemistry of the lungs (A, B) and spleens (C, D) of allogeneic (A, C) and syngeneic (B, D) mice with intra-arterial injection. All the tissues were stained against Alu human repeat sequence. Mice were sacrificed on D7 after BMT. (B) Real time qPCR results. A = allogeneic group with artery injection, B = syngeneic group with artery injection, C = tail vein injection for allogeneic group, D = syngeneic group.
Real time PCR was used to quantify hMSCs presence in the organs of transplant recipients (Figure 6, panel B). Specifically, TaqMan probe specific to the luciferase gene in transduced hMSCs was used. In the lung, both IA and TV injection led to increased hMSC detection. In contrast to TV injection, IA injection resulted in more detectable hMSCs in the small (IA 0.76 ± 0.15% vs. TV 0.10 ± 0.02%, p=0.025) and large intestines (IA 1.49 ± 0.49% vs. TV 0.15 ± 0.007%, p=0.060). Also, IA injection resulted in more detectable hMSCs in the small (allo 0.76 ± 0.15% vs. syn 0.22 ± 0.049%, p=0.040) and large (allo 1.49 ± 0.49 vs. syn 0.093 ± 0.039, p=0.057) intestines of allogeneic (allo) mice in contrast to syngeneic (syn) mice. Taken together, IHC and PCR data correlate with in vivo and ex vivo BLI, confirm the presence of hMSCs in GvHD target tissues, and substantiate that IA injection leads to greater hMSCs migration into the GI tract.
Discussion
Effective and targeted delivery of cells to injured organs is critical to the therapeutic success of cell-based therapies. In this regard, hMSCs have been used to treat acute GvHD[5]. However, clinical results using hMSCs have been inconsistent, likely reflecting, in part, suboptimal and non-standardized delivery with respect to route, timing and frequency of administration[15]. Our previous studies using TV injection for hMSCs delivery showed that the majority of cells remained in the lung with transient migration into GvHD target tissues including intestines and secondary lymphoid organs like the spleen[6,7]. The focus of the current study was to investigate how the method of cell administration might affect in vivo xenogeneic hMSCs biodistribution. To this end, we have demonstrated that IA injection is feasible and enables biodistribution throughout the body with preferential localization to sites of radiation-induced injury[16]. Therefore, the current study applied in vivo BLI and radionuclide imaging to track hMSCs migration following TV and IA injection in the BMT animals after reporter gene-tagged and radiolabeled hMSCs were equally mixed for co-injection. IA injection resulted in widely dispersed initial and sustained hMSCs biodistribution versus TV injection, which remained largely entrapped in the pulmonary circulation. hMSCs biodistribution into GvHD target tissues was verified using ex vivo BLI, IHC and real time PCR. These observations complement our previous mechanistic study in which hMSCs modulated GvHD by decreasing in vivo T-cell proliferation within secondary lymphoid organs through production of immunomodulatory soluble factors like prostaglandin E2 (PGE2)[6]. In addition, radionuclide imaging used in these animal models has potential translational application for studying cellular therapy migration in the clinical setting and is identical to clinical protocols for red and white blood cells using Tc-99m labeling to determine sites of bleeding and infection, respectively [17].
IA injection resulted in earlier, wider hMSC biodistribution in both allogeneic and syngeneic transplant recipients relative to TV injection, which resulted in more localized hMSC retention in the lungs. We[7] and others[18–22] have observed that intravenous injection results in hMSCs entrapment in the mouse lungs. Consequently, hMSCs migration to target organs and tissues is impeded, ultimately hindering their intended clinical effect, particularly if such an effect requires activation by specific microenvironments[5,23] or close proximity to cells that hMSC paracrine soluble factors modulate[24]. In contrast, IA injection bypasses the pulmonary circulation and delivers larger numbers of cells directly to sites of radiation and alloimmune-induced injury as seen in syngeneic and allogeneic transplant recipients, respectively. Therefore, we believe the combination of more effective delivery via IA injection and sustained pro-inflammatory response induced by alloreactivity resulted in greater biodistribution of xenogeneic hMSCs.
BLI signal was initially strong, but declined in TV-injected animals whereas BLI signal remained stronger in IA injected animals, particularly in allogeneic animals (Figure 1). Thus, IA injection clearly prolonged cell retention. Furthermore, in contrast to TV-injected hMSCs, IA-injected hMSCs assumed more extensive biodistribution in both syngeneic and allogeneic animals, which then transitioned to strong BLI signal localizing to the abdomen and lungs in allogeneic animals only (Figure 3). General factors contributing to BLI signal degradation include gradual cell loss or redistribution in organs and cell apoptosis. Irrespective of reasons for attenuated BLI intensity, ex vivo BLI imaging (Figure 4), IHC (Figure 5) and real-time PCR (Figure 6) confirmed the presence of hMSCs in the GI tract of IA-injected allogeneic recipient mice.
The positive IHC stains using mrfp antibody against mrfp, one of the fused reporter gene components, suggested that hMSCs migrate to large intestine (Figure 5) and spleen (Figure 6). The human Alu nuclear staining, which identified human cells in the mouse background[25] of the same intestinal tissue further validates this finding and extends our previous findings for the spleen[6] to now include the large intestine as a site for hMSCs migration and potential attenuation in T-cell activation and proliferation. Similar to the spleen, the intestine is a secondary lymphoid organ (SLO) containing mucosa-associated lymphoid tissue (MALT), which is involved in GvHD pathophysiology[26]. Using BLI in allogeneic transplant recipient mice, Beilhack and colleagues found that alloreactive T-cell proliferation associated with GvHD occurs in secondary lymphoid organs followed by T-cell homing to the intestines[27]. Furthermore, T-cell activation and proliferation occur rapidly within the first week after transplant. Applying this knowledge to our current study, hMSCs migrated to the GI tract in allogeneic recipients, specifically the large intestines, wherein, they may potentially attenuate T-cell proliferation similar to how hMSCs attenuated T-cell proliferation in the white pulp of spleen[6].
Although IA injection delivers more xenogeneic hMSCs throughout the body and prolongs hMSCs retention in allogeneic transplant mice, we did observe that some mice receiving IA injection did not survive as long as TV-injected mice. A potential reason may be the surgery for IA catheter placement, causing vascular occlusion over time in mice as has been observed in other mouse models incorporating stromal cells[18,19].
BLI was primarily used as an imaging tool to track hMSCs distribution following TV and IA injection in syngeneic and allogeneic transplant recipient mice. Advantages to BLI include its simplicity, convenience, real-time imaging and low cost. In particular, ATP-dependent reaction of luciferase with D-luciferin substrate enables imaging only living cells. However, tissue scattering and absorption make BLI only a semi-quantitative imaging method. Radionuclide imaging and BLI were used in combination to study initial hMSC biodistribution. Given the short half-life of 99mTc (6 hours), scintigraphic imaging would provide quantitative information regarding initial deposition of injected cells and their ensuing migration, maximally for one day due to the radio decay of the 99mTc label. However, this radionuclide imaging approach is an established clinical diagnostic imaging modality; therefore, it is readily available for translational applications for in vivo imaging of hMSCs transplantation in the clinical setting. One way to extend radionuclide imaging time is to affix hMSCs with an isotope having a longer half-life. Indium (111In) is a candidate isotope; as it has a longer half-life (2.8 days) and has not been shown to affect viability or functionality of either lymphocytes[28]or hMSCs[8].
Conclusions
Using a murine model of allogeneic BMT, xenogeneic hMSCs biodisbtribution patterns were compared following TV and IA injection, whereby IA injection led to stronger, longer and more dispersed hMSCs biodistribution. In contrast, TV injection resulted in hMSCs entrapped mainly in the lungs and then cleared within one week with smaller fractions of cells transiently migrating to the intestines. Cell deposition in the intestinal area was clearly observed via BLI in the allogeneic mice despite the injection technique, with mice receiving IA injection having significantly stronger signal by day 7. Results herein show that BLI can be easily used to track xenogeneic hMSCs in diseases models and that arterial injection has the potential to effectively alter hMSC distribution patterns by enhancing delivery to target tissues in combination with allo-induced inflammation.
Acknowledgments
The authors wish to acknowledge the Hematopoietic Stem Cell Core Facility of the Case Comprehensive Cancer Center for its collection and processing of patient samples (P30 CA 43703) and the National Center for Stem Cell and Regenerative Medicine at Case Western Reserve University for its continued research support.
Abbreviations
- alloBMT:
Allogeneic BMT
- BLI:
Bioluminescence Imaging
- GvHD:
Graft-versus-Host Disease
- GvL:
Graft-versus-Leukemia
- hMSC:
Human Mesenchymal Stromal Cell
- IA:
Intra-arterial
- IHC:
Immunohistochemistry
- TV:
Tail-vein
Potential Conflicts of Interests
None
References
- 1.Pasquini MC, Wang Z. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR Summary Slides, 2013. http://www.cibmtr.org. http://www.cibmtr.org. Available at:
- 2.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012 11;12(6):443–58. doi: 10.1038/nri3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Lalu MM, McIntyre L, Pugliese C, Fergusson D, Winston BW, Marshall JC, Granton J, Stewart DJ. Canadian Critical Care Trials Group. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS One. 2012710e47559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auletta JJ, Cooke KR, Solchaga LA, Deans RJ, van't Hof W. Regenerative stromal cell therapy in allogeneic hematopoietic stem cell transplantation: current impact and future directions. Biol Blood Marrow Transplant. 2010;16(7):891–906. doi: 10.1016/j.bbmt.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auletta JJ, Eid SK, Wuttisarnwattana P, Silva I, Metheny L, Keller MD, Guardia-Wolff R, Liu C, Wang F, Bowen T, Lee Z, Solchaga LA, Ganguly S, Tyler M, Wilson DL, Cooke KR. Human Mesenchymal Stromal Cells Attenuate Graft-Versus-Host Disease and Maintain Graft-Versus-Leukemia Activity Following Experimental Allogeneic Bone Marrow Transplantation. Stem Cells. 2015;33(2):601–14. doi: 10.1002/stem.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Love Z, Wang F, Dennis J, Awadallah A, Salem N, Lin Y, Weisenberger A, Majewski S, Gerson S, Lee Z. Imaging of mesenchymal stem cell transplant by bioluminescence and PET. J Nucl Med. 2007;48(12):2011–20. doi: 10.2967/jnumed.107.043166. [DOI] [PubMed] [Google Scholar]
- 8.Gao JZ, Dennis JE, Muzic RF, Lundberg M, Caplan AI. The dynamic in vivo distribution of bone marrow-derived mesenchymal stent cells after infusion. Cells Tissues Organs. 2001;169(1):12–20. doi: 10.1159/000047856. [DOI] [PubMed] [Google Scholar]
- 9.Zielske SP, Gerson SL. Lentiviral transduction of P140K MGMT into human CD34(+) hematopoietic progenitors at low multiplicity of infection confers significant resistance to BG/BCNU and allows selection in vitro. Mol Ther. 2002;5(4):381–7. doi: 10.1006/mthe.2002.0571. [DOI] [PubMed] [Google Scholar]
- 10.Auletta JJ, Zale EA, Welter JF, Solchaga LA. Fibroblast Growth Factor-2 Enhances Expansion of Human Bone Marrow-Derived Mesenchymal Stromal Cells without Diminishing Their Immunosuppressive Potential. Stem Cells Int. 2011;2011:235176. [DOI] [PMC free article] [PubMed]
- 11.Ray P, De A, Min JJ, Tsien RY, Gambhir SS. Imaging tri-fusion multimodality reporter gene expression in living subjects. Cancer Res. 2004;64(4):1323–30 . doi: 10.1158/0008-5472.can-03-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, Rossignol DP, Ferrara JL. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J Clin Invest. 2001;107(12):1581–9. doi: 10.1172/JCI12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mouiseddine M, François S, Semont A, Sache A, Allenet B, Mathieu N, Frick J, Thierry D, Chapel A. Human mesenchymal stem cells home specifically to radiation-injured tissues in a non- obese diabetes/severe combined immunodeficiency mouse model. Br J Radiol. 2007;80 Spec No 1:S49–55. [DOI] [PubMed]
- 14.François S, Bensidhoum M, Mouiseddine M, Mazurier C, Allenet B, Semont A, Frick J, Sache A, Bouchet S, Thierry D, Gourmelon P, Gorin NC, Chapel A. Local irradiation not only induces homing of human mesenchymal stem cells at exposed sites but promotes their widespread engraftment to multiple organs: a study of their quantitative distribution after irradiation damage. Stem Cells. 2006;24(4):1020–9. doi: 10.1634/stemcells.2005-0260. [DOI] [PubMed] [Google Scholar]
- 15.Kaipe H, Erkers T, Sadeghi B, Ringden O. Stromal cells-are they really useful for GVHD? Bone marrow transplantation. 2014;49(6):737–43. doi: 10.1038/bmt.2013.237. Epub 2014/01/28. [DOI] [PubMed] [Google Scholar]
- 16.Lin P, Correa D, Kean TJ, Awadallah A, Dennis JE, Caplan AI. Serial transplantation and long-term engraftment of intra-arterially delivered clonally derived mesenchymal stem cells to injured bone marrow. Mol Ther. 2014;22(1):160–8. doi: 10.1038/mt.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Vries EF, Roca M, Jamar F, Israel O, Signore A. Guidelines for the labelling of leucocytes with (99m)Tc-HMPAO. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur J Nucl Med Mol Imaging. 2010;37(4):842–8. doi: 10.1007/s00259-010-1394-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walczak P, Zhang J, Gilad AA, Kedziorek DA, Ruiz-Cabello J, Young RG, Pittenger MF, van Zijl PC, Huang J, Bulte JW. Dual-modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. 2008;39(5):1569–74. doi: 10.1161/STROKEAHA.107.502047. Stroke. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tolar J, O'shaughnessy MJ, Panoskaltsis-Mortari A, McElmurry RT, Bell S, Riddle M, McIvor RS, Yant SR, Kay MA, Krause D, Verfaillie CM, Blazar BR. Host factors that impact the biodistribution and persistence of multipotent adult progenitor cells. Blood. 2006;107(10):4182–8. doi: 10.1182/blood-2005-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hale SL, Dai W, Dow JS, Kloner RA. Mesenchymal stem cell administration at coronary artery reperfusion in the rat by two delivery routes: a quantitative assessment. Life Sci. 2008;83(13-14):511–5. doi: 10.1016/j.lfs.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Togel F, Yang Y, Zhang P, Hu Z, Westenfelder C. Bioluminescence imaging to monitor the in vivo distribution of administered mesenchymal stem cells in acute kidney injury. Am J Physiol Renal Physiol. 2008;295(1):F315–21. doi: 10.1152/ajprenal.00098.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chabner KT, Adams GB, Qiu J, Moskowitz M, Marsters ES, Topulos GP, Scadden DT. Direct vascular delivery of primitive hematopoietic cells to bone marrow improves localization but not engraftment. Blood. 2004;103(12):4685–6. doi: 10.1182/blood-2003-12-4145. [DOI] [PubMed] [Google Scholar]
- 23.Krampera M. Mesenchymal stromal cell ‘licensing’: a multistep process. Leukemia. 2011. [DOI] [PubMed]
- 24.Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, Zhao RC, Shi Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2(2):141–50. doi: 10.1016/j.stem.2007.11.014. Epub 2008/03/29. [DOI] [PubMed] [Google Scholar]
- 25.Just L, Timmer M, Tinius J, Stahl F, Deiwick A, Nikkhah G, Bader A. Identification of human cells in brain xenografts and in neural co-cultures of rat by in situ hybridisation with Alu probe. J Neurosci Methods. 2003;126(1):69–77. doi: 10.1016/s0165-0270(03)00065-7. [DOI] [PubMed] [Google Scholar]
- 26.Silva IA, Olkiewicz K, Askew D, Fisher JM, Chaudhary MN, Vannella KM, Deurloo DT, Choi SW, Pierce EM, Clouthier SG, Liu C, Cooke KR. Secondary lymphoid organs contribute to, but are not required for the induction of graft-versus-host responses following allogeneic bone marrow transplantation: a shifting paradigm for T cell allo-activation. Biol Blood Marrow Transplant. 2010;16(5):598–611. doi: 10.1016/j.bbmt.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, Baker EM, Cao YA, Contag CH, Negrin RS. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. 2005;106(3):1113–22. doi: 10.1182/blood-2005-02-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pittet MJ, Grimm J, Berger CR, Tamura T, Wojtkiewicz G, Nahrendorf M, Romero P, Swirski FK, Weissleder R. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc Natl Acad Sci U S A. 2007;104(30):12457–61. doi: 10.1073/pnas.0704460104. [DOI] [PMC free article] [PubMed] [Google Scholar]