

Abstract
The correct interpretation of microbial sequencing data applied to surveillance and outbreak investigation depends on accessible genomic databases to provide vital genetic context. Our aim was to construct and describe a United Kingdom MRSA database containing over 1000 methicillin-resistant Staphylococcus aureus (MRSA) genomes drawn from England, Northern Ireland, Wales, Scotland, and the Republic of Ireland over a decade. We sequenced 1013 MRSA submitted to the British Society for Antimicrobial Chemotherapy by 46 laboratories between 2001 and 2010. Each isolate was assigned to a regional healthcare referral network in England and was otherwise grouped based on country of origin. Phylogenetic reconstructions were used to contextualize MRSA outbreak investigations and to detect the spread of resistance. The majority of isolates (n = 783, 77%) belonged to CC22, which contains the dominant United Kingdom epidemic clone (EMRSA-15). There was marked geographic structuring of EMRSA-15, consistent with widespread dissemination prior to the sampling decade followed by local diversification. The addition of MRSA genomes from two outbreaks and one pseudo-outbreak demonstrated the certainty with which outbreaks could be confirmed or refuted. We identified local and regional differences in antibiotic resistance profiles, with examples of local expansion, as well as widespread circulation of mobile genetic elements across the bacterial population. We have generated a resource for the future surveillance and outbreak investigation of MRSA in the United Kingdom and Ireland and have shown the value of this during outbreak investigation and tracking of antimicrobial resistance.
Methicillin-resistant Staphylococcus aureus (MRSA) was first isolated in 1961 in the United Kingdom (UK), 1 yr after methicillin was introduced into clinical practice (Jevons 1961). The prevalence of MRSA gradually increased thereafter, and by 1971, 5% of S. aureus isolates referred to the National Staphylococcal Reference Laboratory were MRSA (Marples and Reith 1992). Outbreaks of gentamicin-resistant MRSA in several hospitals during the late 1970s (Shanson 1981) were followed by the emergence of MRSA with potential for epidemic spread (Johnson et al. 2005). By the mid-1980s, MRSA had spread across the UK, and the majority were epidemic (E)MRSA-1, later assigned as sequence type (ST) 239 by multilocus sequence typing (MLST) (Kerr et al. 1990; Johnson et al. 2005). A decline in EMRSA-1 in the late 1980s and early 1990s was associated with an increase in EMRSA-3 (ST 5) (Marples and Reith 1992; Richardson and Reith 1993; Cox et al. 1995; Enright et al. 2002). This dynamic process continued with the emergence in the early 1990s of EMRSA-15 (ST 22) and EMRSA-16 (ST 36) (Richardson and Reith 1993; Cox et al. 1995; Enright et al. 2002), which disseminated across the UK. These two clones continue to predominate, with EMRSA-15 accounting for ∼85% of MRSA bloodstream infections in the UK in 2007 and with trends suggesting that EMRSA-16 is in decline (Ellington et al. 2010; McAdam et al. 2012). Antimicrobial resistance is known to differ between EMRSA-15 and -16, with EMRSA-16 being the more resistant lineage of the two. However, for both lineages, the acquisition of the SCCmec element conferring methicillin resistance and the presence of mutations in gyrA and grlA conferring fluoroquinolone resistance are considered to be major contributors to the success of these epidemic lineages (Knight et al. 2012; McAdam et al. 2012; Holden et al. 2013).
Bacterial genotyping using pulsed-field gel electrophoresis (PFGE), MLST, and spa typing has been used to identify epidemic clones and to give insights into the microevolutionary dynamics of predominant MRSA lineages in the UK. However, these methods have limited resolution and lack discriminatory power when one or a small number of clones predominate (McAdam et al. 2012; Miller et al. 2014; Bartels et al. 2015). This means that once widely established, the subsequent dynamics of clonal MRSA spread within and between healthcare facilities cannot be fully elucidated. As a result, bacterial typing does not form a central component of MRSA transmission and outbreak investigation. Several recent publications have confirmed the ability of whole-genome sequencing (WGS) to define transmission dynamics of a single clone at different geographic and temporal scales. This has identified global and local transmission routes and, when combined with epidemiological data, can confirm or refute putative MRSA outbreaks (Köser et al. 2012; Harris et al. 2013; Nubel et al. 2013; Miller et al. 2014; Török et al. 2014; Bartels et al. 2015). Similarly, while surveillance of MRSA has been carried over several years and a limited number of point prevalence studies of variable methodology have been undertaken in different settings, serial systematic prevalence studies of individual epidemic lineages are lacking (Johnson et al. 2012; Afshinnekoo et al. 2015; Bartels et al. 2015; Peng et al. 2015). WGS could potentially be used for national and local surveillance of MRSA lineages and to enhance the investigation of suspected outbreaks, but comprehensive genomic databases are required to provide the context that would allow robust epidemiological interferences. Here, we describe the analysis of over 1000 MRSA genomes drawn from across the UK and Ireland over a period of a decade and the first evaluation of this rich data set to describe the macroepidemiology of MRSA.
Results
Sampling framework for MRSA genome database
Our first objective was to define the population structure and dynamics of MRSA across England, Northern Ireland, Wales, Scotland (together making up the UK), and the Republic of Ireland over the last decade using WGS. This was achieved by partnering with the British Society for Antimicrobial Chemotherapy (BSAC), which coordinates an antimicrobial resistance surveillance project across this geographical area (for details, see www.bsacsurv.org) (Reynolds et al. 2008). The sampling framework is systematic and unbiased since diagnostic microbiology laboratories submit a defined number of consecutive but nonduplicate S. aureus isolated from blood cultures each year. A total of 47 centers contributed S. aureus bacteremia isolates to the BSAC Bacteremia Resistance Surveillance Program between 2001 and 2010, 24–25 centers each year from 2001–2009, and 38 in 2010 when the network was expanded. Sixteen centers contributed in all 10 yr; 13 contributed only in 2010. The BSAC target was to receive 10 S. aureus isolates per center per year in 2001–2007, 20 in 2008–2009, and 14 in 2010. Actual collections were 97% of target on average, and <5% of 261 center-year collections were <90% complete. MRSA bacteremia isolates were confirmed and sequenced from the 45 laboratories between 2001 and 2010, the geographic locations of which are shown in Figure 1 (for details, see Supplemental Table S1). The number of MRSA isolates sequenced by year is shown in Supplemental Figure S1.
Figure 1.
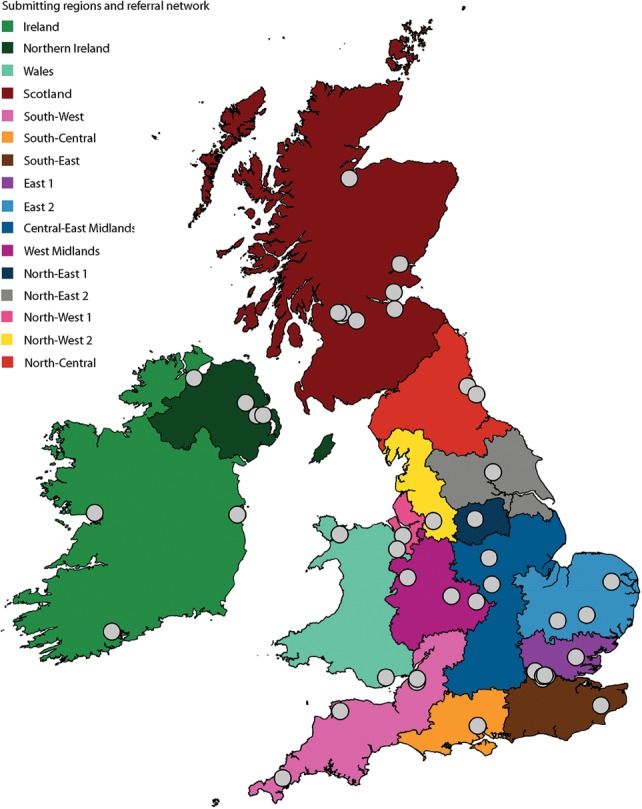
Map of laboratories, submitting regions, and referral networks. Laboratories submitting isolates to the British Society of Antimicrobial Chemotherapy collection are shown as a gray dot. Regions are colored as indicated, and English regions were based on referral networks as previously described (Donker et al. 2014).
Regional healthcare referral networks have been described for England. These consist of hospitals in geographical proximity that frequently exchange patients through referrals and are defined by boundaries beyond which there is a sharp fall in patient movement (Donker et al. 2014). Patient transfer between hospitals is highly relevant to patterns of MRSA transmission, and so we placed our phylogenetic analysis in the context of these referral networks. Each network contained at least one submitting laboratory, with some networks containing multiple laboratories (Fig. 1; Supplemental Table S1). Around 100 MRSA samples were sequenced per year, with an average of six isolates per year per network. Published data are not available on referral networks within Wales, Scotland, Northern Ireland, and the Republic of Ireland, and these were each treated as single entities for the purposes of the analysis (Fig. 1; Supplemental Table S1).
Overview of the population structure of MRSA in the UK
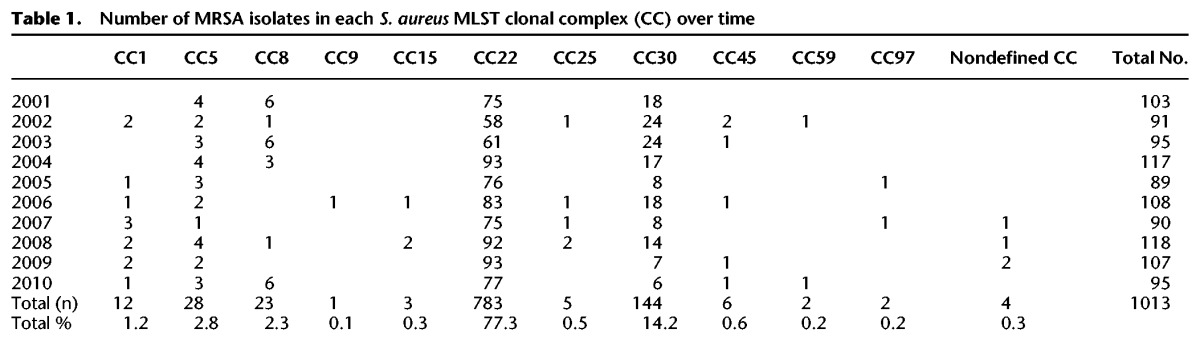
The frequency of each clonal complex (CC) by year is shown in Table 1. CCs are STs that share five or more alleles across the seven loci examined (Cooper and Feil 2004). The majority of isolates (n = 783, 77%) belonged to CC22, which contains the dominant epidemic clone in the UK and Ireland, EMRSA-15. The second most frequent CC was CC30 (n = 144, 14%), containing the epidemic clone EMRSA-16 (ST36). Isolates in CC1, CC5, and CC8 each made up between 1% and 3% of the population, with <1% of isolates assigned to CC9, -15, -25, -45, -59, and -97, as well as previously undefined CCs. Just two isolates were found to belong to the USA300 clone, which is the most dominant MRSA clone in the United States of America (Uhlemann et al. 2014). Of the isolates submitted, only three CCs—CC22, CC30, and CC5—had been retrieved continuously in every year of the study period, whereas all other CCs were found sporadically. These data describe a period of clonal stability with no evidence for newly emerging lineages. Subsequent analyses of the MRSA populations were lineage specific and examined the fine-scale variation within the two major lineages.
Table 1.
Number of MRSA isolates in each S. aureus MLST clonal complex (CC) over time
Lineage-specific phylogenetic analyses
Phylogenetic reconstruction based on 21,848 core SNPs within the 783 CC22 isolates (Supplemental Figs. S2, S3) revealed a tightly related, hospital-adapted lineage containing EMRSA-15 (ST22-A2) and its progenitor lineage (non-ST22-A1) and a genetically more divergent community-associated population (non-ST22-A) as described previously (Holden et al. 2013). Therefore we reconstructed the phylogeny focusing only on EMRSA-15, which gave 20,488 core SNP sites in 775 isolates (Fig. 2; Supplemental Fig. S4). Genome-based studies of global clones of MRSA (e.g., ST239 [Harris et al. 2010], ST22 [Holden et al. 2013]) have demonstrated genetic clustering of isolates according to country/continent of isolation, and therefore, we hypothesized that EMRSA-15 would demonstrate geographical clustering in the UK and Ireland. By color-coding each isolate in the phylogeny according to the referral network, we showed the presence of strong phylogenetic clustering by referral network (Fig. 2). Based on all internal nodes in the phylogeny of isolates from England, quantitative analysis revealed that 44.1% of all internal nodes had descendants from strictly the same referral network, thus clustering within their respective originating network or region. To substantiate this hypothesis, isolates were randomly assigned to referral network locations. The average of these 1000 permutation tests showed on average only 3.0% (IQR: 2.4%–3.6%) of isolates clustered with isolates within their referral network, indicating that the strong geographic signal is not arbitrary.
Figure 2.

Phylogeny of EMRSA-15 and distribution of antibiotic resistance genes. The maximum likelihood tree of ST22-A isolates was based on core SNPs identified by mapping to the reference ST22 genome HO 5095 0412. The total number of SNP sites present in the core genome alignment in this lineage was 20,488 SNPs. Branches are labeled with colors of the referral networks as indicated by the legend. Represented is the year of isolation, as well as the acquisition of fusB or fusC, fusA mutations, acquisition of dfrA, and dfrA mutations, and ermC acquisition as indicated by the legend.
Study isolates were drawn from a period of time that post-dates the dissemination of EMRSA-15 across the UK and Ireland, and the sequence data generated here are consistent with regional diversification following widespread dissemination. In some instances, EMRSA-15 appears to have been introduced into a region on several occasions, as reflected by the existence of several distinct clades (e.g., in referral networks South-West and North-Central) (Fig. 2). We also observed that an introduction event was sometimes followed by this lineage becoming established and coexisting with other distinct clades, for example, in North-Central.
A phylogenetic tree based on 4400 core SNP sites for 140 EMRSA-16 (ST36) isolates revealed a similar structure to the EMRSA-15 (ST22-A) phylogeny. Although EMRSA-16 was more sparsely populated, it was widely disseminated with evidence of geographic structuring (Fig. 3), as reported previously (McAdam et al. 2012). Analysis of less common CCs suggested some clustering; for example, eight of 12 CC1 isolates were from the South-West, and 11 of the 28 CC5 isolates were from Wales.
Figure 3.

Phylogeny of EMRSA-16 and distribution of antibiotic resistance genes. Maximum likelihood tree based on core SNPs identified by mapping to the reference ST36 genome MRSA252. The total number of SNP sites present in the core genome alignment in this lineage was 4400 SNP sites. Branches are labeled with colors of the referral networks as indicated by the legend. Represented is the year of isolation, as well as acquisition of ermA and ermC (erythromycin resistance), ileS-2 (mupirocin resistance), aacA-aphD (aminoglycoside resistance), dfrA and dfrG (trimethoprim resistance), and ant4-1 (aminoglycoside resistance), as indicated by the legend.
Using the genomic framework to contextualize outbreaks
Next, we tested the utility of this genetic database for the investigation of suspected MRSA outbreaks. We have described previously the use of WGS to both confirm and refute local outbreaks at the Cambridge University Hospitals NHS Foundation Trust (Köser et al. 2012; Harris et al. 2013; Török et al. 2014), and we placed these within the context provided by the national BSAC collection. The first MRSA outbreak involved seven infants in a single neonatal intensive care unit (NICU) caused by EMRSA-15, in which one isolate in the cluster was genetically divergent compared with the other isolates as a result of being a hypermutator (Köser et al. 2012). The second MRSA outbreak initially involved a special care baby unit (SCBU), but MRSA was shown through genetic analysis to have spread to mothers on a post-natal ward, to 10 patients in the community who developed skin and soft tissue infections, and to a colonized healthcare worker in the SCBU (Harris et al. 2013). The outbreak MRSA was a novel ST (ST2371), which is highly related to ST22 (a single locus variant). A third investigation of four patients with five episodes of MRSA who had overlapping periods of admission on a hepatology ward showed that they had been infected by their carriage strain and thus refuted an outbreak on the ward (Török et al. 2014). Placing genomes from these three investigations into the phylogenetic tree of the 783 CC22 isolates demonstrated striking demarcation for the two outbreak clusters but scattering throughout the tree for MRSA from the hepatology pseudo-outbreak. This analysis also demonstrated that the two outbreaks in units contained within the same hospital building were caused by phylogenetically distant lineages (Fig. 4).
Figure 4.

Contextualization of outbreak investigations of CC22 MRSA studied at Cambridge University Hospitals. The maximum likelihood tree was based on 22,238 core SNPs for the 783 ST22 genomes, together with seven isolates from an MRSA outbreak on a neonatal intensive care unit (NICU, green) (Köser et al. 2012), 15 isolates from an MRSA outbreak that focused on a special care baby unit (SCBU, orange) but extended to other wards and the community (Harris et al. 2013), and 42 isolates sequenced as part of an MRSA outbreak investigation on a hepatology ward (nine isolates from four patients with bacteremia [P1-4; pink filled dots] and the remainder from patients who were MRSA carriers on the same ward during a comparable timeframe [pink open dots]) (Török et al. 2014).
Determining distribution and spread of resistance at different regional levels
The database could also be used for the surveillance of antimicrobial resistance in MRSA. To test this, we mined the BSAC data set to illustrate the potential to highlight (1) local expansion of resistance, (2) regional expansion of a specific resistance determinant, and (3) resistance determinants that are more widely disseminated. Localized emergence of antimicrobial resistance was observed for isolates in a closely related lineage (pairwise SNP difference of 28–114) belonging to CC5 from a healthcare institution in Wales, which were clearly separate from the rest of the CC5 isolates (Fig. 5). These 11 isolates all contain the Tn554 transposon encoding ermA for erythromycin and clindamycin resistance, as well as type II staphylococcal cassette chromosome mec element encoding methicillin resistance (SCCmec II). Further antibiotic resistance mutations were apparent in this cluster with the presence of both grlA and gyrA mutations for fluoroquinolone resistance. Additionally, 10 out of 11 isolates also contained a mutation in dfrA conferring trimethoprim resistance. This extended spectrum of antibiotic resistance may explain their apparent regional success in a single healthcare institution.
Figure 5.

Phylogeny of CC5. The maximum likelihood tree was based on core SNPs identified by mapping to the reference ST5 genome N315. The total number of SNP sites present in the core genome alignment in this lineage was 3229 SNP sites. Welsh isolates have light green branches, and the branch leading to reference N315 is colored red.
We noted that EMRSA-15 isolates from the Republic of Ireland had a higher prevalence of fusidic acid resistance compared with isolates from the UK (37.5% vs. 10.4%, respectively). A high prevalence of fusidic acid resistance has been noted previously for Ireland and Greece (Castanheira et al. 2010). Fusidic acid resistance can be conferred through gene acquisition (fusB or fusC) or by chromosomal mutations (fusA). Genomic analysis readily demonstrated that mutation dominated over gene acquisition as the basis for resistance (53 vs. four events, respectively) (Fig. 2). Resistance in Irish isolates arose through a single L461K mutation in fusA prior to expansion of this clade and can be found in several contributing centers. Two other Irish clades, found in the same hospitals over the same time period, only showed sporadic acquisition of resistance as singular, independent events (Fig. 2).
Examples of more widely distributed resistance profiles included trimethoprim resistance in EMRSA-15 and gentamicin/mupirocin resistance in EMRSA-16. Acquisition of gentamicin (aacA-aphD), mupirocin (ileS-2), erythromycin (ermC), and aminoglycoside (ant4-1) resistance genes were found to coincide with each other as well as with plasmid-related genes in EMRSA-16 (Fig. 3). The presence of these clustered genes in several referral networks (West Midlands, South-East) suggests a number of circulating plasmids in the EMRSA-16 population. Similarly, in EMRSA-15, acquisition of dfrA was identified to be the genetic basis for trimethoprim resistance in isolates from Northern Ireland (10 isolates), Scotland (seven isolates), and West Midlands/North-Central (four and five isolates, respectively) (Fig. 2). This is consistent with the carriage of this gene on a mobile genetic element, but since dfrA was found separately on its own small contig, we cannot deduce whether it was carried on a plasmid or transposon. Trimethoprim resistance was also encoded by chromosomal mutations in dfrA, with L41F and F99Y being prominent mutations basal to a number of referral network clusters (Fig. 2).
We also observed that the extent of antibiotic resistance within a lineage is governed by the mobile element on which it is carried, an example of which is erythromycin resistance. Contrary to other resistance determinants that appeared to be locally restricted, erythromycin resistance was very unstable within the EMRSA-15 population. This is likely due to the fact that the resistance determinant ermC is carried on a small 2-kb plasmid, which is readily lost and gained. In contrast, erythromycin resistance is ubiquitous in the EMRSA-16 lineage as ermA is carried on transposon Tn554, which stably integrates into the chromosome as part of the SCCmec II element.
Discussion
This evaluation of over 1000 MRSA genomes representing isolates from the UK and Republic of Ireland describes the genetic structure of dominant lineages (EMRSA-15 and EMRSA-16) over the last decade. Parallels were observed between the dominant epidemic clone EMRSA-15 and the declining epidemic clone EMRSA-16, including geographic structuring and clonality. Numerous other MRSA lineages were identified at low prevalence, which mostly lacked specific phylogenetic structuring and showed no evidence of expansion over time. There was strong evidence for region-specific subclones of EMRSA-15, which at a finer level echoes a previous finding of regional and country-specific clustering for this lineage (Holden et al. 2013). Regional clustering was also observed for EMRSA-16, as was previously described (McAdam et al. 2012; Miller et al. 2014). Owing to the lack of network data for Scotland, Ireland, Wales, and Northern Ireland, it was not possible to undertake any further regional stratification within these countries. However, for the English referral network, isolates from a specific referral network were more likely to cluster with isolates from the same region than with isolates from a different network. This could be influenced by the submitting hospitals, since not all hospitals contributed MRSA isolates in every year. The current sampling strategy did, however, achieve good temporal and regional coverage. We also observed the presence of multiple subclones within individual referral networks. This is consistent with a model in which the initial UK-wide dissemination of EMRSA-15 included several introductions into each location with subsequent local diversification.
Local and regional structuring was also observed for antibiotic resistance determinants, with both epidemic clones showing regional clades with specific antibiotic resistance patterns. Country- or region-specific antibiotic treatment regimens impact the evolution of drug resistance. This has been exemplified previously for clindamycin in EMRSA-15, where Germany shows higher prescription levels than the UK and where resistance levels are significantly higher (Holden et al. 2013). Differences in antimicrobial resistance have also been noted for the USA-300 clone that is successful in the United States, with the coexistence of fluoroquinolone-resistant and -susceptible lineages (Alam et al. 2015). Here, we provide evidence for higher levels of resistance to fusidic acid in Ireland compared with the UK. Antimicrobial prescription policies or usage may differ between the UK and the Republic of Ireland, exerting a potential selective pressure leading to higher prevalence of fusidic acid resistance in Irish isolates. Locally successful clones may also differ in antibiotic resistance patterns, such as the expansion of CC5 isolates in a single healthcare institution, or distribution of trimethoprim resistance within referral networks. Wider dissemination of antibiotic resistance determinants can also be facilitated through carriage of multiple plasmids, as shown by linked gentamicin/mupirocin resistance putatively carried on plasmids in the EMRSA-16 population. The fluidity of antimicrobial resistance markers such as ermC in EMRSA-15 affects clinical practice, as the pattern of antimicrobial resistance is often used during infection control investigations as a proxy to determine whether isolates are related and therefore might be part of an outbreak. This was demonstrated during an outbreak on SCBU, where the ermC plasmid was lost in some patients (Harris et al. 2013). By looking at a wider range of isolates within geographically defined regions, it becomes possible to distinguish between widespread resistance markers and locally emerging strains, as well as the stability of mobile genetic elements within a population.
This sequence data set has considerable long-term value, since it provides a genomic framework for the surveillance of MRSA lineages and tracking of MRSA transmission between referral networks in the UK, as well as globally circulating lineages like EMRSA-15. The likely introduction of bacterial genome sequencing to confirm or refute MRSA outbreaks will need to use such data as a source of reference genomes and region-specific comparators, and the availability of this data set in public databases will provide the ability to visualize such comparisons. Equally, it can also be used to discern differences within regional clones relating to antibiotic resistance and will provide valuable information on resistance determinants as well as their horizontal propagation and targeted patient care.
Methods
Isolates, sequencing, and analysis
A total of 1013 MRSA isolates were provided by the BSAC bacteremia resistance surveillance program (Reynolds et al. 2008) from 2001–2010 (Supplemental Table S2; www.bsacsurv.org). DNA extraction was carried out on a QIAxtractor (Qiagen), and library preparation was performed as previously described (Köser et al. 2012). Index-tagged libraries were created, and 96 separated libraries were sequenced in each of eight channels using the Illumina HiSeq platform (Illumina) to generate 100-bp paired-end reads at the Wellcome Trust Sanger Institute. Average sequence depth was 82.32×, and average sequence quality was 39. The ST and CC of each isolate were determined from the sequence (Enright et al. 2000; Cooper and Feil 2004). Sequence reads were then mapped to relevant reference genomes based on the MLST assignment to identify single-nucleotide polymorphisms (SNPs) (Harris et al. 2010). The references genomes used were HO 5096 0412 (Köser et al. 2012) for CC 22 isolates, MRSA252 (Holden et al. 2004) for CC30 isolates, and N315 (Kuroda et al. 2001) for CC5 isolates. SNPs were identified in the derived whole-genome alignments as previously described (Holden et al. 2013), and mobile genetic elements, phage, and repetitive regions were excluded, leaving a core genome size of 2,655,809 bp. Phylogenetic estimation was done using RAxML with the general time reversal model and gamma correction (Stamatakis et al. 2005). Trees were visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/) and iTOL (Letunic and Bork 2011). De novo assembled multicontig draft assemblies were generated running Velvet optimizer and Velvet (Zerbino and Birney 2008). Contigs with <300 bases were removed, and scaffolding software SSPACE (Boetzer et al. 2011) was employed. The assembly was further improved using GapFiller (Boetzer and Pirovano 2012). Antimicrobial susceptibility testing was determined by the BSAC agar dilution method (Reynolds et al. 2008). Clustering of regions was assessed on a phylogeny of English EMRSA-15 isolates by counting the proportion of internal nodes whose descendants were isolated from a single region. In order to compare this proportion with the proportions expected when isolates would be distributed randomly, we randomized the leaves 1000 times and calculated the proportions accordingly.
Data access
All sequences from this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under the study number ERP001012 and individual accession numbers are given in Supplemental Table S2.
Competing interest statement
All authors declare that they have no conflicts of interest. M.E.T. and J.P. have received support for travel and conference expenses from Illumina. M.T.G.H. is a consultant for Pfizer.
Supplementary Material
Acknowledgments
We thank the library construction, sequencing, and core informatics teams at the Wellcome Trust Sanger Institute for assistance. We thank David Harris and Martin Aslett for their help in submitting the sequenced isolates to public databases. The study was supported by grants from the UKCRC Translational Infection Research Initiative and the Medical Research Council (grant no. G1000803) with contributions to the grant from the Biotechnology and Biological Sciences Research Council, the National Institute for Health Research on behalf of the Department of Health, and the Chief Scientist Office of the Scottish Government Health Directorate (to S.J.P.); by Wellcome Trust grant number 098051 awarded to the Wellcome Trust Sanger Institute; and by a Healthcare Infection Society Major Reasearch Grant. M.E.T. is a Clinician Scientist Fellow, supported by the Academy of Medical Sciences and the Health Foundation and the NIHR Cambridge Biomedical Research Centre. B.G.S. was supported by Wellcome Trust grant number 089472. The study was approved by the University of Cambridge Human Biology Research Ethics Committee (reference HBREC.2013.05) and by the Cambridge University Hospitals NHS Foundation Trust Research and Development Department (reference A092869). Isolates were supplied by the BSAC Resistance Surveillance Project.
Author contributions: S.J.P., J.P., M.E.T., S.D.B., D.M.A., H.G., E.J.F., and B.G.S. were involved in the design of the study. R.R. provided bacterial isolates and related data from the BSAC Resistance Surveillance Project. K.E.R. and B.B. managed transfer of sample collections and extracted genomic DNA. T.D. and H.G. provided the healthcare network framework and clustering analysis. S.R. and M.T.G.H. were responsible for bioinformatic analyses of whole-genome sequence data. J.P., M.E.T., and S.J.P. were responsible for supervision and management of the study. All authors were involved in writing the paper and have approved the final version.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.196709.115.
Freely available online through the Genome Research Open Access option.
References
- Afshinnekoo E, Meydan C, Chowdhury S, Jaroudi D, Boyer C, Bernstein N, Maritz JM, Reeves D, Gandara J, Chhangawala S, et al. 2015. Geospatial resolution of human and bacterial diversity with city-scale metagenomics. Cell Syst 1: 72–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam MT, Read TD, Petit RA, Boyle-Vavra S, Miller LG, Eells SJ, Daum RS, David MZ. 2015. Transmission and microevolution of USA300 MRSA in U.S. households: evidence from whole-genome sequencing. MBio 6: e00054-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels MD, Larner-Svensson H, Meiniche H, Kristoffersen K, Schonning K, Nielsen JB, Rohde SM, Christensen LB, Skibsted AW, Jarlov JO, et al. 2015. Monitoring meticillin resistant Staphylococcus aureus and its spread in Copenhagen, Denmark, 2013, through routine whole genome sequencing. Eurosurveillance 20: 20–28. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol 13: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578–579. [DOI] [PubMed] [Google Scholar]
- Castanheira M, Watters AA, Mendes RE, Farrell DJ, Jones RN. 2010. Occurrence and molecular characterization of fusidic acid resistance mechanisms among Staphylococcus spp. from European countries (2008). J Antimicrob Chemother 65: 1353–1358. [DOI] [PubMed] [Google Scholar]
- Cooper JE, Feil EJ. 2004. Multilocus sequence typing: What is resolved? Trends Microbiol 12: 373–377. [DOI] [PubMed] [Google Scholar]
- Cox RA, Conquest C, Mallaghan C, Marples RR. 1995. A major outbreak of methicillin-resistant Staphylococcus aureus caused by a new phage-type (EMRSA-16). J Hosp Infect 29: 87–106. [DOI] [PubMed] [Google Scholar]
- Donker T, Wallinga J, Grundmann H. 2014. Dispersal of antibiotic-resistant high-risk clones by hospital networks: Changing the patient direction can make all the difference. J Hosp Infect 86: 34–41. [DOI] [PubMed] [Google Scholar]
- Ellington MJ, Hope R, Livermore DM, Kearns AM, Henderson K, Cookson BD, Pearson A, Johnson AP. 2010. Decline of EMRSA-16 amongst methicillin-resistant Staphylococcus aureus causing bacteraemias in the UK between 2001 and 2007. J Antimicrob Chemother 65: 446–448. [DOI] [PubMed] [Google Scholar]
- Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38: 1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. 2002. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci 99: 7687–7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, et al. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SR, Cartwright EJ, Török ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, et al. 2013. Whole-genome sequencing for analysis of an outbreak of methicillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 13: 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, et al. 2004. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci 101: 9786–9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Hsu LY, Kurt K, Weinert LA, Mather AE, Harris SR, Strommenger B, Layer F, Witte W, de Lencastre H, et al. 2013. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res 23: 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevons MP. 1961. Celbenin-resistant Staphylococci. Br Med J 1: 124–125. [Google Scholar]
- Johnson AP, Pearson A, Duckworth G. 2005. Surveillance and epidemiology of MRSA bacteraemia in the UK. J Antimicrob Chemother 56: 455–462. [DOI] [PubMed] [Google Scholar]
- Johnson AP, Davies J, Guy R, Abernethy J, Sheridan E, Pearson A, Duckworth G. 2012. Mandatory surveillance of methicillin-resistant Staphylococcus aureus (MRSA) bacteraemia in England: the first 10 years. J Antimicrob Chemother 67: 802–809. [DOI] [PubMed] [Google Scholar]
- Kerr S, Kerr GE, Mackintosh CA, Marples RR. 1990. A survey of methicillin-resistant Staphylococcus aureus affecting patients in England and Wales. J Hosp Infect 16: 35–48. [DOI] [PubMed] [Google Scholar]
- Knight GM, Budd EL, Whitney L, Thornley A, Al-Ghusein H, Planche T, Lindsay JA. 2012. Shift in dominant hospital-associated methicillin-resistant Staphylococcus aureus (HA-MRSA) clones over time. J Antimicrob Chemother 67: 2514–2522. [DOI] [PubMed] [Google Scholar]
- Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Hsu LY, Chewapreecha C, Croucher NJ, Harris SR, et al. 2012. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med 366: 2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, et al. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357: 1225–1240. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2011. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39(Web Server issue): W475–W478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marples RR, Reith S. 1992. Methicillin-resistant Staphylococcus aureus in England and Wales. Commun Dis Rep CDR Rev 2: R25–R29. [PubMed] [Google Scholar]
- McAdam PR, Templeton KE, Edwards GF, Holden MT, Feil EJ, Aanensen DM, Bargawi HJ, Spratt BG, Bentley SD, Parkhill J, et al. 2012. Molecular tracing of the emergence, adaptation, and transmission of hospital-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci 109: 9107–9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RR, Price JR, Batty EM, Didelot X, Wyllie D, Golubchik T, Crook DW, Paul J, Peto TEA, Wilson DJ, et al. 2014. Healthcare-associated outbreak of meticillin-resistant Staphylococcus aureus bacteraemia: role of a cryptic variant of an epidemic clone. J Hosp Infect 86: 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nubel U, Nachtnebel M, Falkenhorst G, Benzler J, Hecht J, Kube M, Brocker F, Moelling K, Buhrer C, Gastmeier P, et al. 2013. MRSA transmission on a neonatal intensive care unit: epidemiological and genome-based phylogenetic analyses. PLoS One 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Ou Q, Lin D, Xu P, Li Y, Ye X, Zhou J, Yao Z. 2015. Metro system in Guangzhou as a hazardous reservoir of methicillin-resistant Staphylococci: findings from a point-prevalence molecular epidemiologic study. Sci Rep 5: 16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds R, Hope R, Williams L; BSAC Working Parties on Resistance Surveillance. 2008. Survey, laboratory and statistical methods for the BSAC Resistance Surveillance Programmes. J Antimicrob Chemother 62Suppl 2: ii15–ii28. [DOI] [PubMed] [Google Scholar]
- Richardson JF, Reith S. 1993. Characterization of a strain of methicillin-resistant Staphylococcus aureus (EMRSA-15) by conventional and molecular methods. J Hosp Infect 25: 45–52. [DOI] [PubMed] [Google Scholar]
- Shanson DC. 1981. Antibiotic-resistant Staphylococcus aureus. J Hosp Infect 2: 11–36. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. 2005. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21: 456–463. [DOI] [PubMed] [Google Scholar]
- Török ME, Harris SR, Cartwright EJ, Raven KE, Brown NM, Allison ME, Greaves D, Quail MA, Limmathurotsakul D, Holden MT, et al. 2014. Zero tolerance for healthcare-associated MRSA bacteraemia: Is it realistic? J Antimicrob Chemother 69: 2238–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlemann AC, Dordel J, Knox JR, Raven KE, Parkhill J, Holden MT, Peacock SJ, Lowy FD. 2014. Molecular tracing of the emergence, diversification, and transmission of S. aureus sequence type 8 in a New York community. Proc Natl Acad Sci 111: 6738–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.