

INTRODUCTION
Acute pancreatitis in a common and potentially fatal disease which is associated with considerable morbidity and a mortality rate of approximately 30%; its path ogenesis remains unknown[1]. The cause of death is often due to multiple organ failure which frequently complicates severe acute pancreatitis. In particular, acute lung injury often occurs at an early stage of severe acute pancreatitis and develops into adult respiratory distress syndrome (ARDS)[2,3].
Numerous studies have been done to clarify the mechanisms of acute lung injury in AHNP. Activated leukocytes and inflammatory mediators, such as interleukin-1 and tumor necrosis factor α have been suggested to play a predominant role in the development of acute lung injury in AHNP[4-6]. Further investigation have found that pretreatment of mice with antineutrophile serum results in reduction of severity of pancreatitis and complete prevention of lung injury[7]. More recently, Hofbauer et al[8-10] postulated that the pancreatitis-associated lung injury (PALI) might be dependent on the continued generation and action of platelet-activating factor (PAF). Yamaguchi et al[11] have suggested that the PAF antagonist TCV-309 effectively prevented cytokine-induced neutrophil chemoattractant expression by bronchoalveolar macrophages and subsequent PALI. Furthermore, attenuation of pancreatitis and prevention of PALI could be achieved with recombinant PAF acetylhydrolase. P-selectin is a key determinant of leukocyte recruitment, which is upregulated in the pulmonary endothelium during acute pancreatitis[12]. However, detailed mechanisms of acute lung injury in AHNP has not been elucidated.
In the current study, sodium taurocholate-induced pancreatitis rat models was used to investigate the apoptosis of alveolar epithelial cells and expression of apoptosis-regulated gene in PALI, and the relationship between TGFβ1 and apoptosis.
MATERIAL AND METHODS
Material and chemicals
Sodium taurocholate was obtained from Sigma Chemical Co. (USA). In situ cell death detection kit, POD was purchased from Borehinger Mahheim Co. (Germany), protinase K was from Merch Co. (USA), the purified goat anti-bax polyclonal antibody and mouse anti-p53 monoclonal antibody were from Santa Cruz Biotechnology Inc. (USA), the goat anti-TGFβ1 polyclonal antibody was from Promega Co. (USA), and immunochemical SP kit was from Biotech Co. (USA). Other materials and chemicals were obtained from commercial sources.
Animals
Male Wistar rats, weighing 200 g-250 g, were used. Animals were bred and housed in standard cages in a climate-controlled room with an ambient temperature of 22 °C ± 2 °C and a 12-h light/dark cycle. They were fed standard laboratory chow, given water at libitum, and fasted overnight before each experiment.
Models for pancreatitis
Male Wistar rats were anesthetized with 2.5% pentobarbital (0.1 mL/100 g body weight intraperitoneally). A midline laparotomy was performed, followed by the ligation of the bile-pancreatic ducts close to the liver and duodenum. Then pancreatic duct was retrogradely injected 5% sodium taurocholate (0.1 mL/100 g body weight) for 1 min, and stagnant for 4 min. For control, sham operation or retrograde infusion of normal saline into the pancreatic duct was performed as described above, 2, 5 and 14 h after sodium taurocholate infusion, laparotomy was performed again and blood samples were collected aseptically from the abdominal aorta. The pancreas and right lung were rapidly removed and fixed in 10% neutral phosphate buffered formalin for histological study. Pancreatitis was confirmed by measuring amylase levels before and after the experiment and by histological examination.
Serum amylase activity
Serum amylase activity was measured by a chromogenic method with the phadeba amylase test.
Detection of apoptosis
Morphological examination Paraffin-embedded pancreas and lung samples were sectioned (4 μm), stained with hematoxylin/eosin (H&E), and examined by an experienced morphologist who was unaware of the sample identity[13,14].
TUNEL on light microscope TdT-mediated dUTP nick-end labeling (TUNEL) was performed according to the method of Gavrieli et al[15,16] with some modification. Briefly, the sections were deparaffinized by heating for 20 minutes at 60 °C, rehydrated in descending concentrations of ethanol (100%, 95%, 90%, 80%) and then immersed in double distilled water (DDW). After rehydration, the sections were incubated with 20 mg/L proteinase K for 15 minutes at room temperature. The slides were washed in DDW for 2 min 4 times and covered with 0.3% hydrogen peroxidase in methanol for 30 minutes at room temperature to inactivate endogenous peroxidase. The slides were rinsed with phosphate-buffered saline (PBS, pH7.4) 5 min three times and incubated the slides to 50 μL TUNEL reaction mixture for 60 min at 37 °C. After washing in PBS, the slides were incubated with 50 μL Converter-POD for 30 min at 37 °C. The reaction products were visualized by immersion in diaminobenzide solution. For positive controls, TUNEL was performed after deoxyribonuclease treatment. For negative controls, TUNEL was performed with labeling solution instead of TUNEL reaction mixture. Observations and photographs were made using an Olympus microscope. TUNEL positive nuclei were counted in fields (× 40) magnification chosen at random, and the number of labeled nuclei per total nuclei in those fields was expressed as the apoptotic index (AI).
Immunohistochemical staining Immunohis-tochemical staining was performed to explore the expression of apoptosis-regulated gene bax and p53, as well as TGFβ1. Consecutive 4 μm paraffin-embedded tissue sections were subjected to immunostaining with streptavidin peroxidase technique. The sections were deparaffinized, rehydrated in descending concentrations of ethanol and then digested by incubation with 0.1% typsin for 20 min at 37 °C. Tissue sections were submerged for 15 min in 0.1 M PBS containing 0.1% Triton X-100. Endogenous peroxidase activity was blocked by incubating the slides with 0.3% (vol/vol) H2O2 in methanol, followed by washing in PBS. The sections were incubated for 30 min at room temperature with normal goat serum before overnight incubation at 4 °C with either goat anti-bax polyclonal antibody diluted 1∶50 in PBS, mouse anti-p53 monoclonal antibody diluted 1∶50 in PBS or goat anti-TGFβ1 polyclonal antibody diluted 1∶100 in PBS. After washing in PBS, slides were treated with biotinylated link immnoglobulins biotinylated goat anti-mouse IgG antibody was used for bax and p53 staining. The slides were then incubated for 30 min with streptavidin-biotin-peroxidase complex and were visualized by a 10 min application of diaminobenzidine substrate. The sections were counterstained with hematoxylin to identify nuclei and observed under a light microscope. It was judged that positive staining nuclei were brown, and immunolocalized nuclei. The positive rate of bax and p53 were counted in fields (40 ×) magnification chosen at random, and the number of positive staining nuclei per total nuclei in those fields was expressed as the positive rate.
Statistical analysis
Results were expressed as the mean and standard error of the mean (SEM). The significance of changes was evaluated by using Student’s t test when the data consisted of only two groups or by analysis of variance (ANOVA) when comparing three or more groups. A P value < 0.05 indicated a significant difference.
RESULTS
Acute hemorrhagic necrotizing pancreatitis
AHNP was manifested by a rise in serum amylase activity and morphological evidence. In all animals a marked elevation of serum amylase levels was observed at 2, 5, and 14 h after sodium taurocholate infusion. The morphological changes were observed after sodium taurocholate infusion and included obvious acinar cell necrosis, fat necrosis, extensive neutrophils infiltration, microthrombosis of pancreatic vessels and massive intralobular hemorrhage. In addition, 1 rat (25%) died before the end of the experiment (14 h after sodium taurocholate infusion), whereas other animals survived the entire observation period.
Histological findings
The morphological changes observed in lung included pronounced interstitial edema associated with massive neutrophils infiltration, alveolar wall thickening and foci of vascular thrombosis in large vessels. Simultaneously, increase of pulmonary microvascular permeability was found.
Evidence of apoptosis
Morphological examination In the H&E staining, alveolar epithelial cells presented pyknotic nuclei, cell shrinkage, condensation of chromatin and formation of apoptotic bodies, etc. which were the typical morphological features of apoptosis.
TUNEL staining As shown in Figure 1, TUNEL selectively labeled alveolar epithelial cells nuclei. The apoptotic index of alveolar epithelial cells at 2, 5, and 14 h after induction of AHNP were 0.33% ± 0.7%, 0.89% ± 0.06% and 0.54% ± 0.08%, respectively. Moreover, the apoptotic index of alveolar epithelial cells at 5 h after induction of AHNP was significantly higher than those of 2 and 14 h (P < 0.01).
Figure 1.
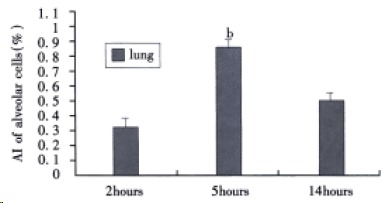
AI of alveolar epithelial cells in sodium taurocholate-induced pancreatitis-associated lung injury. AHNP was induced by retrograde injections into the pancreatic ducts of 5% sodium taurocholate (0.1 mL/100 g body wt) in rats. Results shown are the MEAN ± SEM for four or more animals in each group. Asterisks indicates bP < 0.01 when the AI of alveolar epithelial cells at 5 h after induction of AHNP compared with 2 and 14 h after induction of AHNP.
Immunohistochemical detection of bax and p53 protein
As shown in Figure 2, the positive rate of bax protein in alveolar epithelial cells at 2, 5 and 14 h after induction of AHNP were 0.38% ± 0.11%, 0.67% ± 0.13% and 0.39% ± 0.03%, respectively. Similar to AI of alveolar epithelial cells, the positive rate of bax protein in alveolar epithelial cells at 5 h after induction of AHNP was similarly markedly higher than those of 2 and 14 h (P < 0.05). In this study, the positive rate of p53 protein in alveolar epithelial cells at 2, 5, and 14 h after induction of AHNP were 0.72% ± 0.29%, 0.28% ± 0.08% and 0.32% ± 0.04%, respectively. The positive rate of p53 protein in alveolar epithelial cells significantly decreased at 5 and 14 h after induction of AHNP compared with that of 2 h (P < 0.05).
Figure 2.
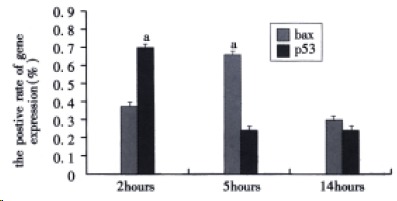
The positive rate of bax and p53 protein in alveolar epithelial cells after sodium taurocholate-induced pancreatitis-associated lung injury. AHNP was induced by retrograde injections into the pancreatic ducts of 5% sodium taurocholate (0.1 mL/100 g body wt) in rats. Results shown were the MEAN ± SEM for four or more animals in each group. Asterisks indicates aP < 0.05 when the positive rate of bax protein in alveolar epithelial cells at 5 h after induction of AHNP compared with 2 and 14 h and the positive rate of p53 protein in alveolar epithelial cells at 2 h after induction of AHNP compared with 5 and 14 h.
Immunohistochemical detection of TGFβ-1 Immun-ohistochemical staining of TGFβ1 was scarcely detected in normal lung tissues. As shown in Figure 3, although the positive rate of TGFβ1 in alveolar epithelial cells at 2, 5, and 14 h after induction of AHNP were 0.35% ± 0.14%, 0.39% ± 0.15% and 0.48% ± 0.12%, respectively, no significance difference was observed among them.
Figure 3.
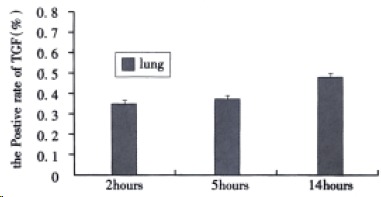
The positive rate of TGFβ1 in alveolar epithelial cells after sodium taurocholate-induced pancreatitis-associated lung injury. AHNP was induced by retrograde injections into the pancreatic ducts of 5% sodium taurocholate (0.1 mL/100 g body wt) in rats. Results shown were the MEAN ± SEM for four or more animals in each group. Therewas no significant difference among the positive rate of TGFβ1 at 2, 5 and 14 h after induction of AHNP.
DISCUSSION
Acute lung injury and adult respiratory distress syndrome (ARDS) are frequently observed during the course of severe acute pancreatitis. The pathogenesis of PALI remained unclear. Bhatia et al[17] reported that neurogenic factors, such as substance P, increased release of proinflammatory midiators from the pancreas leading to increase lung injury by acting via NK1R on acinar cells. Recent studies[18] have suggested that intercellular adhesion molecule 1 (ICAM-1) expression on pulmonary microva scular endothelial cells during pancreatitis contributed to the evolution of PALI by promoting neutrophil sequestration within the lung. Moreover, treatment with monoclonal antibodies against ICAM-1 significantly reduced the lung injury in severe acute pancreatitis[19].Very recently, Tsukahara et al[20] noted that alveolar macrophages were activated by PLA2, mainly by sPLA2 type II, and produced a large amount of NO that contributed to lung injury in acute pancreatitis. Further studies had suggested that the lung injury was reduced by the administration of the PLA2 inhibitor. Recent studies showed that the absence of T and B lymphocytes could prevent severe pulmonary injury resulting from acute pancreatitis, this indicated that systemic lymphocyte activation modulated the systemic response, in particular, pulmonary injury caused by acute pancreatitis[21]. Apoptotic cell death of renal tubules and hepatocytes have been reported to be involved in the mechanisms of renal failure and liver failure during severe acute pancreatitis[22,23]. Although numerous trials had been done to clarify the mechanisms of pancreatitis-associated lung injury, there was no literature referring to involvement of apoptosis in pancreatitis-associated lung injury.
Apoptosis was initially confirmed as a specific form of cell death that served to eliminate excessive or unwanted cell types during embryogenesis and normal tissue growth[24], but had been recently clarified to be induced in cellular injury with inflammatory disease[25]. It was generally believed that apoptosis was a genetically regulated form of cell death, and pyknotic nuclei and formation of apoptotic bodies were the typical features of apoptosis[26,27]. In the current work, we selected two apoptosis-regulated gene bax and p53, which were confirmed to be involved in the control of apoptosis. Proapoptotic gene bax, one of the members of apoptosis-regulated gene bcl-2 family, was a dominant inhibitor of bcl-2 and promoted cell apoptosis[28-30]. Tumor supressor gene p53 was a special related gene with activation of cell-cycle arrest and apoptosis[31,32]. A very important role of normal or wild type p53 as a “guardian of the genome” was capable of inducing apoptosis upon DNA damage. Recent studies suggested that activation of NF-kappa B activity augmented p53-induced apoptosis[32]. On the contrary, mutant p53 failed to initiate apoptosis process, so mutant p53 was thought to have effect on apoptosis inhibition. More recently, Buckley et al[33] demonstrated that apoptosis via induction of p53, p21, and bax proteins was seen in alveolar epithelial cells cultured from hyperoxic rats, and apoptosis might be a protective michanism that limits lung injury[34].
TGFβ family in mammalian cells was comprised of at least three homologous polypeptides that could regulate a variety of cellular functions, including proliferation and differentiation processes[35,36]. Recently, some investigators reported that TGFβ1 could induce apoptosis in normal and malignant cells[37,38]. It was demonstrated that the induction of apoptosis by TGFβ1 was due to the regeneration of reactive oxygen species in the cells, the involvement of caspase family proteases[39-41], induction of p53 and bax[37], or expression of TIEG (TGFβ-inducible early gene)[42-44]. In this study, we sought to explore the potential relationship between TGFβ1 and apoptosis of alveolar epithelial cells in pancreatitis-associated lung injury.
We found that the apoptotic index of alveolar epithelial cells at 5 h after induction of AHNP was significantly higher than those of 2 and 14 h (P < 0.01), and the positive rate of bax protein in alveolar epithelial cells at 5 h was markedly higher than those of 2 and 14 h after induction of AHNP (P < 0.05). Interestingly, the positive rate of bax protein in alveolar epithelial cells was paralleled to the AI of alveolar epithelial cells in pancreatitis-associated lung injury. The positive rate of p53 protein in alveolar epithelial cells dramaticaly decreased at 5 and 14 h after induction of AHNP compared with 2 h (P < 0.05). Similarly, the positive rate of p53 protein in alveolar epithelial cells was intended to parallel to the AI of alveolar epithelial cells in PALI. Immunostaining of TGFβ1 was detected at 2, 5 and 14 h after induction of AHNP, no significant difference was observed among them. Our data demonstrated that apoptosis of alveolar epithelial cells might be mediated by apoptosis-regulated gene bax and p53, but TGFβ1 was not implicated in apoptosis of alveolar epithelial cells.
In summary, apoptosis of alveolar epithelial cells might be involved in PALI, and the mechanisms of apoptosis probably correlates with the expression of apoptosis-regulated gene bax and p53 but is not related with the expression of TGFβ1. Recent studies[45] have suggested that mild pancreatitis was found to be associated with extensive apoptotic acinar cell death while severe pancreatitis was noted to involve extensive acinar cell necrosis but very little acinar cell apoptosis. More recently, some investigators[46-48] have found that induction of apoptosis in pancreatic acinar cells attenuates the severity of experimental acute pancreatitis. Furthermore, Kaiser et al[45] have demonstrated that inhibition of apoptosis by administration of cyclohexamide could enhance the severity of pancreatitis. These data have led to the concept that pharmacological induction of apoptosis in pancreatic acinar cell injury to reduce inflammatory reaction provides a new therapeutic strategy for the treatment of acute pancreatitis. Very recently, we found that the mechanisms of SSa in treating acute pancreatitis might be induction of apoptosis in pancreatic acinar cell injury to reduce inflammatory response[49].
Future studies for clarifying the initiators, regulators, genetic control and signal transductal pathway of apoptosis of alveolar epithelial cells, would be able to reveal the pathogenesis of PALI, and further elucidate the role of apoptosis of alveolar epithelial cells in PALI.
Footnotes
Edited by Wu XN
Proofread by Ma JY
Presented at the meeting of 11th Asian Pacific Congress of Gastroenterology, 8th Asian Pacific Congress of Digestive Endoscopy, Hong Kong, 10-14 March, 2000
References
- 1.Steinberg W, Tenner S. Acute pancreatitis. N Engl J Med. 1994;330:1198–1210. doi: 10.1056/NEJM199404283301706. [DOI] [PubMed] [Google Scholar]
- 2.de Beaux AC, Goldie AS, Ross JA, Carter DC, Fearon KC. Serum concentrations of inflammatory mediators related to organ failure in patients with acute pancreatitis. Br J Surg. 1996;83:349–353. doi: 10.1002/bjs.1800830317. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein A, Milani R, Fernezlian SM, Leme AS, Capelozzi VL, Martins MA. Acute lung injury in two experimental models of acute pancreatitis: infusion of saline or sodium taurocholate into the pancreatic duct. Crit Care Med. 2000;28:1497–1502. doi: 10.1097/00003246-200005000-00040. [DOI] [PubMed] [Google Scholar]
- 4.Kusske AM, Rongione AJ, Reber HA. Cytokines and acute pancreatitis. Gastroenterology. 1996;110:639–642. doi: 10.1053/gast.1996.v110.agast960639. [DOI] [PubMed] [Google Scholar]
- 5.Pezzilli R, Billi P, Miniero R, Fiocchi M, Cappelletti O, Morselli-Labate AM, Barakat B, Sprovieri G, Miglioli M. Serum interleukin-6, interleukin-8, and beta 2-microglobulin in early assessment of severity of acute pancreatitis. Comparison with serum C-reactive protein. Dig Dis Sci. 1995;40:2341–2348. doi: 10.1007/BF02063235. [DOI] [PubMed] [Google Scholar]
- 6.Pezzilli R, Billi P, Miniero R, Barakat B. Serum interleukin-10 in human acute pancreatitis. Dig Dis Sci. 1997;42:1469–1472. doi: 10.1023/a:1018814710291. [DOI] [PubMed] [Google Scholar]
- 7.Bhatia M, Saluja AK, Hofbauer B, Lee HS, Frossard JL, Steer ML. The effects of neutrophil depletion on a completely noninvasive model of acute pancreatitis-associated lung injury. Int J Pancreatol. 1998;24:77–83. doi: 10.1007/BF02788564. [DOI] [PubMed] [Google Scholar]
- 8.Hofbauer B, Saluja AK, Bhatia M, Frossard JL, Lee HS, Bhagat L, Steer ML. Effect of recombinant platelet-activating factor acetylhydrolase on two models of experimental acute pancreatitis. Gastroenterology. 1998;115:1238–1247. doi: 10.1016/s0016-5085(98)70096-4. [DOI] [PubMed] [Google Scholar]
- 9.Formela LJ, Wood LM, Whittaker M, Kingsnorth AN. Amelioration of experimental acute pancreatitis with a potent platelet-activating factor antagonist. Br J Surg. 1994;81:1783–1785. doi: 10.1002/bjs.1800811224. [DOI] [PubMed] [Google Scholar]
- 10.Galloway SW, Kingsnorth AN. Lung injury in the microembolic model of acute pancreatitis and amelioration by lexipafant (BB-882), a platelet-activating factor antagonist. Pancreas. 1996;13:140–146. doi: 10.1097/00006676-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Yamaguchi Y, Matsumura F, Liang J, Okabe K, Matsuda T, Ohshiro H, Ishihara K, Akizuki E, Yamada S, Ogawa M. Platelet-activating factor antagonist (TCV-309) attenuates the priming effects of bronchoalveolar macrophages in cerulein-induced pancreatitis rats. Pancreas. 1999;18:355–363. doi: 10.1097/00006676-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Folch E, Salas A, Panés J, Gelpí E, Roselló-Catafau J, Anderson DC, Navarro S, Piqué JM, Fernández-Cruz L, Closa D. Role of P-selectin and ICAM-1 in pancreatitis-induced lung inflammation in rats: significance of oxidative stress. Ann Surg. 1999;230:792–78; discussion 792-78;. doi: 10.1097/00000658-199912000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt J, Lewandrowski K, Fernandez-del Castillo C, Mandavilli U, Compton CC, Warshaw AL, Rattner DW. Histopathologic correlates of serum amylase activity in acute experimental pancreatitis. Dig Dis Sci. 1992;37:1426–1433. doi: 10.1007/BF01296014. [DOI] [PubMed] [Google Scholar]
- 14.Lei WZ, Wei JJ, Shen WL, Jin LR. The relationship between multiple organ damages and endotoxemia in experimental acute pancreatitis. Zhonghua Shiyan Waike Zazhi. 1995;12:131–134. [Google Scholar]
- 15.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Facchinetti A, Tessarollo L, Mazzocchi M, Kingston R, Collavo D, Biasi G. An improved method for the detection of DNA fragmentation. J Immunol Methods. 1991;136:125–131. doi: 10.1016/0022-1759(91)90258-h. [DOI] [PubMed] [Google Scholar]
- 17.Bhatia M, Saluja AK, Hofbauer B, Frossard JL, Lee HS, Castagliuolo I, Wang CC, Gerard N, Pothoulakis C, Steer ML. Role of substance P and the neurokinin 1 receptor in acute pancreatitis and pancreatitis-associated lung injury. Proc Natl Acad Sci USA. 1998;95:4760–4765. doi: 10.1073/pnas.95.8.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frossard JL, Saluja A, Bhagat L, Lee HS, Bhatia M, Hofbauer B, Steer ML. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology. 1999;116:694–701. doi: 10.1016/s0016-5085(99)70192-7. [DOI] [PubMed] [Google Scholar]
- 19.Werner J, Z'graggen K, Fernández-del Castillo C, Lewandrowski KB, Compton CC, Warshaw AL. Specific therapy for local and systemic complications of acute pancreatitis with monoclonal antibodies against ICAM-1. Ann Surg. 1999;229:834–40; discussion 841-2. doi: 10.1097/00000658-199906000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsukahara Y, Morisaki T, Horita Y, Torisu M, Tanaka M. Phospholipase A2 mediates nitric oxide production by alveolar macrophages and acute lung injury in pancreatitis. Ann Surg. 1999;229:385–392. doi: 10.1097/00000658-199903000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer J, Laine VJ, Rau B, Hotz HG, Foitzik T, Nevalainen TJ, Beger HG. Systemic lymphocyte activation modulates the severity of diet-induced acute pancreatitis in mice. Pancreas. 1999;19:62–68. doi: 10.1097/00006676-199907000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Takase K, Takeyama Y, Nishikawa J, Ueda T, Hori Y, Yamamoto M, Kuroda Y. Apoptotic cell death of renal tubules in experimental severe acute pancreatitis. Surgery. 1999;125:411–420. [PubMed] [Google Scholar]
- 23.Takeyama Y, Hori Y, Takase K, Ueda T, Yamamoto M, Kuroda Y. Apoptotic cell death of hepatocytes in rat experimental severe acute pancreatitis. Surgery. 2000;127:55–64. doi: 10.1067/msy.2000.99875. [DOI] [PubMed] [Google Scholar]
- 24.Williams GT. Programmed cell death: apoptosis and oncogenesis. Cell. 1991;65:1097–1098. doi: 10.1016/0092-8674(91)90002-g. [DOI] [PubMed] [Google Scholar]
- 25.Haslett C. Resolution of acute inflammation and the role of apoptosis in the tissue fate of granulocytes. Clin Sci ( Lond) 1992;83:639–648. doi: 10.1042/cs0830639. [DOI] [PubMed] [Google Scholar]
- 26.Stewart BW. Mechanisms of apoptosis: integration of genetic, biochemical, and cellular indicators. J Natl Cancer Inst. 1994;86:1286–1296. doi: 10.1093/jnci/86.17.1286. [DOI] [PubMed] [Google Scholar]
- 27.Wyllie AH. Apoptosis: cell death in tissue regulation. J Pathol. 1987;153:313–316. doi: 10.1002/path.1711530404. [DOI] [PubMed] [Google Scholar]
- 28.Gukovskaya AS, Perkins P, Zaninovic V, Sandoval D, Rutherford R, Fitzsimmons T, Pandol SJ, Poucell-Hatton S. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology. 1996;110:875–884. doi: 10.1053/gast.1996.v110.pm8608898. [DOI] [PubMed] [Google Scholar]
- 29.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 30.Kimura K, Shimosegawa T, Sasano H, Abe R, Satoh A, Masamune A, Koizumi M, Nagura H, Toyota T. Endogenous glucocorticoids decrease the acinar cell sensitivity to apoptosis during cerulein pancreatitis in rats. Gastroenterology. 1998;114:372–381. doi: 10.1016/s0016-5085(98)70490-1. [DOI] [PubMed] [Google Scholar]
- 31.Bennett MR. Mechanisms of p53-induced apoptosis. Biochem Pharmacol. 1999;58:1089–1095. doi: 10.1016/s0006-2952(99)00153-7. [DOI] [PubMed] [Google Scholar]
- 32.Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000;404:892–897. doi: 10.1038/35009130. [DOI] [PubMed] [Google Scholar]
- 33.Buckley S, Barsky L, Driscoll B, Weinberg K, Anderson KD, Warburton D. Apoptosis and DNA damage in type 2 alveolar epithelial cells cultured from hyperoxic rats. Am J Physiol. 1998;274:L714–L720. doi: 10.1152/ajplung.1998.274.5.L714. [DOI] [PubMed] [Google Scholar]
- 34.Ortiz LA, Moroz K, Liu JY, Hoyle GW, Hammond T, Hamilton RF, Holian A, Banks W, Brody AR, Friedman M. Alveolar macrophage apoptosis and TNF-alpha, but not p53, expression correlate with murine response to bleomycin. Am J Physiol. 1998;275:L1208–L1218. doi: 10.1152/ajplung.1998.275.6.L1208. [DOI] [PubMed] [Google Scholar]
- 35.Menke A, Yamaguchi H, Gress TM, Adler G. Extracellular matrix is reduced by inhibition of transforming growth factor beta1 in pancreatitis in the rat. Gastroenterology. 1997;113:295–303. doi: 10.1016/s0016-5085(97)70107-0. [DOI] [PubMed] [Google Scholar]
- 36.Riesle E, Friess H, Zhao L, Wagner M, Uhl W, Baczako K, Gold LI, Korc M, Büchler MW. Increased expression of transforming growth factor beta s after acute oedematous pancreatitis in rats suggests a role in pancreatic repair. Gut. 1997;40:73–79. doi: 10.1136/gut.40.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teramoto T, Kiss A, Thorgeirsson SS. Induction of p53 and Bax during TGF-beta 1 initiated apoptosis in rat liver epithelial cells. Biochem Biophys Res Commun. 1998;251:56–60. doi: 10.1006/bbrc.1998.9411. [DOI] [PubMed] [Google Scholar]
- 38.Huang YL, Shen CK, Luh TY, Yang HC, Hwang KC, Chou CK. Blockage of apoptotic signaling of transforming growth factor-beta in human hepatoma cells by carboxyfullerene. Eur J Biochem. 1998;254:38–43. doi: 10.1046/j.1432-1327.1998.2540038.x. [DOI] [PubMed] [Google Scholar]
- 39.Chen RH, Chang TY. Involvement of caspase family proteases in transforming growth factor-beta-induced apoptosis. Cell Growth Differ. 1997;8:821–827. [PubMed] [Google Scholar]
- 40.Brown TL, Patil S, Cianci CD, Morrow JS, Howe PH. Transforming growth factor beta induces caspase 3-independent cleavage of alphaII-spectrin (alpha-fodrin) coincident with apoptosis. J Biol Chem. 1999;274:23256–23262. doi: 10.1074/jbc.274.33.23256. [DOI] [PubMed] [Google Scholar]
- 41.Saltzman A, Munro R, Searfoss G, Franks C, Jaye M, Ivashchenko Y. Transforming growth factor-beta-mediated apoptosis in the Ramos B-lymphoma cell line is accompanied by caspase activation and Bcl-XL downregulation. Exp Cell Res. 1998;242:244–254. doi: 10.1006/excr.1998.4096. [DOI] [PubMed] [Google Scholar]
- 42.Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99:2365–2374. doi: 10.1172/JCI119418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friess H, Lu Z, Riesle E, Uhl W, Bründler AM, Horvath L, Gold LI, Korc M, Büchler MW. Enhanced expression of TGF-betas and their receptors in human acute pancreatitis. Ann Surg. 1998;227:95–104. doi: 10.1097/00000658-199801000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 45.Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol. 1995;269:C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]
- 46.Hahm KB, Kim JH, You BM, Kim YS, Cho SW, Yim H, Ahn BO, Kim WB. Induction of apoptosis with an extract of Artemisia asiatica attenuates the severity of cerulein-induced pancreatitis in rats. Pancreas. 1998;17:153–157. doi: 10.1097/00006676-199808000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Bhatia M, Wallig MA, Hofbauer B, Lee HS, Frossard JL, Steer ML, Saluja AK. Induction of apoptosis in pancreatic acinar cells reduces the severity of acute pancreatitis. Biochem Biophys Res Commun. 1998;246:476–483. doi: 10.1006/bbrc.1998.8519. [DOI] [PubMed] [Google Scholar]
- 48.Fujimoto K, Hosotani R, Doi R, Wada M, Lee JU, Koshiba T, Miyamoto Y, Imamura M. Role of neutrophils in cerulein-induced pancreatitis in rats: possible involvement of apoptosis. Digestion. 1997;58:421–430. doi: 10.1159/000201478. [DOI] [PubMed] [Google Scholar]
- 49.Gong ZH, Yuan YZ, Lou KX, Tu SP, Zhai ZK, Xu JY. Effects and mechanisms of somatostatin analogues on apoptosis of pancreatic acinar cells in acute pancreatitis in mice. Shijie Huaren Xiaohua Zazhi. 1999;7:964–966. [Google Scholar]