

Abstract
The extent to which histone modifying enzymes contribute to DNA methylation in mammals remains unclear. Previous studies suggested a link between the lysine methyltransferase EHMT2 (also known as G9A and KMT1C) and DNA methylation in the mouse. Here, we used a model of knockout mice to explore the role of EHMT2 in DNA methylation during mouse embryogenesis. The Ehmt2 gene is expressed in epiblast cells but is dispensable for global DNA methylation in embryogenesis. In contrast, EHMT2 regulates DNA methylation at specific sequences that include CpG-rich promoters of germline-specific genes. These loci are bound by EHMT2 in embryonic cells, are marked by H3K9 dimethylation, and have strongly reduced DNA methylation in Ehmt2−/− embryos. EHMT2 also plays a role in the maintenance of germline-derived DNA methylation at one imprinted locus, the Slc38a4 gene. Finally, we show that DNA methylation is instrumental for EHMT2-mediated gene silencing in embryogenesis. Our findings identify EHMT2 as a critical factor that facilitates repressive DNA methylation at specific genomic loci during mammalian development.
Cytosine methylation plays diverse roles in mammalian development. It contributes to genomic imprinting, X Chromosome inactivation, and the stable repression of retroelements and developmental genes (Smith and Meissner 2013). During development, most CpG island promoters remain protected from DNA methylation, except for a small set associated with germline-specific genes (Borgel et al. 2010; Auclair et al. 2014). The pluripotency genes Pou5f1 (also known as Oct4) and Dppa3 also acquire CpG methylation in the post-implantation embryo, which stabilizes the exit from pluripotency (Feldman et al. 2006; Borgel et al. 2010). This process requires the de novo methyltransferases DNMT3A/B, whereas the subsequent maintenance of DNA methylation through cell divisions is ensured by DNMT1.
While the targets of DNA methylation are well characterized, little is known about the molecular determinants of DNA methylation in mammals. In plants and filamentous fungi, a large portion of DNA methylation is directed by histone H3 methylated on lysine 9, and deletion of H3K9 methyltransferases has a major impact on DNA methylation (Saze et al. 2012). A link between H3K9 methylation and DNA methylation has been documented also in mammalian cells (for review, see Rose and Klose 2014). SUV39H1 (also known as KMT1A), SUV39H2 (also known as KMT1B), and SETDB1 (also known as KMT1E), which mediate H3K9 trimethylation (H3K9me3) at pericentric heterochromatin and ERV retrotransposons, interact with DNMTs (Fuks et al. 2003; Li et al. 2006) and modulate DNA methylation at pericentric satellite repeats and ERV retrotransposons in mouse embryonic stem cells (ESCs) (Lehnertz et al. 2003; Matsui et al. 2010).
On the other hand, the lysine methyltransferase EHMT2 (also known as G9A and KMT1C) and its closely related partner EHMT1 (also known as GLP and KMT1D) catalyze H3K9 mono- and dimethylation (H3K9me1 and me2) in euchromatin (Tachibana et al. 2002, 2005). EHMT2 and EHMT1 play pivotal roles during early mouse development (Tachibana et al. 2002, 2005). They exist mostly as an EHMT2/EHMT1 heterodimeric complex, which is the main functional H3K9 methyltransferase because the absence of either EHMT2 or EHMT1 strongly affects global H3K9me1/2 in embryonic cells (Tachibana et al. 2005). EHMT2 interacts and colocalizes with DNMT1 and UHRF1 at sites of DNA replication (Esteve et al. 2006; Kim et al. 2009). Inversely, UHRF1 binds to chromatin containing H3K9me2/3, which may facilitate the maintenance of DNA methylation at genomic sites containing methylated H3K9 (Karagianni et al. 2008; Rothbart et al. 2012; Liu et al. 2013). In mouse ESCs, EHMT2 controls DNA methylation at germline differentially methylated regions (gDMRs) of imprinted loci (Xin et al. 2003; Dong et al. 2008), class I and II ERV retrotransposons, LINE1 elements, satellite repeats, and CpG-rich promoters of germline and developmental genes (Ikegami et al. 2007; Dong et al. 2008; Tachibana et al. 2008; Myant et al. 2011). EHMT2 also interacts with the de novo methyltransferases DNMT3A and DNMT3B (Epsztejn-Litman et al. 2008; Kotini et al. 2011) and participates in the de novo methylation of newly integrated retroviruses (Leung et al. 2011) and pluripotency genes in ESCs (Feldman et al. 2006; Epsztejn-Litman et al. 2008; Athanasiadou et al. 2010). Several studies suggest that the influence of EHMT2 on DNA methylation in ESCs is independent of its catalytic activity (Dong et al. 2008; Epsztejn-Litman et al. 2008; Tachibana et al. 2008).
These cell-based studies suggested that EHMT2 is an important regulator of DNA methylation in mammals, yet the contribution of EHMT2 to DNA methylation in mammalian embryogenesis is unknown. This prompted us to explore the role of EHMT2 in DNA methylation during mouse embryogenesis using mice deficient for EHMT2 and a combination of locus-specific and genome-wide approaches. Our in vivo studies show that EHMT2 plays a role in the deposition of repressive DNA methylation at specific genomic sites during embryonic development.
Results
EHMT2 has a global influence on the DNA methylome of murine ES cells
Previous studies reported reduced DNA methylation in cultured murine Ehmt2−/− ESCs (Ikegami et al. 2007; Dong et al. 2008; Epsztejn-Litman et al. 2008; Tachibana et al. 2008; Myant et al. 2011). To extend these findings and quantify the influence of EHMT2 on the methylome of ESCs genome-wide, we generated single-base resolution methylomes by reduced representation bisulfite sequencing (RRBS) in WT TT2 ESCs, Ehmt2−/− TT2 ESCs, and Ehmt2−/− cells rescued with a WT Ehmt2 transgene (Tachibana et al. 2002). As expected, WT ESCs show a bimodal distribution of CpG methylation with most of the hypomethylated CpGs residing in CpG islands located proximal to transcription start sites (TSSs) (Fig. 1A,B). The inactivation of EHMT2 in ESCs leads to a global and uniform decrease of CpG methylation over all sequences of the genome (Fig. 1A,B). In agreement with the earlier studies (Dong et al. 2008; Tachibana et al. 2008; Myant et al. 2011), hypomethylation also affects the promoters of germline and developmental genes such as the Dazl, Wfdc15a, Brdt, Tuba3a, and Rhox genes (Fig. 1C). In addition, all classes of transposable elements (TEs) lose on average ∼40% CpG methylation in Ehmt2−/− compared with the parental TT2 ESCs (Fig. 1D). The reintroduction of a WT Ehmt2 transgene in Ehmt2−/− ESCs restores DNA methylation at all sequences, including TEs (Fig. 1A–D). The overall methylation is slightly higher in EHMT2-rescued cells compared with the parental cell line, which may be attributed to the higher levels of the EHMT2 protein in rescued cells (Mozzetta et al. 2014).
Figure 1.
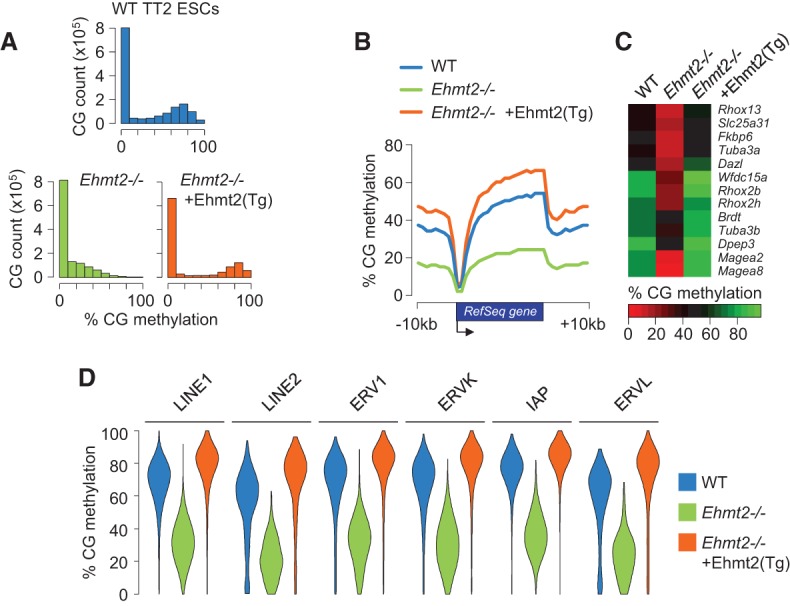
Global impact of EHMT2 on the DNA methylome of mouse ESCs. (A) Density histograms of RRBS methylation scores at CpGs in WT ESCs, Ehmt2−/− ESCs, and Ehmt2−/− ESCs rescued with a WT Ehmt2 transgene (Tg). (B) Average distribution of RRBS methylation over RefSeq genes and flanking sequences in WT, Ehmt2−/−, and Ehmt2 rescued ESCs. (C) Heatmap representation of the RRBS methylation scores measured in the promoters (−1000 to +1000 bp) of selected germline and developmental genes. (D) Violin plots of RRBS methylation scores in retrotransposons.
Role of EHMT2 in DNA methylation at candidate genes in embryos
We next investigated the role of EHMT2 in DNA methylation during embryogenesis in vivo. We first followed the expression of Ehmt2 mRNAs in mouse embryos by RT-qPCR and found that it is expressed at the time of de novo methylation of DNA between embryonic day (E) 4.5 and E8.5 (Fig. 2A). To study the role of EHMT2 in DNA methylation, we used an Ehmt2 knockout line in which a LacZ cassette inserted after exon 11 results in truncated transcripts lacking the Ankyrin (ANK) repeats and the catalytic SET domain (Supplemental Fig. S1A–D; Wagschal et al. 2008). In agreement with an earlier Ehmt2 knockout (Tachibana et al. 2002), Ehmt2−/− mice show a developmental delay and mid-gestation lethality around E10.5 (Wagschal et al. 2008). Using this model, we first analyzed DNA methylation at candidate genes by combined bisulfite restriction analysis (COBRA) and bisulfite sequencing. We assessed DNA methylation of the promoters of the germline genes Dazl, Dpep3, and Tex12 and observed no hypomethylation in Ehmt2−/− embryos (Fig. 2B). Since EHMT2 controls the de novo methylation of pluripotency genes in differentiating ESCs (Epsztejn-Litman et al. 2008), we investigated DNA methylation at the Pou5f1 and Dppa3 promoters but found no evidence for reduced DNA methylation in E9.5 Ehmt2−/− embryos (Fig. 2C) or in trophoblast cells of E9.5 Ehmt2−/− animals (Supplemental Fig. S2A). Finally, because Ehmt2 cooperates with Dnmt3 to regulate eye development in zebrafish (Rai et al. 2010), we measured methylation in the promoters of eye-specific genes (Rho, Crygd, Cplx4, Mfrp) and found no sign of hypomethylation at these either (Fig. 2B).
Figure 2.

Impact of EHMT2 on the methylome in mouse embryos. (A) Expression of Ehmt2 mRNAs measured by RT-qPCR in early mouse embryos, depicted as a ratio relative to the expression of two housekeeping genes (Actb and Rpl13a; mean ± SEM, n = 2 technical replicates on five to 10 pooled embryos). (B) Promoter DNA methylation of candidate genes measured by COBRA in WT and Ehmt2−/− E9.5 embryos. The restriction fragments marked with an asterisk are end products of the digestion (indicating DNA methylation). (C) Promoter DNA methylation of the Pou5f1 and Dppa3 genes measured by bisulfite sequencing in WT and Ehmt2−/− E9.5 embryos. Circles represent methylated (black) or unmethylated (white) CpG dinucleotides; each horizontal line is one sequenced clone. (D) Pairwise comparison of methylated DNA immunoprecipitation (MeDIP) log2 ratios at individual oligos in WT and Ehmt2−/− E9.5 embryos. The density of data points increases from blue to dark red. The Pearson correlation coefficient (r) is indicated on the graph. (E) Density histograms of RRBS CpG methylation scores in WT and Ehmt2−/− E8.5 embryos. (F) Average distribution of RRBS methylation over RefSeq genes and flanking sequences in WT and Ehmt2−/− embryos. (G) Violin plots of RRBS methylation measured in CpG islands (CGIs) and retrotransposons. (H) Example of methylation profiles in the promoter of the Dazl gene in WT and Ehmt2−/− embryos. The upper tracks depict smoothed MeDIP log2 ratios of individual oligonucleotides, and the bottom tracks depict RRBS methylation scores at individual CpGs. The CpG density is shown in black.
Methylome profiling reveals a global conservation of DNA methylation in Ehmt2−/− embryos
Having found no changes in DNA methylation at candidate genes in Ehmt2−/− embryos, we generated genome-scale methylomes using two approaches. We first profiled 5-methylcytosine (5mC) with methylated DNA immunoprecipitation (MeDIP) coupled to microarrays covering on average 11 kb at all gene promoters. A direct comparison showed that the 5mC log2 values correlate strongly between WT and Ehmt2−/− embryos (Fig. 2D). In parallel, we generated methylomes at single-base resolution by RRBS in WT and Ehmt2−/− E8.5 embryos (Supplemental Fig. S3A). The RRBS data were highly reproducible between replicate embryos (Supplemental Fig. S3B) and revealed no signs of hypomethylation at the genome level in Ehmt2−/− embryos (Fig. 2E,F). To extend this finding, we assessed the methylation status of TEs by averaging methylation scores in TEs and found no effects on the overall methylation of TEs in Ehmt2−/− embryos except a minor decrease at LINE2 and ERVL elements (Fig. 2G). Moreover we found no evidence of overall hypomethylation at TE families when we mapped RRBS sequencing reads to Repbase consensus sequences (data not shown). This was confirmed by restriction analysis and conventional bisulfite sequencing at the IAP, RLTR4, and ETnERV elements (Supplemental Fig. S2B,C). The promoters of most germline and developmental genes, including those hypomethylated in Ehmt2−/− ESCs such as Dazl, Wfdc15a, Brdt, Tuba3a, and Rhox, are methylated at the same levels in Ehmt2−/− and WT embryos (Fig. 2H; Supplemental Fig. S4A,B). Finally, the absence of EHMT2 does not impair CpG island methylation on the inactive X Chromosome in female embryos (Supplemental Fig. S4C,D; Ohhata et al. 2004). Combined, these data indicate that in contrast to ESCs, EHMT2 is dispensable for genome-wide DNA methylation in embryogenesis.
Identification of hypomethylated sequences in Ehmt2−/− embryos
Despite global conservation, the methylomes of Ehmt2−/− embryos are distinct and clustered separately from WT embryos (Supplemental Fig. S3B,C). RRBS revealed that the knockout of Ehmt2 leads to focal changes, with 956 regions losing >20% and 517 regions gaining >20% methylation in Ehmt2−/− embryos (Fig. 3A; Supplemental Table S1). Hypomethylation occurs in promoters, gene bodies, intergenic regions, and occasionally in transposons of the L1Md_T and LTR-ERVK families (Fig. 3B; Supplemental Table S1). The Ehmt2 gene body itself is hypomethylated in Ehmt2−/− embryos after the site of LacZ insertion where transcription is aborted (Supplemental Fig. S5), supporting the model that transcription promotes gene body methylation (Baubec et al. 2015). Strikingly, the absence of EHMT2 also led to hypermethylation at a small set of CpG islands, mostly within genes (Fig. 3B; Supplemental Fig. S6A). Because hypomethylation occurs more frequently and with higher amplitude, we focused on the regions losing methylation, which we called hypomethylated regions (HMRs).
Figure 3.

Identification of hypomethylated regions (HMRs) in Ehmt2−/− embryos. (A) Histogram of the number of sequences that gain or lose >20% methylation in the RRBS from Ehmt2−/− compared with WT embryos. (B) Pie charts representing the percentages of hypomethylated and hypermethylated sequences mapping to promoters, gene bodies, intergenic regions, and transposable elements (TEs). (C) Boxplot showing the dynamics of methylation at HMRs (filtered to lose >30% methylation in Ehmt2−/− embryos) in early embryos, Ehmt2−/− E8.5 embryos (green), and Dnmt3b−/− E8.5 embryos (red). (D) Preferential site of expression of genes with a promoter-proximal HMR (−1000 to +1000 bp relative to the TSS). The dashed line represents the position for a P-value of 0.05. (E) Examples of promoter-proximal HMRs. The sites of hypomethylation identified by MeDIP (gray boxes) confirm the differences measured by RRBS. (F) Bisulfite cloning and sequencing of selected promoter HMRs in WT and Ehmt2−/− E9.5 embryos. The values below the sequencing data indicate the percentage of methylated CpGs in the amplicon.
HMRs undergo de novo methylation at implantation (Fig. 3C), which excludes that their hypomethylation reflects the developmental delay of Ehmt2−/− embryos. The extent of DNA hypomethylation at HMRs in Ehmt2−/− embryos is similar to that caused by inactivation of the DNA methyltransferase DNMT3B (Fig. 3C). Notably, promoter HMRs are enriched for promoters of germline-specific genes (Fig. 3D), including Cyct, Naa11, Asz1, Hormad2, Morc2b, Pdha2, Ptpn20, and Abca16 (Fig. 3E; Supplemental Fig. S7). Several other germline genes show a reduction of promoter methylation just below the threshold of 20% (Wdr20rt, Rpl10l, 1700019A02Rik) or, when not covered by RRBS, were identified as hypomethylated in the MeDIP data (Slc9b1, 4933427D06Rik). HMRs also occur in promoter regions of somatic genes such as Trim13, Tff3, Aplnr, Tenm2, Sh3tc2, Ano5 and genes of the Xlr imprinted cluster (Xlr3a/b, Xlr4a/b/c) (Fig. 3E; Supplemental Fig. S7). To validate HMRs, we performed COBRA on nine genes and confirmed the hypomethylation in Ehmt2−/− embryos compared with WT and Ehmt2+/− littermates (Supplemental Fig. S8A,B). Bisulfite sequencing confirmed a decrease in methylation ranging from 32% to 84% over multiple contiguous CpGs (Fig. 3F; Supplemental Fig. S8C). Taken together, our data show that EHMT2 is required for the deposition of DNA methylation at specific genomic sites during embryogenesis, including the CpG-rich promoters of several germline genes.
EHMT2 participates in the maintenance of imprinted DNA methylation at the Slc38a4 locus
We also examined the contribution of EHMT2 to the maintenance of DNA methylation imprints in vivo. RRBS quantification of DNA methylation at 16 imprinted gDMRs revealed a methylation level close to 50% in WT embryos, and this is unchanged at all but one gDMR in Ehmt2−/− embryos (Fig. 4A; Supplemental Fig. S9A). This is confirmed by bisulfite sequencing and McrBC digestion at four gDMRs (H19, Kcnq1ot1, Snrpn, Peg3) in Ehmt2−/− embryos (Fig. 4B; Supplemental Fig. S9B). Remarkably, the absence of EHMT2 leads to the specific hypomethylation of the Slc38a4 gDMR, which carries allele-specific methylation inherited from the oocyte (Fig. 4A; Proudhon et al. 2012). Unlike most other HMRs, the RRBS data indicate that the extent of hypomethylation at the Slc38a4 gDMR is variable between individual embryos (Fig. 4C), suggesting a partially penetrant effect at this locus. Thus, EHMT2 plays a role in the maintenance of the DNA methylation imprint at the Slc38a4 locus during embryogenesis.
Figure 4.

Locus-specific control of DNA methylation imprints by EHMT2 at the Slc38a4 gDMR. (A) Methylation scores measured by RRBS in 16 known imprinted gDMRs in WT and Ehmt2−/− embryos. (B) Bisulfite cloning and sequencing analysis of four gDMRs in WT and Ehmt2−/− E9.5 embryos. (C) MeDIP profiles at the imprinted Slc38a4 locus (top) and RRBS scores in four WT and three Ehmt2−/− embryos (bottom). The orange rectangle marks the position of the gDMR.
The influence of EHMT2 on DNA methylation is direct
We next investigated the mechanisms by which EHMT2 controls DNA methylation. RNA-seq indicates that the expression of genes encoding DNMTs, UHRF1, and TETs is not perturbed in Ehmt2−/− embryos (Fig. 5A), which was confirmed by RT-qPCR (Supplemental Fig. S10A). We detected a mild up-regulation of Dnmt3b only, which likely is a consequence of the developmental delay of Ehmt2−/− embryos. This finding agrees with data in ESCs (Dong et al. 2008; Tachibana et al. 2008) and suggests that EHMT2 does not influence DNA methylation indirectly by modulating the expression of the DNA methylation machinery. To determine if EHMT2 binds to HMRs, we performed chromatin immunoprecipitation (ChIP) on primary mouse embryonic fibroblasts (MEFs) and found EHMT2 enriched at several HMRs (Fig. 5B). Importantly, we were also able to detect an enrichment of EHMT2 at HMRs by ChIP in vivo in E8.5 embryos (Fig. 5C).
Figure 5.

HMRs are bound by EHMT2 and marked by H3K9me2. (A) RNA-seq quantification of the expression of genes encoding components of the DNA methylation machinery in Ehmt2−/− and WT E9.5 embryos (mean FPKM ± SEM, n = 2 embryos). (B) ChIP-qPCR analysis of EHMT2 binding at HMRs in primary mouse embryonic fibroblasts (MEFs), represented as the percentage of input (mean ± SEM, n = 5). ChIP assays were performed with an antibody against EHMT2 and a control rabbit IgG. Actb served as a negative control, and Rhox11 was chosen as a positive control (Myant et al. 2011). (C) ChIP-qPCR analysis of EHMT2 binding at HMRs in E8.5 embryos (mean ± SEM, n = 4). (D) Browser views of EHMT2 ChIP-seq profiles in ESCs (Mozzetta et al. 2014) reveal that peaks of EHMT2 binding colocalize with promoter-proximal HMRs (red bars) at three germline genes. (E) Heatmap representation of the distribution of EHMT2 and H3K9 mono- and dimethylation at HMRs. The data represent the average density of ChIP-seq reads for EHMT2 in ESCs, H3K9me1/2 in ESCs, and H3K9me2 in E8.5 embryos normalized by the density of reads in the input control. (F) ChIP-qPCR analysis of H3K9me2 and H3K4me3 at HMRs in E8.5 embryos, represented as the percentage of input (mean ± SEM, n = 4). The promoters of the housekeeping genes Actb and Ube2f served as controls. (G) ChIP-qPCR analysis of H3K9me2 at four gene promoters in WT and Ehmt2−/− E8.5 embryos (mean ± SEM, n = 6 embryos for WT, n = 4 embryos for Ehmt2−/−). The heatmap on the bottom indicates CpG methylation measured by RRBS in the same promoters. (**) P < 0.01 (t-test).
To further confirm the binding of EHMT2 at HMRs in embryonic cells, we explored a published EHMT2 ChIP-seq data set in ESCs (Mozzetta et al. 2014) and found that peaks of EHMT2 frequently colocalize with HMRs, in particular in the promoters of germline genes (Cyct, Naa11, Asz1, Hormad2, Morc2b, Pdha2, Pgam2, Ptpn20, Abca16, Slc9b1, Wdr20rt, 4933427D06Rik, 1700019A02Rik) (Fig. 5D; Supplemental Fig. S11A). Similarly, the ChIP-seq data indicate a higher binding of EHMT2 in the vicinity of the Slc38a4 gDMR compared with other imprinted gDMRs (Supplemental Fig. S11B). To explore this in a systematic way, we computed the distribution of EHMT2 ChIP-seq signals around all HMRs and observed that HMRs contain higher EHMT2 signal than the surrounding regions (Fig. 5E). Conversely, hypermethylated DMRs rarely match peaks of EHMT2, suggesting that a proportion of them might result from indirect effects (Supplemental Fig. S6B). Thus, EHMT2 is frequently enriched in the proximity of HMRs in embryonic cells, suggesting that it influences DNA methylation in cis at these targets.
Interplay between H3K9me2 and DNA methylation in embryos
We then investigated if the influence of EHMT2 on DNA methylation is related to its activity as a methylase of H3K9. We performed ChIP-qPCR of H3K9me2 on WT E8.5 embryos and found that all the tested HMRs are strongly enriched for H3K9me2 (Fig. 5F). Conversely, they are not marked by the active histone mark H3K4me3 except for the Slc38a4 imprinted gene (Fig. 5F). A similar pattern of histone marks was observed in primary embryonic fibroblasts (Supplemental Fig. S12A). For two HMRs (Naa11 and Asz1), we confirmed that H3K9me2 is reduced in Ehmt2−/− embryos, demonstrating that EHMT2 exerts its catalytic activity at these sites in vivo (Fig. 5G). To extend these findings, we analyzed ChIP-seq profiles of H3K9me1/2 in ESCs (Liu et al. 2015) and also performed H3K9me2 ChIP-seq in E8.5 embryos. According to the ChIP-seq data, HMRs are marked by H3K9 mono- and dimethylation in ESCs and E8.5 embryos, but the enrichment for these marks is only slightly higher than that of the surrounding sequences (Fig. 5E). In fact, the ChIP-seq signals indicate that H3K9me2 exists in large blocks of chromatin covering most of the genome, suggesting that the presence of high levels of H3K9me2 is not a hallmark of HMRs. ChIP-qPCR in E8.5 embryos confirmed that other gene promoters carry H3K9me2 at levels comparable to HMRs and lose this mark in Ehmt2−/− embryos without any detectable effect on their DNA methylation (Fig. 5G). Thus, HMRs are marked by H3K9me2, but there is no consistent correlation between the loss of H3K9me2 and DNA methylation in Ehmt2−/− embryos.
DNA methylation is instrumental for EHMT2-mediated repression of germline genes in embryos
Finally, we explored the impact of EHMT2 on gene expression by conducting RNA-seq in WT versus Ehmt2−/− E8.5 embryos. As expected (Tachibana et al. 2002), Magea genes are reactivated in Ehmt2−/− embryos (Fig. 6A). We identified 253 genes as more than threefold up-regulated and 181 genes as more than threefold down-regulated in Ehmt2−/− embryos (Fig. 6A; Supplemental Table S2). The down-regulated genes are enriched for genes involved in neuronal and muscle morphogenesis (data not shown). The up-regulated genes contain genes of the Rhox and Xlr clusters on the X Chromosome and are enriched for testis-specific genes (Fig. 6B). Indeed, several of the most up-regulated genes correspond to germline genes with reduced promoter DNA methylation: Cyct, Naa11, Asz1, Pdha2, Ptpn20, Abca16, Hormad2, and Slc9b1 (Fig. 6C,D; Supplemental Table S2). Notably, several genes with a promoter HMR (Morc2b, Trim13, Tenm2, Sh3tc2) are modestly or not overexpressed in Ehmt2−/− embryos, indicating that promoter DNA hypomethylation is not a consequence of gene reactivation. This pattern of gene reactivation upon inactivation of the Ehmt2 gene was validated by RT-qPCR on independent embryos (Supplemental Fig. S10B).
Figure 6.

EHMT2 represses germline genes via DNA methylation in mouse embryos. (A) Comparison of RNA-seq expression levels for RefSeq genes in WT and Ehmt2−/− embryos. Genes of the Magea family and differentially expressed genes are highlighted in colors. (B) Preferential tissue of expression of genes up-regulated at least threefold in Ehmt2−/− embryos. (C) Examples of RNA-seq profiles at the Cyct and Asz1 genes in two biological replicates of WT and Ehmt2−/− embryos. (D) Activation of germline genes in Ehmt2−/− and Dnmt3b−/− E8.5 embryos. The heatmap on the bottom indicates CpG methylation measured by RRBS in the corresponding promoters in Ehmt2−/− and Dnmt3b−/− E8.5 embryos. (E) ChIP-qPCR analysis of H3K9me2 in WT and Dnmt3b−/− E8.5 embryos (mean ± SEM, n = 3 embryos for WT, n = 4 embryos for Dnmt3b−/−), showing that the reduced DNA methylation does not impact the deposition of H3K9me2. (F) Model: EHMT2 deposits H3K9me2 and facilitates cytosine methylation at a subset of gene promoters in embryos. The inactivation of EHMT2 inhibits H3K9me2 and leads to reduced cytosine methylation, leading to aberrant gene activation. In Dnmt3b−/− embryos, EHMT2 is able to bind to its target promoters but can no longer recruit cytosine methylation, which leads to incomplete gene silencing.
To further test the role of DNA methylation in the derepression of EHMT2 targets, we compared RNA-seq from Ehmt2 and the DNA methyltransferase Dnmt3b (Auclair et al. 2014) knockout embryos and found a highly significant overlap (P = 5.9156 × 10−22, hypergeometric test) between the genes up-regulated in Ehmt2−/− and Dnmt3b−/− at E8.5 (Supplemental Fig. S13A,B). Several of the germline genes up-regulated in Ehmt2−/− embryos (including Cyct, Naa11, Asz1) are also overexpressed when hypomethylated in Dnmt3b mutants, albeit not to the extent seen in Ehmt2 mutants (Fig. 6D). We monitored H3K9me2 by ChIP in Dnmt3b−/− embryos and found that the reduced DNA methylation does not impair the deposition of H3K9me2 at HMRs (Fig. 6E; Supplemental Fig. S12A). Furthermore, we found no influence of the reduced DNA methylation on the binding of EHMT2 at promoter HMRs in Dnmt3b−/− fibroblasts (Supplemental Fig. S12B). We conclude that the dependence of CpG methylation on EHMT2 is not accompanied by a reciprocal dependence of EHMT2 on CpG methylation and that H3K9me2 alone is not sufficient to induce an efficient silencing of the germline genes targeted by EHMT2. Collectively, these results demonstrate that EHMT2 acts upstream of DNA methylation, and that EHMT2-guided DNA methylation is instrumental for the repression of these EHMT2 targets in vivo (Fig. 6F).
Discussion
The molecular pathways guiding DNA methylation in mammalian genomes remain poorly understood. Here we addressed the role of the lysine methyltransferase EHMT2 in the control of DNA methylation in the mouse in vivo. EHMT2 controls DNA methylation at specific sites, including the CpG-rich promoters of germline-specific genes. The inactivation of EHMT2 also affects the maintenance of the DNA methylation imprint at the Slc38a4 gDMR, but not at gDMRs of other imprinted loci, which highlights the existence of locus-specific mechanisms of maintenance of imprinted DNA methylation. Slc38a4 differs from other imprinted gDMRs in that it shows no ZFP57 binding (Saadeh and Schulz 2014; Strogantsev et al. 2015), displays no allelic ATRX binding and H3.3 incorporation (Voon et al. 2015), and also lacks H3K9me3 in ES cells. These marked differences could explain why this gDMR uniquely relies on alternative, EHMT2-dependent mechanisms for the maintenance of its germline-derived allelic DNA methylation.
Does EHMT2 promote DNA methylation in cis? Several arguments suggest that this is the case. First, EHMT2 binding is detected by ChIP at many of the regions that show DNA hypomethylation. Second, we did not observe a deregulation of genes encoding DNMTs or TETs in Ehmt2−/− embryos. Third, several of the hypomethylated genes are not overexpressed in Ehmt2−/− embryos, indicating that their hypomethylation is not a secondary consequence of gene activation. We therefore speculate that the absence of EHMT2 impairs the recruitment of DNA methylation in cis at these targets. This scenario, however, does not exclude that DNA methylation may be affected indirectly at some sites.
What mechanisms link EHMT2 to DNA methylation? H3K9me2 is recognized by UHRF1, a cofactor for DNMT1, suggesting that EHMT2-mediated H3K9me2 could stimulate the maintenance of DNA methylation by the UHRF1–DNMT1 complex (Karagianni et al. 2008; Rothbart et al. 2012; Liu et al. 2013). HMRs are marked by H3K9me2, and the loss of DNA methylation at HMRs correlates with a decrease in H3K9me2 in Ehmt2−/− embryos. However this model is in contradiction with the observations that H3K9me2 covers a large part of the genome and that DNA methylation at other sites of H3K9me2 loss is unaffected in Ehmt2−/− embryos. Thus, our results are in favor of a role of EHMT2 in DNA methylation that is independent of H3K9 methylation. EHMT2 methylates other histone residues such as H3K56 (Yu et al. 2012) and lysines in H1 variants (Trojer et al. 2009; Weiss et al. 2010) and has several nonhistone protein substrates (Rathert et al. 2008), including DNMT3A (Chang et al. 2011), which could be necessary for effective methylation of DNA. Alternatively, as supported by studies in ESCs (Dong et al. 2008; Epsztejn-Litman et al. 2008; Tachibana et al. 2008), EHMT2 could regulate DNA methylation independently of its catalytic activity. EHMT2 physically interacts with the DNA methyltransferases DNMT1, DNMT3A, and DNMT3B (Esteve et al. 2006; Epsztejn-Litman et al. 2008; Kotini et al. 2011) and could stimulate their recruitment at specific genomic sites. It also remains to be studied if the EHMT2 heterodimeric partner EHMT1 functionally overlaps with EHMT2 in the control of embryonic DNA methylation.
Our data shed new light on the role of EHMT2 during embryogenesis. RNA-seq demonstrated that the inactivation of EHMT2 leads to the reactivation of a few hundred genes. Besides known targets of EHMT2 such as the genes of the Magea family, many of the most up-regulated genes correspond to germline genes and genes of the imprinted Xlr cluster harboring reduced promoter DNA methylation. By comparing embryos deficient for EHMT2 or the DNA methyltransferase DNMT3B, we provide evidence that the recruitment of DNA methylation is important for the biological functions of EHMT2. Indeed, germline genes are reactivated in Dnmt3b−/− embryos even though the binding of EHMT2 and the levels of H3K9me2 are unchanged, indicating that DNA methylation is important to achieve efficient silencing of these EHMT2 target genes. We note, however, that the extent of gene reactivation in Dnmt3b−/− embryos does not reach the levels seen in Ehmt2−/− embryos, suggesting that H3K9 and DNA methylation cooperatively silence these genes. Moreover, other genes like Magea2 and Wfdc15a are reactivated in Ehmt2−/− embryos without a reduction in their promoter DNA methylation, illustrating that EHMT2 also regulates genes independently of DNA methylation.
The mechanisms that specify the genes repressed by EHMT2 are still unclear. One hypothesis is that EHMT2 is recruited by long noncoding RNAs (lncRNAs). This model has emerged from studies on imprinted domains where specific recruitment of histone lysine methyltransferases controls the allelic repression at placenta-specific genes of the Kcnq1 and Igf2r domains in the mouse (Nagano et al. 2008; Wagschal et al. 2008). In humans, similarly, expression of an antisense lncRNA mediates EHMT2 recruitment and local DNA methylation at the DHRS4 gene locus (Li et al. 2012). Another recent study shows that chromatin at the Slc38a4 locus is controlled by a lncRNA (Monnier et al. 2013), again establishing a possible link between noncoding RNA and EHMT2 recruitment. Alternatively, it is known that EHMT2 is part of larger protein complexes with sequence-specific DNA binding factors that could guide EHMT2 to specific sites. In particular, EHMT2 was identified as a member of the E2F6 complex (Ogawa et al. 2002), which has been linked to the control of DNA methylation in mouse cells (Velasco et al. 2010).
We show that the influence of EHMT2 on DNA methylation is much more restricted in embryos compared with cultured ESCs. This is in line with single-gene studies showing that Wfdc15a, Magea2, and Snrpn are hypomethylated in Ehmt2−/− ESCs but not embryos (Xin et al. 2003; Tachibana et al. 2008). In addition, the inactivation of EHMT2 has little effect on DNA methylation in differentiated cells (Link et al. 2009; Sharma et al. 2012). One hypothesis to explain this discrepancy is that EHMT2 could have a greater influence on the kinetics of DNA methylation at earlier timepoints of embryogenesis. Unfortunately, we failed to test this hypothesis because Ehmt2−/− embryos implant much later than WT embryos, precluding us from dissecting mutant and control embryos from the same mothers at E6.5 and making it difficult to compare the kinetics of methylation because of the confounding effect of the developmental delay. Alternatively, the absence of EHMT2 in ESCs could lead to indirect effects on DNA methylation, for example, by promoting a naïve pluripotent state associated with a global reduction in DNA methylation (Ficz et al. 2013; Habibi et al. 2013). Interestingly, studies on other epigenetic regulators also revealed that knockout embryos have milder methylation defects compared with ESCs. For example, loss of SUV39H1/2 leads to a reduction in DNA methylation at satellite DNA in ESCs (Lehnertz et al. 2003), but this is not apparent in embryo-derived fibroblasts (Pannetier et al. 2008). This indicates that the mechanisms controlling DNA methylation are less robust in cultured stem cells and illustrates the remarkable epigenetic robustness of the mammalian embryo.
Methods
Mouse embryos
The morning of the vaginal plug was designated E0.5. Preimplantation blastocysts (E3.5–E4.5) were collected by flushing the uteri with M2 medium (Sigma-Aldrich). Post-implantation epiblasts (E5.5–E7.5) and embryos (E8.5–E9.5) were manually dissected in M2 medium. We prepared genomic DNA samples by proteinase K digestion, phenol/chloroform extraction and precipitation with ethanol. We maintained Ehmt2 knockout mice on a C57BL/6J background and obtained Ehmt2−/− embryos by natural mating of Ehmt2+/− females and males. Genotyping was performed by two PCR reactions using primers that amplify the LacZ reporter cassette and the exons 11–12 (Supplemental Fig. S1B). Dnmt3b−/ embryos were obtained as previously described (Auclair et al. 2014).
Isolation and culture of MEFs
We generated primary MEFs from E13.5 embryos. The embryos were mechanically dissociated and incubated with trypsin 0.25% for 10 min at 37°C. MEFs were isolated by differential attachment and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 50 µg/mL gentamycine in a humidified incubator at 37°C with 5% CO2. We used MEFs at low passage (<p4).
DNA methylation analysis by restriction enzyme digestion and bisulfite treatment
We digested 1 μg of genomic DNA with 20 U of McrBC (New England BioLabs), an endonuclease that digests methylated DNA in a sequence-independent manner, followed by qPCR to measure the percentage of digestion relative to the undigested control DNA. We normalized values to those of an intergenic control sequence containing no CpGs. Bisulfite conversion of genomic DNA was performed with the EpiTect bisulfite kit (Qiagen). We performed PCR amplification of converted DNA followed by COBRA or cloning as previously described (Borgel et al. 2010). Cloned PCR products were sequenced, aligned with the BISMA software, and filtered to remove clonal biases. Primer sequences for qPCR and PCR amplification of bisulfite-treated DNA are provided in the Supplemental Table S3.
MeDIP-chip
We performed MeDIP on 200 ng genomic DNA prepared from WT and Ehmt2−/− littermate embryos collected at E9.5. Precipitates from three replicates of MeDIP and input samples were hybridized to Roche Nimblegen mm9 HD2 2.1M deluxe promoter arrays covering −8200 to +3000 bp from 23,517 TSSs. Sample preparation and bioinformatic treatment of microarray data were performed as previously described (Borgel et al. 2010). For data representation, we smoothed log2 ratios over 200-bp windows using the Ringo R package (Toedling et al. 2007).
RRBS
We performed RRBS on single embryos for four WT and three Ehmt2−/− littermate embryos collected at E8.5. RRBS libraries were produced as previously described (Auclair et al. 2014). Briefly, we digested 70 ng of genomic DNA 5 h with MspI (Thermo Scientific), performed end-repair and A-tailing (with 5 U Klenow-fragment, Thermo Scientific), and ligated to methylated adapters (with 30 U T4 DNA ligase, Thermo Scientific) in Tango 1× buffer. Fragments between 150 and 400 bp were excised from a 3% agarose 0.5× TBE gel with the MinElute gel extraction kit (Qiagen) and bisulfite-converted with the EpiTect bisulfite kit (Qiagen) with two consecutive rounds of conversion. Final RRBS libraries were amplified by PCR with the PfUTurbo Cx hotstart DNA polymerase (Agilent) using the following PCR conditions: 2 min at 95°C; 16 cycles of 30 sec at 95°C, 30 sec at 65°C, and 45 sec at 72°C; and 7 min at 72°C. We purified the libraries with AMPure magnetic beads (Beckman Coulter) and performed paired-end sequencing (0032 × 75 bp) on an Illumina HiSeq 2500 at Integragen SA. ESCs lines were cultivated as previously described (Mozzetta et al. 2014), and RRBS was performed with 100 ng starting DNA and 14 cycles for the final PCR. We cleaned the sequencing reads with Trim Galore (v0.2.1, parameters --rrbs --paired -r1 30 -r2 30 -q 20 --length 20 --retain_unpaired; http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and aligned to the mm10 genome with BSMAP (v2.74, parameters -v 2 -w 100 -r 1 -x 400 -m 30 -D C-CGG -n 1) (Xi and Li 2009). Percentage of methylation values were calculated as the ratio of the number of Cs over the total number of Cs and Ts with methratio.py in BSMAP (parameters -z -u -g). The bisulfite conversion efficiency was estimated by calculating the C-to-T conversion in non-CpG sites. For all data analysis, we filtered CpGs to have a minimum sequencing depth of 8× and visualized methylation values with the IGV browser (Robinson et al. 2011).
RT-qPCR analysis
Total RNAs were extracted from embryos with TRIzol (Invitrogen), treated with the RQ1 DNase, and reverse transcribed with the QuantiTect reverse transcription kit (Qiagen). We quantified the expression of target cDNAs by qPCR with the KAPA SYBR FASR qPCR kit on a StepOnePlus real-time PCR system (Life Technologies) using the standard curve method. qPCR reactions were performed in triplicates, and expression was normalized to the expression of two housekeeping genes (Rpl13a, Actb). In parallel, we amplified no-RT controls to rule out the presence of contaminating DNA. Primer sequences for qPCR are provided in the Supplemental Table S3.
RNA-seq
We performed RNA-seq on single embryos for two WT and two Ehmt2−/− littermate embryos collected at E8.5. Total RNAs were extracted with the RNeasy protect mini kit (Qiagen). RNA-seq libraries were prepared with the Ovation RNA-seq System V2 (NuGEN) and sequenced on a HiSeq 2500 (1 × 50 bp). Raw sequences were aligned to the mouse mm10 genome by TopHat (v2.0.13, default parameters plus --no-coverage-search --library-type fr-firststrand) (Kim et al. 2013) using a transcriptome index built from RefSeq mm10. Browser tracks in bigWig format were generated with bam2wig.py from the RSeQC package (v2.4, parameters -u -t 5000000000) (Wang et al. 2012) and visualized with the IGV browser. Read counts in genes were computed with htseq-count (v0.6.0, parameters –t exon –s no) (Anders et al. 2015) using a RefSeq mm10 GTF file. We tested for differential gene expression using DESeq2 (v1.4.5) (Love et al. 2014) and defined differentially expressed genes as having a fold change greater than three and an adjusted P-value <0.01. Genes on the Y Chromosome were excluded. Normalized counts and FPKM scores were calculated with the “counts” and “fpkm” functions of DESeq2. For the comparison with genes up-regulated in Dnmt3b−/− embryos (GEO accession no. GSE60334) (Auclair et al. 2014), we selected genes up-regulated at least threefold with an adjusted P-value below 0.01.
ChIP of EHMT2
We cross-linked 5.106 MEFs with 2 mM EGS (Life Technologies) for 45 min and then with 1% formaldehyde (Thermo Scientific) for 10 min in DPBS. The excess formaldehyde was quenched with 125 mM glycine for 5 min at RT. The cells were scraped and lysed in 200 µL lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris at pH 8.1) supplemented with protease inhibitors (Diagenode) for 10 min on ice. We sonicated the chromatin using a Bioruptor waterbath sonicator (Diagenode) to obtain fragments in the range of 0.2 to 1.5 kb. The sheared chromatin was cleared by centrifugation at 14,000 rpm for 20 min at 4°C and diluted 1:10 in dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl at pH 8.1, 167 mM NaCl, protease inhibitors). Ten percent of the chromatin was set aside as the input. The chromatin was immunoprecipitated overnight at 4°C with DiaMag protein A-coated magnetic beads (Diagenode) prebound to a mouse monoclonal EHMT2 antibody (R&D Systems no. PP-A8620A-00, Clone A8620A) or an IgG2a isotype control (R&D Systems no. MAB0031). For the prebinding of beads, 5 μg antibody were coupled to 17 µL magnetic beads in 90 µL of dilution buffer for 3 h at 4°C. The beads–antibody complexes were washed once in low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl at pH 8.1, 150 mM NaCl), once in high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl at pH 8.1, 500 mM NaCl), once in LiCl buffer (0.25 M LiCl, 1% IGEPAL-CA630, 1% sodium deoxycholiate, 1 mM EDTA, 10 mM Tris-HCl pH 8.1), and twice in TE buffer (10 mM Tris-HCl at pH 8.1, 1 mM EDTA). The beads were eluted twice with the elution buffer (1% SDS, 0.1 M NaHCO3) for 15 min at RT, and the two eluates were combined. The cross-linking was reversed in 200 mM NaCl for 4 h at 65°C. We isolated DNA by proteinase K digestion for 1 h at 45°C followed by phenol/chloroform extraction and precipitation in EtOH. For ChIP on E8.5 embryos, we dissociated eight embryos in trypsin 0.25% EDTA 1 mM for 5 min at RT, and cross-linked the cells in suspension with 1.5 mM EGS for 30 min followed with 1% formaldehyde for 10 min in 1 mL of DPBS. Subsequent steps are the same as described above. The qPCR analysis was done with the KAPA SYBR FAST qPCR kit on a StepOnePlus real-time PCR system (Life Technologies), and we calculated for each gene the percentage of recovery in the ChIP relative to the input. Primer sequences for qPCR are provided in the Supplemental Table S3.
ChIP of histone methylation
Five to eight embryos collected at E8.5 were dissociated in trypsin 0.25% EDTA 1 mM for 5 min at RT, washed in PBS, and cross-linked with 1% formaldehyde for 10 min at RT. The excess formaldehyde was quenched with 125 mM glycine for 5 min at RT. The embryos were washed in PBS supplemented with protease inhibitors (Diagenode), and ChIP was performed with a Bioruptor waterbath sonicator (Diagenode) and the LowCell ChIP kit (Diagenode). We used antibodies against H3K9me2 (Abcam ab1220), H3K4me3 (Abcam ab8580), and an IgG2a control (R&D Systems no. MAB0031). ChIP on MEFs was performed on 200,000 cells with the LowCell ChIP kit (Diagenode). For experiments on Ehmt2−/− and Dnmt3b−/− embryos, we performed ChIP on single embryos. The embryos were dissociated and cross-linked as described above, and ChIP was performed with the true MicroChIP kit (Diagenode). Primer sequences for qPCR are provided in the Supplemental Table S3.
H3K9me2 ChIP-seq
ChIP was performed as described above on three pools of eight embryos collected at E8.5. ChIP-seq libraries were prepared with the NEXTflex ChIP-seq Kit (Bioo Scientific) and sequenced on a HiSeq 2500 (1 × 50 bp). Because of the broad distribution of H3K9me2, we merged the FASTQ files from the three experiments. Raw reads were aligned to the mm10 genome with Bowtie 2 (v2.2.4, default parameters) (Langmead and Salzberg 2012). We flagged PCR duplicates with Picard MarkDuplicates (v1.136) (http://broadinstitute.github.io/picard) and generated density tracks in WIG format with igvtools count (v2.3.3.2, parameters -w 25 -e 300 --minMapQuality 30) (Thorvaldsdottir et al. 2013).
Data analysis and characterization of HMRs
Data processing and representation were performed with the R software (R Core Team 2015) using custom developed scripts. To represent the distribution of methylation over genes (Fig. 1F), we calculated the average methylation in 20 equal-sized windows of each protein-coding RefSeq gene and in 10 1-kb windows of flanking sequences. To calculate methylation of TEs, we averaged the methylation scores of all the CpGs contained in annotated repeats with a size >400 bp. We used the eDMR algorithm from the methylKit R package (Li et al. 2013) to identify RRBS DMRs between WT and Ehmt2−/− embryos with at least three CpGs, a difference in methylation >20%, and an adjusted P-value <0.01. We annotated DMRs relative to exons, introns, and repeats using the UCSC RefSeq and Repeatmasker tracks (repeats with a size >400 bp). DMRs were considered promoter-proximal when the center of the DMR is distant <1000 bp from a RefSeq TSS. We performed gene ontology and tissue expression analyses with DAVID (Huang da et al. 2009). HMRs were compared with ChIP-seq data for EHMT2 in ESCs (GEO accession no. GSE46545) (Mozzetta et al. 2014), H3K9me1/2 in ESCs (GEO accession no. GSE54412) (Liu et al. 2015), H3K4me2 in ESCs (GEO accession no. GSE30203) (Stadler et al. 2011), and H3K9me2 in E8.5 embryos (this study). To generate the heatmaps, we counted the density of reads from the ChIP-seq WIG files in 200-bp windows and divided by the density of reads in the same windows in the corresponding input sample.
Data access
The MeDIP-chip, RRBS, ChIP-seq, and RNA-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE71500.
Supplementary Material
Acknowledgments
We thank Dr. Yoichi Shinkai for the authorization to use Ehmt2 KO cell lines. We thank Lauriane Fritsch, Julien Pontis, and Slimane Ait-Si-Ali for providing cell lines and sharing EHMT2 ChIP-seq data and for scientific discussions. We also thank the staff of the IGBMC high-throughput sequencing facility. This work was supported by the Agence National de Recherche (ANR Blanc “EMPREINTE”), the Institut National du Cancer (INCa_5960, INCa_7889), the Association pour la Recherche contre le Cancer (ARC SFI20101201555, SFI20121205729), the Fondation pour la Recherche Médicale (FRM DPM20121125544 and DEQ20150331703), the ITMO Cancer (EPIG201416), the Agency for International Cancer Research (UK), the MEDDTL (11-MRES-PNRPE-9-CVS-072), the LABEX EpiGenMEd, the ATIP-AVENIR program, the EpiGeneSys Network of Excellence, and the European Research Council (ERC Consolidator grant no. 615371). G.A. was supported by fellowships from the French Ministère de la Recherche and from the Ligue Contre le Cancer.
Author Contributions: G.A., J.B., L.A.S., J.V., P.C., and M.G. conducted the experiments. S.G. and M.D. processed the sequencing data and performed data analysis. R.F. and M.W. designed the study and, together with T.F., participated in data analysis. R.F. and M.W. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.198291.115.
References
- Anders S, Pyl PT, Huber W. 2015. HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiadou R, de Sousa D, Myant K, Merusi C, Stancheva I, Bird A. 2010. Targeting of de novo DNA methylation throughout the Oct-4 gene regulatory region in differentiating embryonic stem cells. PLoS One 5: e9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair G, Guibert S, Bender A, Weber M. 2014. Ontogeny of CpG island methylation and specificity of DNMT3 methyltransferases during embryonic development in the mouse. Genome Biol 15: 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, Schubeler D. 2015. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520: 243–247. [DOI] [PubMed] [Google Scholar]
- Borgel J, Guibert S, Li Y, Chiba H, Schubeler D, Sasaki H, Forne T, Weber M. 2010. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet 42: 1093–1100. [DOI] [PubMed] [Google Scholar]
- Chang Y, Sun L, Kokura K, Horton JR, Fukuda M, Espejo A, Izumi V, Koomen JM, Bedford MT, Zhang X, et al. 2011. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat Commun 2: 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, Yang HW, Lam LL, Mager DL, Schubeler D, et al. 2008. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J 27: 2691–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H, et al. 2008. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol 15: 1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. 2006. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 20: 3089–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, Cedar H, Bergman Y. 2006. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol 8: 188–194. [DOI] [PubMed] [Google Scholar]
- Ficz G, Hore TA, Santos F, Lee HJ, Dean W, Arand J, Krueger F, Oxley D, Paul YL, Walter J, et al. 2013. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell 13: 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Hurd PJ, Deplus R, Kouzarides T. 2003. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res 31: 2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HH, Matarese F, Lepikhov K, Gut M, Brun-Heath I, Hubner NC, et al. 2013. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell 13: 360–369. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Ikegami K, Iwatani M, Suzuki M, Tachibana M, Shinkai Y, Tanaka S, Greally JM, Yagi S, Hattori N, Shiota K. 2007. Genome-wide and locus-specific DNA hypomethylation in G9a deficient mouse embryonic stem cells. Genes Cells 12: 1–11. [DOI] [PubMed] [Google Scholar]
- Karagianni P, Amazit L, Qin J, Wong J. 2008. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol Cell Biol 28: 705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Esteve PO, Jacobsen SE, Pradhan S. 2009. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res 37: 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini AG, Mpakali A, Agalioti T. 2011. Dnmt3a1 upregulates transcription of distinct genes and targets chromosomal gene clusters for epigenetic silencing in mouse embryonic stem cells. Mol Cell Biol 31: 1577–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH. 2003. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13: 1192–1200. [DOI] [PubMed] [Google Scholar]
- Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, Tachibana M, Shinkai Y, Lehnertz B, Mager DL, et al. 2011. Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc Natl Acad Sci 108: 5718–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. 2006. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem 281: 19489–19500. [DOI] [PubMed] [Google Scholar]
- Li Q, Su Z, Xu X, Liu G, Song X, Wang R, Sui X, Liu T, Chang X, Huang D. 2012. AS1DHRS4, a head-to-head natural antisense transcript, silences the DHRS4 gene cluster in cis and trans. Proc Natl Acad Sci 109: 14110–14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Garrett-Bakelman FE, Akalin A, Zumbo P, Levine R, To BL, Lewis ID, Brown AL, D'Andrea RJ, Melnick A, et al. 2013. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics 14Suppl 5: S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link PA, Gangisetty O, James SR, Woloszynska-Read A, Tachibana M, Shinkai Y, Karpf AR. 2009. Distinct roles for histone methyltransferases G9a and GLP in cancer germ-line antigen gene regulation in human cancer cells and murine embryonic stem cells. Mol Cancer Res 7: 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J, Koseki H, Wong J. 2013. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun 4: 1563. [DOI] [PubMed] [Google Scholar]
- Liu N, Zhang Z, Wu H, Jiang Y, Meng L, Xiong J, Zhao Z, Zhou X, Li J, Li H, et al. 2015. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev 29: 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. 2010. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464: 927–931. [DOI] [PubMed] [Google Scholar]
- Monnier P, Martinet C, Pontis J, Stancheva I, Ait-Si-Ali S, Dandolo L. 2013. H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc Natl Acad Sci 110: 20693–20698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, Margueron R, Ait-Si-Ali S. 2014. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell 53: 277–289. [DOI] [PubMed] [Google Scholar]
- Myant K, Termanis A, Sundaram AY, Boe T, Li C, Merusi C, Burrage J, de Las Heras JI, Stancheva I. 2011. LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res 21: 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. 2008. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322: 1717–1720. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. 2002. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296: 1132–1136. [DOI] [PubMed] [Google Scholar]
- Ohhata T, Tachibana M, Tada M, Tada T, Sasaki H, Shinkai Y, Sado T. 2004. X-inactivation is stably maintained in mouse embryos deficient for histone methyl transferase G9a. Genesis 40: 151–156. [DOI] [PubMed] [Google Scholar]
- Pannetier M, Julien E, Schotta G, Tardat M, Sardet C, Jenuwein T, Feil R. 2008. PR-SET7 and SUV4-20H regulate H4 lysine-20 methylation at imprinting control regions in the mouse. EMBO Rep 9: 998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudhon C, Duffie R, Ajjan S, Cowley M, Iranzo J, Carbajosa G, Saadeh H, Holland ML, Oakey RJ, Rakyan VK, et al. 2012. Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol Cell 47: 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Rai K, Jafri IF, Chidester S, James SR, Karpf AR, Cairns BR, Jones DA. 2010. Dnmt3 and G9a cooperate for tissue-specific development in zebrafish. J Biol Chem 285: 4110–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, Jeltsch A. 2008. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol 4: 344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR, Klose RJ. 2014. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta 1839: 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI, Barsyte-Lovejoy D, Martinez JY, Bedford MT, Fuchs SM, et al. 2012. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol 19: 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadeh H, Schulz R. 2014. Protection of CpG islands against de novo DNA methylation during oogenesis is associated with the recognition site of E2f1 and E2f2. Epigenetics Chromatin 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H, Tsugane K, Kanno T, Nishimura T. 2012. DNA methylation in plants: relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol 53: 766–784. [DOI] [PubMed] [Google Scholar]
- Sharma S, Gerke DS, Han HF, Jeong S, Stallcup MR, Jones PA, Liang G. 2012. Lysine methyltransferase G9a is not required for DNMT3A/3B anchoring to methylated nucleosomes and maintenance of DNA methylation in somatic cells. Epigenetics Chromatin 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. 2013. DNA methylation: roles in mammalian development. Nat Rev Genet 14: 204–220. [DOI] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495. [DOI] [PubMed] [Google Scholar]
- Strogantsev R, Krueger F, Yamazawa K, Shi H, Gould P, Goldman-Roberts M, McEwen K, Sun B, Pedersen R, Ferguson-Smith AC. 2015. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol 16: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, et al. 2002. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16: 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. 2005. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 19: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. 2008. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 27: 2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toedling J, Skylar O, Krueger T, Fischer JJ, Sperling S, Huber W. 2007. Ringo: an R/Bioconductor package for analyzing ChIP-chip readouts. BMC Bioinformatics 8: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. 2009. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem 284: 8395–8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco G, Hube F, Rollin J, Neuillet D, Philippe C, Bouzinba-Segard H, Galvani A, Viegas-Pequignot E, Francastel C. 2010. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc Natl Acad Sci 107: 9281–9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon HP, Hughes JR, Rode C, De La Rosa-Velazquez IA, Jenuwein T, Feil R, Higgs DR, Gibbons RJ. 2015. ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep 11: 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagschal A, Sutherland HG, Woodfine K, Henckel A, Chebli K, Schulz R, Oakey RJ, Bickmore WA, Feil R. 2008. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol Cell Biol 28: 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W. 2012. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28: 2184–2185. [DOI] [PubMed] [Google Scholar]
- Weiss T, Hergeth S, Zeissler U, Izzo A, Tropberger P, Zee BM, Dundr M, Garcia BA, Daujat S, Schneider R. 2010. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin 3: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Li W. 2009. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Z, Tachibana M, Guggiari M, Heard E, Shinkai Y, Wagstaff J. 2003. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J Biol Chem 278: 14996–15000. [DOI] [PubMed] [Google Scholar]
- Yu Y, Song C, Zhang Q, DiMaggio PA, Garcia BA, York A, Carey MF, Grunstein M. 2012. Histone H3 lysine 56 methylation regulates DNA replication through its interaction with PCNA. Mol Cell 46: 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.