

Abstract
The CRISPR/Cas technology enables targeted genome editing and the rapid generation of transgenic animal models for the study of human genetic disorders. Here we describe an autosomal recessive human disease in two unrelated families characterized by a split-foot defect, nail abnormalities of the hands, and hearing loss, due to mutations disrupting the SAM domain of the protein kinase ZAK. ZAK is a member of the MAPKKK family with no known role in limb development. We show that Zak is expressed in the developing limbs and that a CRISPR/Cas-mediated knockout of the two Zak isoforms is embryonically lethal in mice. In contrast, a deletion of the SAM domain induces a complex hindlimb defect associated with down-regulation of Trp63, a known split-hand/split-foot malformation disease gene. Our results identify ZAK as a key player in mammalian limb patterning and demonstrate the rapid utility of CRISPR/Cas genome editing to assign causality to human mutations in the mouse in <10 wk.
Split-hand/split-foot malformation (SHFM) is a limb anomaly characterized by median clefts with missing or malformed central rays (Elliott et al. 2005). SHFM is clinically and genetically heterogeneous and represents a paradigmatic genetic disorder displaying different modes of inheritance, variable expressivity, and incomplete penetrance (Birnbaum et al. 2012; Klopocki et al. 2012). Submicroscopic duplications at 10q24 and 17p13.3, TP63 mutations, and deletions of exonic enhancers in DYNC1I1 represent major SHFM-causing mechanisms (Ianakiev et al. 2000; de Mollerat et al. 2003; Birnbaum et al. 2012; Klopocki et al. 2012). Mutations in other genes, including WNT10B in an autosomal recessive form, have been reported. However, in up to two thirds of affected individuals, the causative mutation remains unknown (Ugur and Tolun 2008; Tayebi et al. 2014). One of the key challenges in rare Mendelian disorders is to identify additional disease alleles in unrelated families. CRISPR/Cas genome editing can now be used to create a large number of new alleles in the mouse within a few weeks by creating specific mutations and deletions in a gene of interest (Wang et al. 2013; Kraft et al. 2015). Here we report on the combination of whole-exome sequencing in patients with CRISPR/Cas genome editing in mice to identify and validate a novel disease-causing gene and to assign an unexpected role to the protein kinase ZAK in mammalian limb development.
Results
Identification of ZAK mutations in split-foot malformation with mesoaxial polydactyly
We studied an autosomal recessive disorder characterized by a split-foot phenotype, nail abnormalities of the hands, and hearing impairment. In a multiplex consanguineous Pakistani family with four affected individuals (family F1) (Fig. 1; Supplemental Figs. 1, 2), autozygosity mapping following single-nucleotide polymorphism (SNP) array genotyping identified a disease locus on Chromosome 2q31 with a maximum LOD score of 3.5 (Fig. 2A; Supplemental Fig. 1A). The 9.1-Mb linkage region does not contain any known gene that is mutated in limb malformations. After performing exome sequencing of one affected individual (F1-VI:3) and filtering for rare potentially damaging variants, we detected two homozygous missense variants in the linkage region: c.1247T > A (p.Phe416Tyr) in ZNF385B and c.1103T > G (p.Phe368Cys) in ZAK (Fig. 2B; Supplemental Fig. 1B). Both variants cosegregated with the disease, were predicted to be damaging, and were absent or very rare in controls (Supplemental Table 1). As neither of the two genes had a known function in limb development, we performed whole-mount in situ hybridization (ISH) on mouse embryos for Znf385b and Zak to prioritize these candidate genes for further studies. While Znf385b was mainly expressed in the developing brain (Supplemental Fig. 3), Zak was expressed in the heart and the developing limbs (Fig. 3), making it a plausible candidate gene for limb defects. Furthermore in an RNA-seq data set of human embryonic hindlimbs published by Cotney et al. (2013), ZAK was shown to be expressed in human limbs, while ZNF585B expression was not detected. The zinc finger protein 385B (ZNF385B, formerly known as ZNF533) is also expressed in the developing brain (http://www.brain-map.org/), lungs, and ovaries (http://www.proteinatlas.org/), but its function is currently unknown. Overlapping deletions of ZNF385B have been identified in patients with mental retardation, high palate, and micrognathia (Mencarelli et al. 2007; Monfort et al. 2008). Therefore ZNF385B was suggested to be a candidate gene for mental retardation. We next sequenced ZAK in 106 unrelated patients representing the broad clinical SHFM spectrum. In an affected boy from Tunisia (F2-IV:2) (Fig. 1B,D,F), we used PCR, array-CGH, and qPCR to detect a homozygous intragenic deletion of ZAK (Fig. 2B; Supplemental Fig. 4A,B) that was heterozygous in his unaffected first-cousin parents (Supplemental Fig. 4B). The boy presented with a very similar split-foot phenotype, including distal duplication of digit II and unilateral syndactyly I/II and IV/V (Fig. 1C–F). The hands were normal, but the nails of both digits IV showed a duplication of the nail bed (Fig. 1F). The patient had normal newborn hearing screening, and at the age of 2 yr, the psychomotor and language development was normal. As shown by sequencing of a junction PCR fragment, this 14.7-kb deletion removed exons 12–16 of ZAK (Supplemental Fig. 4C); it was absent from in-house (n = >600) and public CNV databases (Firth et al. 2009; MacDonald et al. 2014).
Figure 1.

Split-foot malformation with mesoaxial polydactyly (SFMMP) in two families. (A) Pedigree of family F1. Black symbols indicate individuals with a split-foot phenotype and/or hearing loss. (+) c.1103T reference allele of ZAK; (−) c.1103T > G (p.Phe368Cys) mutation of ZAK. Autozygosity mapping was performed with DNA from individuals designated with an asterisk; and exome sequencing was performed with DNA from individual VI:3, designated with an arrowhead. (B) Pedigree of family F2. IV:2 is affected by the split-foot phenotype. (+) Reference allele; (−) genomic deletion of exons 12–16 of ZAK. (C) Pictures of the feet of affected individuals from family F1 showing no split foot in V:2, bilateral split-foot malformation in VI:3 and VI:4, and unilateral split-foot malformation in VI:1. (D) Hands and feet of the affected child in family F2. The nail anomaly is boxed. (E) Radiographs of the feet of individual VI:4 in family 1 showing bilateral split-foot malformation, syndactyly between the first and rudimentary second toes, duplication of the left proximal phalanx of the second toe corresponding to mesoaxial polydactyly (arrowhead), Y-shaped appearance of the right metatarsal bone II due to synostosis between the horizontally placed remnant of the proximal phalanx II and the hypoplastic metatarsal II, and hypoplastic and misshaped phalangeal bones of toes III to V with fusions of some interphalangeal joints. In addition to split-foot malformation with missing third toe, the radiograph of the left foot of individual F2-IV:2 shows cutaneous syndactyly between the first and second toes and hypoplasia of the distal phalanx of the first ray and of the middle and distal phalanges of the second toe. A second rudimentary phalangeal bone of toe II is present (arrowhead). There is hypoplasia of middle and distal phalanges of toes IV and V, as well as syndactyly between these toes. (F) Clinical pictures of individual F1-IV:4 and individual F2-IV:2 showing a duplication of the nail bed on digit IV of both hands.
Figure 2.

Identification of homozygous ZAK mutations in SFMMP. (A) Representation of genome-wide homozygosity in family F1 following SNP array genotyping with 250k arrays. Homozygosity scores with respect to the maximum LOD score of 3.5 are plotted against the physical position genome-wide. The linkage region on Chromosome 2 is indicated by the red peak; it is 9.1 Mb in size and delimited by recombinant markers rs836624 and rs6433931. (B) Schematic representation of ZAK with the missense mutation and the intragenic deletion shown in red. The ZAK protein contains a kinase domain, as well as a leucine zipper (LZ) and a sterile alpha motif (SAM) domain. Multiple protein sequence alignment; the Phe368Cys mutation site is boxed in red.
Figure 3.
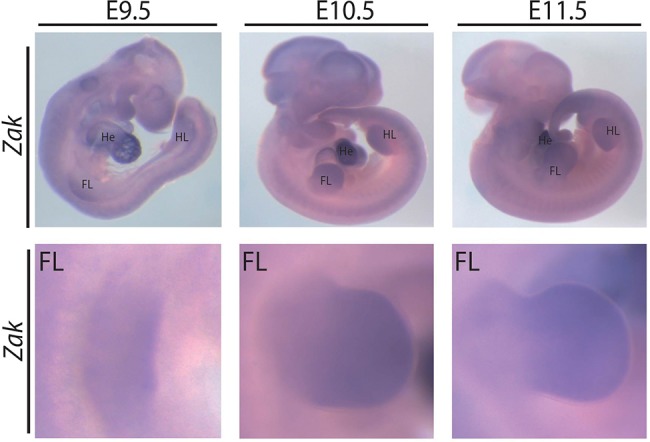
Expression of Zak in mouse embryos. In situ hybridization using a probe for Zak on mouse embryos at the indicated embryonic days. (Top) Whole embryos; (bottom) magnifications of the forelimb (FL).
As typically observed in SHFM, there was marked phenotypic variability in both affected families with foot defects in affected individuals ranging from bilateral to unilateral foot involvement to no split foot in one individual (F1-V:2) (Fig. 1C). Individuals F1-VI:4 and F2-VI:2 showed partial duplication of digit II in combination with syndactyly I/II (Fig. 1C–E). The patients also had duplications of the nail bed on the hands (Fig. 1F). In addition to limb defects, all patients in family 1 had bilateral sensorineural hearing impairment (Supplemental Fig. 2), consistent with Zak expression in hair cells of the mouse cochlea (Francis et al. 2013). We refer to this distinct phenotype as “split-foot malformation with mesoaxial polydactyly” (SFMMP).
ZAK is a member of the MAPKKK family that is involved in the control of cell growth and differentiation, the regulation of cytoskeletal changes, and gene expression (Yang 2003; Cheng et al. 2009). No role had been assigned to ZAK in vertebrate limb development, but knockdown of its Xenopus laevis ortholog mltk was previously shown to result in loss of craniofacial cartilage (Gotoh et al. 2001; Suzuki et al. 2012). ZAK was shown to be a direct target of TP63 binding, and a functional link of both proteins was suggested; ZAK has also been shown to suppress the JNK pathway (Yang et al. 2006; Wang et al. 2014a). The ZAK gene encodes two isoforms, both containing an N-terminal protein kinase domain followed by a leucine zipper (LZ). Notably, the missense alteration and the intragenic deletion both affect the SAM domain, which is only present in the longer ZAK-α isoform. While the deletion is predicted to remove the complete SAM domain, Phe368Cys affects a highly conserved residue (Fig. 2B).
Generation and analysis of Zak−/− mice using CRISPR/Cas genome editing in mice
The recent development of the CRISPR/Cas technology has led to a wider use of genome editing and opens new possibilities to create mutations and structural variations within weeks in model systems for the investigation of rare Mendelian disorders (Wang et al. 2013). We therefore aimed to study the role of mutations in Zak in transgenic mice using CRISPR/Cas genome editing. To create a complete knockout of both Zak isoforms, we designed one single guide RNA (sgRNA) in exon 2 and transfected mouse embryonic stem cells (ESCs) with the CRISPR construct. We created homozygous frameshift mutations in exon 2 (Supplemental Fig. 5A) and performed diploid ESC aggregation to produce highly chimeric animals. The CRISPR/Cas-mediated inactivation of Zak resulted in fully penetrant lethality at E9.5 due to severe cardiac edema and growth retardation (Supplemental Fig. 5B). No viable homozygous mice were obtained from two independent clones. In contrast, heterozygous mice were morphologically indistinguishable from their wild-type littermates.
Since both the missense mutation and the intragenic deletion affect the SAM domain of ZAK-α, we hypothesized that mutations exclusively in the SAM domain might be responsible for the split-foot phenotype. We therefore aimed to genocopy the intragenic deletion of family 2 by specifically deleting the SAM domain using an adapted CRISPR/Cas protocol for the introduction of structural variants (Kraft et al. 2015; Lupianez et al. 2015). We transfected two CRISPR/Cas sgRNAs flanking the deletion and created ESCs with homozygous deletions of the 12-kb fragment. The deletion was predicted to be in frame, and expression analysis confirmed normal Zak mRNA levels in the embryonic limbs (Supplemental Fig. 6A). No predicted off-target mutation was found via Sanger sequencing (Supplemental Fig. 6B). In highly (>95%) chimeric animals generated from two independent homozygous clones, we observed a spectrum of unilateral complex hindlimb duplication phenotypes with low penetrance (four out of 64 animals) (Fig. 4A–D). In one mouse at E18.5, one supernumerary hindlimb with normal polarity was connected to an imperfectly duplicated pelvic girdle (Fig. 4A). The duplicated hindlimb showed a shortened femur, fibula, and tibia, and a normal sized foot. The forelimbs were normal. At later stages, variable splitting of the distal skeleton of the hindlimbs was observed. One mouse showed a partial duplication of the right hindlimb and pelvic structures (Fig. 4B; Supplemental Video 1). While the foot was completely duplicated, the fibula and tibia were partially duplicated (Fig. 4C), and the femur shaft was split showing duplications of the distal parts (Fig. 4D). Two mice showed polydactyly of the feet, representing a minimal version of a distal duplication defect (Supplemental Fig. 7). The mutant mice did not show any obvious ear abnormalities, and the Preyer's reflex as screening to identify profound sensorineural hearing loss did not show differences between mutants, heterozygous littermates, or wild-type mice (Jero et al. 2001).
Figure 4.

Limb defects in CRISPR/Cas mice with a deletion of the ZAK SAM domain. (A) At E18.5, one supernumerary hindlimb with normal polarity was connected to an imperfectly duplicated pelvic girdle. The duplicated hindlimb showed a shortened femur, fibula, and tibia and normal-sized digits. (B) A 5-wk-old mouse showed a partial duplication of the right hindlimb and pelvic structures. (C) The foot was completely duplicated (blue) and connected by a common digit V with a distal duplication (red). The fibula and tibia were only partially duplicated. (D) The right femur shaft was split, and the distal and proximal parts (red) were duplicated. The left femur appeared normal (blue). (E) Expression analysis of Zakdel/del homozygous mutant (MT) hindlimbs at E11.5 showed a 60% decrease of Trp63 expression compared with wild-type (WT) hindlimbs. Error bars, SD.
To identify regulators or interaction partners of ZAK, we performed whole-mount ISHs and expression analysis for selected key limb development genes. ISH in homozygous Zak−/− embryos revealed normal staining patterns for Fgf8, Shh, and Trp63 at E11.5 (Supplemental Fig. 8). We noted that the mutant mice looked similar to mice treated in utero with retinoic acid, showing lower limb duplications and hindlimb oligodactyly (Niederreither et al. 1996). Treatment of limb bud cells with retinoic acid results in a down-regulation of Trp63 (Wang et al. 2014b). We therefore performed quantitative reverse transcription–PCR (qRT-PCR) for Trp63 in Zak-mutant limb tissue (Yang et al. 2006) and detected a 60% decrease of Trp63 expression in homozygous versus wild-type hindlimbs (Fig. 4E).
Functional analysis of the Phe368Cys missense substitution affecting the SAM domain of ZAK
To finally understand how the Phe368Cys substitution affects the structure of the ZAK SAM domain, we generated a 3D model (Fig. 5A; Harada et al. 2008; Kelley and Sternberg 2009). The model predicts that Phe368 is located in the hydrophobic core of the protein. Mutation to Cys might therefore disrupt the hydrophobic core and lead to SAM domain instability. To test this hypothesis, we purified both the wild-type and mutant ZAK SAM domain and assessed the secondary structure of each using circular dichroism (CD). The CD spectrum of Phe368Cys showed a ∼30% loss of alpha-helicity compared with the wild-type (Fig. 5B). Moreover, thermal unfolding experiments showed a cooperative, albeit irreversible, unfolding transition for the wild-type ZAK SAM domain, while the mutant showed a complete loss of cooperative unfolding transition (Supplemental Fig. 9). These results indicate that the Phe368Cys mutation strongly destabilizes the ZAK SAM domain.
Figure 5.

Functional effects of ZAK SAM domain missense mutation. (A) Phyre model of the ZAK SAM domain based on the structure of the SAM domain of DGKD (30% identity, PDB ID 3BQ7). Phe368 resides in the hydrophobic core of the SAM domain and is shown as spheres highlighted in red. (B) Circular dichroism spectra of wild-type ZAK-SAM and the Phe368Cys mutant. The altered spectrum of ZAK-SAM Phe368Cys correlates with a loss of alpha-helicity. (C) Purified ZAK-SAM and ZAK-LZ-SAM were assessed by SEC-MALS. The SAM domain alone is monomeric with an observed molecular weight (MW) of 10.2 kDa (predicted MW = 8.9 kDa). The LZ-SAM construct has an observed MW of 32.3 kDa, corresponding to a homogenous population of dimer (predicted dimer MW = 29.6 kDa). (D) negGFP fusions of wild-type and Phe368Cys mutants of ZAK-SAM and ZAK-LZ-SAM assessed by native gel electrophoresis. In this assay, described previously (Liu et al. 2000), proteins are fused to a highly negative green fluorescent protein (negGFP). The negative charges on negGFP cause reliable migration toward the cathode in a native gel, and the relative mobility reflects oligomerization. Monomeric SAM domains migrate as discrete bands and with higher mobility than polymeric SAM domains, which migrate slower and with a smeared character. negGFP fusions of a monomeric SAM (ANKS6) and a polymeric SAM (DGKD) are shown as controls. ZAK-SAM appears monomeric, and addition of the LZ causes a significant gel shift. The Phe368Cys (F368C) substitution in the ZAK SAM domain causes a retarded migration, indicating protein aggregation.
ZAK self-associates in vivo and this may be mediated by the SAM domain and/or the LZ (Liu et al. 2000). Size exclusion chromatography–multi-angle light scattering (SEC-MALS) analysis of a purified ZAK SAM domain and a LZ-SAM fusion revealed that the SAM domain alone is monomeric while the LZ drives dimerization (Fig. 5C). The Phe368Cys mutant caused retarded migration on a native gel assay (Knight et al. 2011), indicating that it is aggregation prone (Fig. 5D), consistent with protein unfolding and pathogenicity of the mutation.
Discussion
Our data show that ZAK-α has a role in limb development and that the deletion of its SAM domain is associated with down-regulation of Trp63 in the limb bud. Mutations affecting the expression of TRP63 are associated with a wide spectrum of limb phenotypes: While the knockout of Trp63 results in severe truncation of both limbs, knock-in of an SHFM mutation Arg279His causes a complete reduction of hindlimbs and milder reduction of forelimbs (Yang et al. 1999; Vanbokhoven et al. 2011). In contrast, treatment of mice with retinoic acid was shown to cause lower limb duplications, polydactyly, and oligodactyly, at least in part due to down-regulation of Trp63 (Niederreither et al. 1996; Wang et al. 2014b). Therefore we speculate that mutations in ZAK causing a down-regulation of Trp63 in the hindlimb give rise to a wide spectrum of lower limb phenotypes with reduced penetrance and variable expressivity. Genetically unresolved cases of SHFM and other complex limb defects may be caused by mutations in other members of the ZAK pathway. However, much more experimental work is needed to investigate a potential functional link between TP63 and ZAK.
In this study, we exemplarily show that the CRISPR/Cas9 system can be used to efficiently reengineer human variants in mice with the purpose to test their pathogenicity. This approach holds the promise to contribute to a paradigmatic shift in the investigation of Mendelian disorders. In fact, for many years the confirmation and functional validation of causative mutations in previously unreported disease genes has relied on conducting time-consuming experiments directed at recapitulating key features of the disease in cell systems or animal models. The generation of such animal models, however, was expensive and time-consuming (Ruf et al. 2011; Sternberg and Doudna 2015). By taking advantage of the CRISPR/Cas9 genome editing technology to induce site-specific genomic double-strand breaks in the mouse that are repaired by the nonhomologous end-joining (NHEJ) pathway, we show that several mouse models for a novel human disease can be generated in <10 wk (Lieber 2010; Kraft et al. 2015; Lupianez et al. 2015). The CRISPR/Cas9 system in combination with a template sequence can also be used for the introduction of precise genetic modifications through an alternative homology-directed repair (HDR) pathway (Chu et al. 2015). This will be extremely useful in the future to investigate the pathogenicity of rare missense alleles that might occur only in single families. Thus, beyond the identification of ZAK as a key player in mammalian limb patterning, our results demonstrate the utility of CRISPR/Cas9 genome editing to assign causality to human mutations in the mouse.
In conclusion, we describe a syndrome characterized by a split-foot phenotype, nail abnormalities, and hearing loss due to isoform-specific mutations of ZAK. Our results identify ZAK-α as a key player in limb patterning. Furthermore we demonstrate that CRISPR/Cas genome editing can be used for the fast and efficient creation of new associated alleles in the mouse to study the pathogenicity of mutations and deletions in a gene of interest in Mendelian disorders.
Methods
Subjects and ethics approval
The study was performed with the approval of the University of Ulm ethics committee and the institutional review board (IRB) of the Balochistan University of Information Technology, Engineering, and Management Sciences (BUITEMS). Patients were enrolled with written informed consent for participation in the study. The clinical evaluation included medical history interviews, a physical examination, radiographs of hands and feet, and review of medical records. Blood samples were obtained from each participating individual, and DNA was extracted by standard procedures.
SNP-array genotyping and autozygosity mapping
We performed a genome-wide linkage analysis with autozygosity mapping by using Affymetrix genome-wide human SNP 6.0 arrays and genomic DNA samples from five members of family 1, namely, the four affected individuals V:2, VI:1, VI:3, and VI:4, as well as the unaffected individual VI:2. Autozygosity mapping was performed as previously described (Borck et al. 2011).
Exome sequencing
Exome sequencing was performed in individual VI:3 in family 1. Briefly, genomic DNA was enriched for exonic and adjacent splice site sequences with the SeqCap EZ human exome library v2.0 kit, and libraries were run on an Illumina HiSeq 2000 sequencer via a paired-end 100-bp protocol (Hussain et al. 2013). For data analysis, the Cologne Center for Genomics (CCG) Varbank pipeline v2.6 and user interface was used (Kawalia et al. 2015). Primary data were filtered according to signal purity by the Illumina realtime analysis (RTA) software v1.8. Subsequently, the reads were mapped to the human genome reference build GRCh37/hg19 (http://www.genome.ucsc.edu/) using the BWA-SW alignment algorithm. Mean coverage was 100× in the exome, and 95.6% and 86.4% of target bases were covered more than 10× and 30×, respectively. Further annotation and filtering for high-quality rare variants (MAF < 0.1%) with a predicted impact on protein sequence or splicing was performed as previously described (Borck et al. 2015). We also filtered against an inhouse database containing variants from 511 exomes from individuals with epilepsy to exclude pipeline-related artifacts.
Sanger sequencing
In family 1, validation and cosegregation analyses were performed by Sanger sequencing on an ABI 3730 DNA analyzer with BigDye chemistry v3.1. In family 2, the complete coding region of ZAK (NM_016653.2), including splice sites, was sequenced (for primer sequences and positions, see Supplemental Table 2).
Microarray-based comparative genomic hybridization
Microarray-based comparative genomic hybridization (array CGH) for family 2 was carried out using a whole-genome 1-M oligonucleotide array (Agilent); 1 M arrays were analyzed by feature extraction v9.5.3.1 and CGH analytics v3.4.40 software or cytogenomics v2.5.8.11, respectively (Agilent). Analysis settings were as follows: aberration algorithm, ADM-2; threshold, 6.0; window size, 0.2 Mb; filter, 5probes; log2ratio = 0.29. The genomic profile was visualized by the SignalMap software (SignalMap v1.9.0.03, NimbleGen Systems).
Quantitative real-time PCR (qPCR)
qPCR was performed as previously described (Kraft et al. 2015) using genomic DNA of the index patients and further family members to confirm the deletions and to show segregation with the phenotype (for primer sequences, see Supplemental Table 3).
Breakpoint analysis
The exact determination of the deletion size was done by breakpoint spanning PCR following the qPCR analyses and sequencing of the junction fragment (primer sequences and positions in Supplemental Table 2). For the sequencing of the PCR products, the BigDye v3.1 (Applied Biosystems) sequencing kit was used. PCR products were analyzed by capillary automat ABI3730 (Applied Biosystems). The sequencing results were processed by DNA-STAR software (DNA-Star).
Cloning and mutagenesis
negGFP-fusions of hZAK (UniProt:Q9NYL2) encompassing the SAM domain (residues 333–410; “ZAK-SAM”) and including the upstream LZ region (residues 286–410; “ZAK-LZ-SAM”) were generated by cloning into a negGFP vector described previously (Knight et al. 2011). Hexahistidine small ubiquitin-like modifier (SUMO) tagged constructs were generated by cloning the above sequences into a pHis-SUMO vector (Senturia et al. 2010). Site-directed mutagenesis was performed using the Quickchange method (Agilent). All plasmid sequences were verified by DNA sequencing (Genewiz).
negGFP native gel assay
negGFP-human-ZAK fusions were transformed into ARI814 cells and expressed, harvested, and lysed as described previously (Knight et al. 2011; Leettola et al. 2014). Expression levels were determined by fluorescence intensity, and based on fluorescence, equal amounts of protein were loaded on a 20% RunBlue 12-well native gel (Expedeon). Gels were run at 90 V for 16 h at 4°C and visualized on a Bio-Rad molecular imager FX pro-plus using an excitation wavelength of 488 nm and an emission wavelength of 510 nm. In an attempt to identify ZAK hetero–SAM interactions, negGFP-ZAK-SAM was mixed in a 1:1 ratio based on fluorescence, and negGFP-ZAK-LZ-SAM was mixed in a 3:1 ratio based on fluorescence with 54 other negGFP fusions of hSAM domains. Mixes were allowed to equilibrate for 3 h at 4°C before running on native gels. Neither screen detected any hetero interactions.
Protein expression and purification
pHis-SUMO constructs (ZAK-SAM, ZAK-SAM F368C, and ZAK-LZ-SAM) were transformed into Rosetta(DE3) pLysS cells (Novagen), and 4 L of culture was grown to an OD600 of 0.6, at which point cells were induced with 1 mM isopropyl β-d-galactopyranoside and grown for an additional 16 h at 18°C. Harvested cells were lysed, and pHis-SUMO tagged proteins were purified using Ni-NTA (Qiagen) as described previously (Leettola et al. 2014). Proteins were dialyzed into 20 mM NaHPO4 (pH 8), 0.5 M NaCl, and 2 mM βME and digested with the His6-tagged catalytic domain of SUMO protease 1 (ULP1) at a 50:1 protein:protease molar ratio for 16 h at 4°C (Malakhov et al. 2004). The cleaved His6-SUMO tag and ULP1 protease were removed by subtractive Ni-NTA. Proteins were further purified by dialysis into either 20 mM Tris (pH 7.5; ZAK-SAM and ZAK-SAM F368C) or 20 mM Tris (pH 7.0; ZAK-LZ-SAM), 50 mM NaCl, 2 mM βME and bound to a HiTrap Q HP column (GE). Elution was achieved using a shallow gradient of NaCl (0.05–1 M across 20 column volumes) dissolved in either 20 mM Tris (pH 7.0 or pH 7.5) or 2 mM βME. The proteins eluted between 0.3 and 0.4 M NaCl.
Circular dichroism
Spectra were collected for protein samples at 0.2 mg/mL in 10 mM Tris (pH 7.5), 75 mM NaCl, 1 mM DTT at 25°C using a 1-mm path-length cuvette on a JASCO J-715 CD spectrophotometer equipped with a Peltier temperature control. Spectra were analyzed for secondary structure content using the Neural Network algorithm available in SoftSec (Softwood). Thermal melts were performed by monitoring the change in CD signal at 222 nm across a temperature range of 25°C–80°C, with ramping of 1°C per minute. Wild-type ZAK-SAM and ZAK-SAM F368C thermal denaturation was not reversible.
SEC-MALS
Purified ZAK-SAM and ZAK-LZ-SAM were dialyzed into 20 mM Tris (pH 7.5), 0.15 M NaCl, 2 mM TCEP. One hundred twenty microliters of protein at 10 mg/mL was loaded onto a WTC-030S5 analytical size-exclusion column (Wyatt Technology) equilibrated in 20 mM Tris (pH 7.5), 0.15 M NaCl, 2 mM TCEP using an AKTA purifier (GE) with a flow rate of 0.7 mL/min and analyzed using a miniDAWN TREOS (Wyatt Technology). Eluted protein peaks were analyzed for calculated molecular weight and monodispersity using ASTRA software (Wyatt Technology).
CRISPR sgRNAs selection and cloning
SgRNAs were designed to induce point mutations or flanking the regions to rearrange. We used the http://crispr.mit.edu/ platform to obtain candidate sgRNA sequences with little off-targets specificity. Complementary strands were annealed, phosphorylated, and cloned into the BbsI site of pX459 or pX330 CRISPR/Cas vector (for CRISPR sgRNAs sequences and positions, see Supplemental Table 3).
ESC culture and transfection
We seeded 300,000 G4 cells (129xC57BL/6 F1 hybrid ESCs) on CD1 feeders and transfected them with 8 μg of each CRISPR construct using FuGENE technology (Promega). When the construct originated from the pX330 vector, cells were cotransfected with a puromycine-resistant plasmid. PX459, in contrast, already contains a puromycine-resistant cassette. After 24 h, cells were split and transferred onto DR4 puro-resistant feeders and selected with puromycine for 2 d. Clones were then grown for 5–6 more days, picked, and transferred into 96-well plates on CD-1 feeders. After 2 d of culture, plates were split in triplicates, two for freezing and one for growth and DNA harvesting. Positive clones identified by PCR or Sanger sequencing were thawed and grown on CD-1 feeders until they reached an average of 4 million cells. Three vials were frozen, and DNA was harvested from the rest of the cells to confirm genotyping. PCR-based genotyping and qPCR were performed as previously described (Kraft et al. 2015).
To create a complete knockout of both Zak isoforms, we designed one sgRNA in exon 2. We detected compound and homozygous mutations in 83 clones (out of 96) and selected two clones with homozygous frameshift mutations in exon 2 for diploid ESC aggregation. To genocopy the intragenic deletion of family 2, an adapted CRISPR/Cas protocol for the introduction of structural variants was used (Kraft et al. 2015; Lupianez et al. 2015). We designed two sgRNAs located in the introns centromeric and telomeric to exons 12 and 16 of Zak. We then cotransfected the two sgRNAs in mouse ESCs to induce double-strand breaks at the desired positions. We detected 17 heterozygous and two clones (out of 288) with a homozygous deletion of the 12-kb fragment (Supplemental Fig. 10).
Mouse aggregation
A frozen ESC vial was seeded on CD-1 feeders, and cells were grown for 2 d. Mice were generated by diploid or tetraploid ESC aggregation (Artus and Hadjantonakis 2011). All animal procedures were in accordance with institutional, state, and government regulations (Berlin: LAGeSo).
ISH and skeletal preparations
ISH for Zak, Fgf8, Trp63, and Shh was carried out on wild-type embryos (C57BL/6J) and mutant embryos at embryonic stages E11.5. Skeletal preparations and alizarin red staining of E18.5 wild-type and mutant embryos were performed as previously described (Albrecht et al. 2002).
Data access
ZAK mutation data from this study have been submitted to the NCBI ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/) under accession number SCV000256897. Array-CGH data from this study have been submitted to the DECIPHER database (http://decipher.sanger.ac.uk) under patient ID: 321328. Sequencing data from this study have been submitted to the European Genome-phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted at the EBI, under accession number EGAS00001001633.
Supplementary Material
Acknowledgments
We thank the families for their collaboration and contribution to this project. M.S. was supported by a fellowship of the Berlin-Brandenburg School for Regenerative Therapies (BSRT), Berlin, Germany.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.199430.115.
References
- Albrecht AN, Schwabe GC, Stricker S, Boddrich A, Wanker EE, Mundlos S. 2002. The synpolydactyly homolog (spdh) mutation in the mouse: a defect in patterning and growth of limb cartilage elements. Mech Dev 112: 53–67. [DOI] [PubMed] [Google Scholar]
- Artus J, Hadjantonakis AK. 2011. Generation of chimeras by aggregation of embryonic stem cells with diploid or tetraploid mouse embryos. Methods Mol Biol 693: 37–56. [DOI] [PubMed] [Google Scholar]
- Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, et al. 2012. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res 22: 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, Ur Rehman A, Lee K, Pogoda HM, Kakar N, von Ameln S, Grillet N, Hildebrand MS, Ahmed ZM, Nurnberg G, et al. 2011. Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am J Hum Genet 88: 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, Hog F, Dentici ML, Tan PL, Sowada N, Medeira A, Gueneau L, Thiele H, Kousi M, Lepri F, et al. 2015. BRF1 mutations alter RNA polymerase III–dependent transcription and cause neurodevelopmental anomalies. Genome Res 25: 609. [PMC free article] [PubMed] [Google Scholar]
- Cheng YC, Kuo WW, Wu HC, Lai TY, Wu CH, Hwang JM, Wang WH, Tsai FJ, Yang JJ, Huang CY, et al. 2009. ZAK induces MMP-2 activity via JNK/p38 signals and reduces MMP-9 activity by increasing TIMP-1/2 expression in H9c2 cardiomyoblast cells. Mol Cell Biochem 325: 69–77. [DOI] [PubMed] [Google Scholar]
- Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R. 2015. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33: 543–548. [DOI] [PubMed] [Google Scholar]
- Cotney J, Leng J, Yin J, Reilly SK, DeMare LE, Emera D, Ayoub AE, Rakic P, Noonan JP. 2013. The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mollerat XJ, Gurrieri F, Morgan CT, Sangiorgi E, Everman DB, Gaspari P, Amiel J, Bamshad MJ, Lyle R, Blouin JL, et al. 2003. A genomic rearrangement resulting in a tandem duplication is associated with split hand-split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet 12: 1959–1971. [DOI] [PubMed] [Google Scholar]
- Elliott AM, Evans JA, Chudley AE. 2005. Split hand foot malformation (SHFM). Clin Genet 68: 501–505. [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. 2009. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. Am J Hum Genet 84: 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SP, Katz J, Fanning KD, Harris KA, Nicholas BD, Lacy M, Pagana J, Agris PF, Shin JB. 2013. A novel role of cytosolic protein synthesis inhibition in aminoglycoside ototoxicity. J Neurosci 33: 3079–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh I, Adachi M, Nishida E. 2001. Identification and characterization of a novel MAP kinase kinase kinase, MLTK. J Biol Chem 276: 4276–4286. [DOI] [PubMed] [Google Scholar]
- Harada BT, Knight MJ, Imai S, Qiao F, Ramachander R, Sawaya MR, Gingery M, Sakane F, Bowie JU. 2008. Regulation of enzyme localization by polymerization: polymer formation by the SAM domain of diacylglycerol kinase δ1. Structure 16: 380–387. [DOI] [PubMed] [Google Scholar]
- Hussain MS, Baig SM, Neumann S, Peche VS, Szczepanski S, Nurnberg G, Tariq M, Jameel M, Khan TN, Fatima A, et al. 2013. CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum Mol Genet 22: 5199–5214. [DOI] [PubMed] [Google Scholar]
- Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P. 2000. Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet 67: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jero J, Coling DE, Lalwani AK. 2001. The use of Preyer's reflex in evaluation of hearing in mice. Acta Otolaryngol 121: 585–589. [PubMed] [Google Scholar]
- Kawalia A, Motameny S, Wonczak S, Thiele H, Nieroda L, Jabbari K, Borowski S, Sinha V, Gunia W, Lang U, et al. 2015. Leveraging the power of high performance computing for next generation sequencing data analysis: tricks and twists from a high throughput exome workflow. PLoS One 10: e0126321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4: 363–371. [DOI] [PubMed] [Google Scholar]
- Klopocki E, Lohan S, Doelken SC, Stricker S, Ockeloen CW, Soares Thiele de Aguiar R, Lezirovitz K, Mingroni Netto RC, Jamsheer A, Shah H, et al. 2012. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J Med Genet 49: 119–125. [DOI] [PubMed] [Google Scholar]
- Knight MJ, Leettola C, Gingery M, Li H, Bowie JU. 2011. A human sterile α motif domain polymerizome. Protein Sci 20: 1697–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft K, Geuer S, Will AJ, Chan WL, Paliou C, Borschiwer M, Harabula I, Wittler L, Franke M, Ibrahim DM, et al. 2015. Deletions, inversions, duplications: engineering of structural variants using CRISPR/Cas in mice. Cell Rep pii: S2211-1247(15)00029-7. [DOI] [PubMed] [Google Scholar]
- Leettola CN, Knight MJ, Cascio D, Hoffman S, Bowie JU. 2014. Characterization of the SAM domain of the PKD-related protein ANKS6 and its interaction with ANKS3. BMC Struct Biol 14: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79: 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TC, Huang CJ, Chu YC, Wei CC, Chou CC, Chou MY, Chou CK, Yang JJ. 2000. Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-α motif. Biochem Biophys Res Commun 274: 811–816. [DOI] [PubMed] [Google Scholar]
- Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161: 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. 2014. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 42: D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. 2004. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics 5: 75–86. [DOI] [PubMed] [Google Scholar]
- Mencarelli MA, Caselli R, Pescucci C, Hayek G, Zappella M, Renieri A, Mari F. 2007. Clinical and molecular characterization of a patient with a 2q31.2-32.3 deletion identified by array-CGH. Am J Med Genet A 143A: 858–865. [DOI] [PubMed] [Google Scholar]
- Monfort S, Rosello M, Orellana C, Oltra S, Blesa D, Kok K, Ferrer I, Cigudosa JC, Martinez F. 2008. Detection of known and novel genomic rearrangements by array based comparative genomic hybridisation: deletion of ZNF533 and duplication of CHARGE syndrome genes. J Med Genet 45: 432–437. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Ward SJ, Dolle P, Chambon P. 1996. Morphological and molecular characterization of retinoic acid-induced limb duplications in mice. Dev Biol 176: 185–198. [DOI] [PubMed] [Google Scholar]
- Ruf S, Symmons O, Uslu VV, Dolle D, Hot C, Ettwiller L, Spitz F. 2011. Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nat Genet 43: 379–386. [DOI] [PubMed] [Google Scholar]
- Senturia R, Faller M, Yin S, Loo JA, Cascio D, Sawaya MR, Hwang D, Clubb RT, Guo F. 2010. Structure of the dimerization domain of DiGeorge critical region 8. Protein Sci 19: 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Doudna JA. 2015. Expanding the biologist's toolkit with CRISPR-Cas9. Mol Cell 58: 568–574. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kusakabe M, Nakayama K, Nishida E. 2012. The protein kinase MLTK regulates chondrogenesis by inducing the transcription factor Sox6. Development 139: 2988–2998. [DOI] [PubMed] [Google Scholar]
- Tayebi N, Jamsheer A, Flottmann R, Sowinska-Seidler A, Doelken SC, Oehl-Jaschkowitz B, Hulsemann W, Habenicht R, Klopocki E, Mundlos S, et al. 2014. Deletions of exons with regulatory activity at the DYNC1I1 locus are associated with split-hand/split-foot malformation: array CGH screening of 134 unrelated families. Orphanet J Rare Dis 9: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugur SA, Tolun A. 2008. Homozygous WNT10b mutation and complex inheritance in Split-Hand/Foot malformation. Hum Mol Genet 17: 2644–2653. [DOI] [PubMed] [Google Scholar]
- Vanbokhoven H, Melino G, Candi E, Declercq W. 2011. p63, a story of mice and men. J Invest Dermatol 131: 1196–1207. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Weerapana E, Blewett MM, Cravatt BF. 2014a. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 11: 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YG, Xie P, Wang YG, Li XD, Zhang TG, Liu ZY, Hong Q, Du SX. 2014b. All-trans-retinoid acid (ATRA) suppresses chondrogenesis of rat primary hind limb bud mesenchymal cells by downregulating p63 and cartilage-specific molecules. Environ Toxicol Pharmacol 38: 460–468. [DOI] [PubMed] [Google Scholar]
- Yang JJ. 2003. A novel zinc finger protein, ZZaPK, interacts with ZAK and stimulates the ZAK-expressing cells re-entering the cell cycle. Biochem Biophys Res Commun 301: 71–77. [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, et al. 1999. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398: 714–718. [DOI] [PubMed] [Google Scholar]
- Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K. 2006. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell 24: 593–602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.