

Abstract
Background and Purpose
Meningeal blood flow is controlled by CGRP released from trigeminal afferents and NO mainly produced in arterial endothelium. The vasodilator effect of NO may be due to the NO–derived compound, nitroxyl (HNO), generated through reaction with endogenous H2S. We investigated the involvement of HNO in CGRP release and meningeal blood flow.
Experimental Approach
Blood flow in exposed dura mater of rats was recorded by laser Doppler flowmetry. CGRP release from the dura mater in the hemisected rat head was quantified using an elisa. NO and H2S were localized histochemically with specific sensors.
Key Results
Topical administration of the NO donor diethylamine‐NONOate increased meningeal blood flow by 30%. Pretreatment with oxamic acid, an inhibitor of H2S synthesis, reduced this effect. Administration of Na2S increased blood flow by 20%, an effect abolished by the CGRP receptor antagonist CGRP 8‐37 or the TRPA1 channel antagonist HC030031 and reduced when endogenous NO synthesis was blocked. Na2S dose‐dependently increased CGRP release two‐ to threefold. Co‐administration of diethylamine‐NONOate facilitated CGRP release, while inhibition of endogenous NO or H2S synthesis lowered basal CGRP release. NO and H2S were mainly localized in arterial vessels, HNO additionally in nerve fibre bundles. HNO staining was lost after treatment with L‐NMMA and oxamic acid.
Conclusions and Implications
NO and H2S cooperatively increased meningeal blood flow by forming HNO, which activated TRPA1 cation channels in trigeminal fibres, inducing CGRP release. This HNO‐TRPA1‐CGRP signalling pathway may be relevant to the pathophysiology of headaches.
Abbreviations
- CBS
cystathionine β‐synthase
- CSE
cystathionine γ‐lyase
- Cy3
cyanine dye 3
- DAF
4‐amino‐5‐methylamino‐2′,7′‐difluoresceine diacetate
- HNO
nitroxyl
- iNOS
inducible NOS
- L‐NMMA
L‐NG‐monomethylarginine acetate
- MMA
middle meningeal artery
- MST
mercaptopyruvate sulfurtransferase
- nNOS
neuronal NOS
- NONOate
diethylamine‐NONOate, DEANONOate
- ODQ
1H‐[1,2,4]oxadiazole[4,3‐a]quinoxalin‐1‐one
- sGC
soluble GC
- SIF
synthetic interstitial fluid
- TRPA1
transient receptor potential ankyrin 1 channel
Tables of Links
| TARGETS | |
|---|---|
| Ion channels a | Enzymes c |
| TRPA1 channel | CBS, cystathionine β‐synthase |
| GPCRs b | CSE, cystathionine γ‐lyase |
| CGRP receptor | MST, mercaptopyruvate sulfur transferase |
| nNOS | |
| eNOS |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a, 2013b, 2013c).
Introduction
Neurovascular regulation in the meninges involving CGRP and NO
Meningeal blood flow is regulated by neuronal (Owman, 1990) and epithelium‐dependent metabolic mechanisms (Faraci and Heistad, 1991) acting primarily on arterial blood vessels. Meningeal arteries are surrounded by a dense network of autonomic and trigeminal sensory nerve fibres, a considerable proportion of which contains vasoactive peptides (Edvinsson and Uddman, 1981; Keller and Marfurt, 1991; Messlinger et al., 1993). Upon their activation, trigeminal afferents secrete vasodilatory neuropeptides like CGRP, the most potent intracranial vasodilator (Edvinsson et al., 1987). The vasodilatory function of CGRP release in the dura mater has been shown by many experiments in vitro (Gupta et al., 2006) and in vivo employing video microscopy (Williamson et al., 1997) or laser Doppler flowmetry (Kurosawa et al., 1995). Arterial vasodilatation and blood flow increase caused by CGRP release have therefore widely been used to assess the activation of the trigeminal system in animal experiments (Williamson et al., 1997; Dux et al., 2003; Messlinger et al., 2011). CGRP binds to GPCRs of arterial smooth muscle cells inducing muscular relaxation through intracellular cAMP and cGMP increase (Gray and Marshall, 1992; Brain and Grant, 2004).
The putatively most important metabolic factor mediating vasodilatation in the dura mater is NO, which in arterial smooth muscle cells binds to soluble GC (sGC) increasing intracellular cGMP levels (Brian et al., 1996). Under normal conditions, the main source of NO is most likely the arterial endothelium, which can be demonstrated by NOS immunohistochemistry or NADPH‐diaphorase activity (Hope et al., 1991). The continuous production of endothelial NO seems to be important for the basal relaxation of arterial vessels maintaining the basal blood flow in the meninges (Messlinger et al., 2000). The inducible NOS (iNOS) is expressed, for example, during inflammation by macrophages invading the dura mater (Reuter et al., 2001). Furthermore, some nerve fibres accompanying intracranial arteries, both of trigeminal and parasympathetic origin, have been identified by immunohistochemistry as positive for the neuronal NOS (nNOS) (Nozaki et al., 1993).
Interaction of CGRP and NO in meningeal blood flow
In earlier experiments, we and other groups recognized that the vasodilatory effects of NO clearly depended partly on the action of CGRP and vice versa (Messlinger et al., 2000; Akerman et al., 2002). Electrical or chemical stimulation of meningeal afferents increasing meningeal blood flow through CGRP release was less effective when the production of NO was inhibited, and the blood flow increasing effect of NO donors was decreased when CGRP receptors were inhibited (Strecker et al., 2002a). This indicated a synergistic or permissive effect of NO, the mechanism of which was not clear. We concluded that CGRP receptor activation is necessary to fully enable the vasodilatory function of NO but we could not adequately explain the mechanisms behind this synergistic effect.
However, NO can also be converted to nitroxyl (HNO), a protonated, one electron‐reduced derivative of NO (Fukuto et al., 2005; Fukuto and Carrington, 2011), which is also a vasodilator (Ellis et al., 2000). In addition to its vasodilator effects, HNO exerts positive inotropic and lusitropic effects on heart, initially assigned to CGRP release (Paolocci et al., 2001; 2003). Recently, we have demonstrated that HNO activated the transient receptor potential ankyrin 1 (TRPA1) cation channel which, on opening, allows calcium influx and consequent exocytosis of CGRP from primary afferent terminals (Eberhardt et al., 2014). Using Angeli's salt as an HNO donor, we observed Angeli's salt‐induced CGRP release from the cranial dura mater and increased meningeal blood flow.
The pathway(s) for endogenous generation of HNO has long been a matter of debate (Fukuto et al., 2005; Flores‐Santana et al., 2011; Fukuto and Carrington, 2011) but recent studies of the crosstalk between NO and H2S suggested their potential role in endogenous HNO generation (Yong et al., 2010; 2011; Filipovic et al., 2012; 2013; Miljkovic et al., 2013). H2S has emerged as a new endogenous gasotransmitter that can regulate vascular tone (Yang et al., 2008). H2S is produced by the action of at least three enzymes: cystathionine β‐synthase (CBS), cystathionine γ‐lyase (CSE) and mercaptopyruvate sulfurtransferase (MST) (Kabil and Banerjee, 2014; Kabil et al., 2014). Differently expressed in different tissues (and in different intracellular compartments), these enzymes control H2S production with different efficiency. Most recently, we unambiguously showed that NO and H2S do react directly and that this reaction represents an endogenous source of HNO (Eberhardt et al., 2014), which then activates a HNO‐TRPA1‐CGRP signalling cascade. This is in agreement with previous observations of H2S‐induced TRPA1‐dependent effects on skin blood flow, mesenteric artery dilatation and CGRP release from the trachea (Pozsgai et al., 2010; 2012).
The experiments described here were designed to explain our previous observations of NO‐dependent CGRP release (Messlinger et al., 2000; Akerman et al., 2002; Strecker et al., 2002a) to extend our recent findings about the HNO‐TRPA1‐ CGRP signal cascade, activated by the reaction of NO with H2S (Eberhardt et al., 2014) and to dissect this cascade in the trigeminovascular system of the cranial meninges. To measure changes in meningeal blood flow, we used laser Doppler flowmetry to determine NO‐ and HNO‐induced arterial dilatation. We employed a selective elisa method to directly measure CGRP release from the rat dura mater in vitro. In addition, we utilized different histochemical methods using immunofluorescence and new fluorescence sensors to demonstrate the presence of NO, HNO and H2S and sources of NO involved in the physiological HNO production in the dura mater. We describe, step‐by‐step, the whole cascade that could underlie the NO‐induced activation of trigeminal afferents and the consequent increase in meningeal blood flow resulting from neurogenic vasodilatation of arterial blood vessels. Beyond these vasodilatory processes induced by this HNO‐TRPA1‐CGRP signalling pathway, we discuss its possible role in meningeal nociception and the generation of headaches.
Methods
Animals
All animal care and experimental procedures complied with the guidelines for the welfare of experimental animals of the Federal Republic of Germany and the European Commission (Directive 2010/63/EU), and those of the International Association for the Study of Pain. They were reviewed and approved by the animal protection authorities of the District Government of Mittelfranken (54‐2532.1‐21/12). Animal studies are described in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 96 animals were used in the experiments described here.
For in vivo and in vitro experiments, adult male Wistar rats weighing 300–350 g were used. The animals were bred and housed in the animal facility of the Institute of Physiology under a 12‐12 h light‐dark circle with food and water ad libitum.
Blood flow recordings in vivo
Rats were anaesthetized initially by inhalation of 4% isoflurane (Forene; Abbott, Wiesbaden, Germany) in a closed box. Then they were tracheotomized and artificially ventilated throughout the experiment with 2% isoflurane in oxygen‐enriched room air. End‐expiratory CO2 was monitored and held at 3–3.5%. Systemic blood pressure was recorded with a pressure transducer via a cannula inserted into the femoral artery. A venous catheter introduced into the femoral vein served for the systemic administration of substances. The body temperature of the animals was recorded with a thermoprobe inserted into the rectum, and was kept at 37–37.5°C with a feedback‐controlled heating pad. A cranial window for the measurement of dural blood flow was prepared according to previous studies (Kurosawa et al., 1995; Dux et al., 2002; see Figure 1B). The head of the animal was fixed in a stereotaxic frame, the scalp was removed, and the parietal bone was exposed. The skull was trepanized using a saline‐cooled drill. The experiments were terminated using an overdose of i.v. thiopentone (Trapanal®; Altana, Konstanz, Germany).
Figure 1.
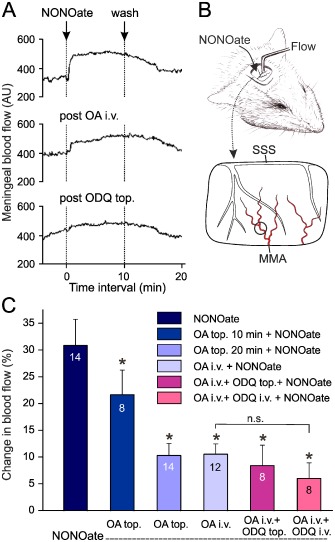
Increases in meningeal blood flow within 10 min periods following topical application of DEANONOate (NONOate, 200 μM) onto the dura mater. (A) Segments of an original recording showing increases in blood flow induced by NONOate before and after i.v. administration of oxamic acid (post OA i.v.) and, in addition, of ODQ topically applied onto the dura mater (post ODQ top.). (B) Experimental set‐up showing the position of the recording probe on the MMA visible in the cranial window (same experiment as in A). SSS, superior sagittal sinus with cortical veins visible through the dura. (C) Increases in meningeal blood flow relative to the baseline prior to substance application; numbers of experiments are indicated in the bars. Effect of NONOate following topical (top., 2 mM) and systemic (i.v., 10 mg·kg−1) pre‐administration of oxamic acid (OA) and the sGC inhibitor ODQ (50 μM or 1 mg·kg−1 respectively). *P < 0.05, significantly different from NONOate‐evoked flow increase without pretreatment; n.s., not significant.
The cranial recording window was filled with 50 μL of synthetic interstitial fluid (SIF) containing (in mM): 108 NaCl, 3.48 KCl, 3.5 MgSO4, 26 NaHCO3, 11.7 NaH2PO4, 1.5 CaCl2, 9.6 Na+ gluconate, 5.55 glucose and 7.6 sucrose; pH 7.4. Meningeal blood flow was measured with needle‐type probes of a laser Doppler flowmeter (Moore DRT4; Moore Instruments, Axminster, UK). Probes were positioned over branches of the middle meningeal artery (MMA) lying distant from visible cortical blood vessels (see Figures 1B and 2A right hand). Perfusion of the dura mater (measured in perfusion units) and systemic blood pressure of the animals were stored and processed with the DRT4 program. Substances were applied either topically with a micropipette onto the exposed dura mater or i.v. through the femoral catheter. Changes in meningeal blood flow after topical or i.v. application of agonists [Na2S and diethylamine‐NONOate (DEANONOate)] were determined by comparing the mean flow values within the 10 min application period with the baseline measured as the mean flow during the 5 min period prior to drug application and expressed as % change. The effects of antagonists (oxamic acid, HC030031, CGRP8‐37 and L‐NMMA, see below) were determined by comparing the blood flow response to the agonists before and after the application of antagonist in each experiment. Effective doses and application periods of drugs were chosen according to previous or pilot experiments.
Figure 2.
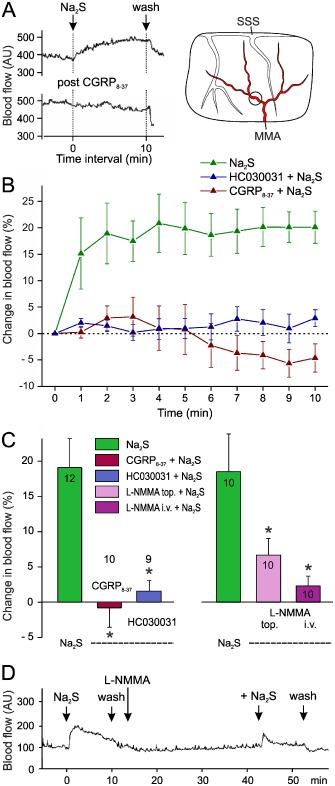
Changes in meningeal blood flow within 10 min periods following topical application of Na2S (300 μM) onto the dura mater. (A) Segments of an original recording showing an increase in blood flow induced by Na2S, which is abolished after topical administration of CGRP 8‐37. The position of the flow probe is indicated in the sketch of the cranial window. (B) Time course of changes in blood flow following application of Na2S with and without preapplication of CGRP 8‐37 (100 μM) or the TRPA1 antagonist HC030031 (50 μM). The flow after Na2S is significantly different from the baseline from the second minute (repeated measures anova and LSD post hoc test, P < 0.01) and different from the Na2S‐evoked flow after pre‐administration of HC030031 or CGRP 8‐37 (two‐way anova, P < 0.001). (C) Changes in blood flow relative to the baseline induced by Na2S prior to and after pre‐administration of CGRP 8‐37 or HC030031 (left panel, same measurements as in B) and after topical (1 mM) and i.v. (12 mg·kg−1) pre‐administration of L‐NMMA (right panel). Mean values from the numbers of experiments indicated within the bars. *P < 0.05, significantly different from effects of Na2S alone. (D) Original recording of an experiment showing the effect of Na2S prior to and after pre‐administration of L‐NMMA.
Measurement of CGRP release in vitro
Measurement of CGRP released from dural afferents was performed by the method described by Ebersberger et al. (1999). Rats were killed by CO2 inhalation and decapitated. After removal of the skin and muscles, the skull was divided into halves along the midline and the cerebral hemispheres were removed (see Figure 3). Skull halves with intact dura mater were superfused at room temperature for 30 min with SIF. Skull halves were placed in a humid chamber to maintain their temperature at 37°C. The cranial fossae were filled with 500 μL SIF. Consecutive samples of the superfusate were collected at periods of 5 min by carefully removing the content of the skull halves with a micropipette. All samples were transferred to plastic cups (Eppendorf, Hamburg, Germany), 100 μL of each sample was taken, diluted with 20 μL EIA buffer (SPIbio, Paris, France), and immediately processed for analysis of the CGRP content using a specific enzyme‐linked immunoassay (CGRP‐EIA; SPIbio). The absorbance of the reaction product representing the CGRP concentration of the sample was determined photometrically using a microplate reader (Opsys MR; Dynex, Denkendorf, Germany). The minimum detection limit of the assay is specified as 5 pg·mL−1 by the manufacturer. The CGRP concentration was calculated in pg·mL−1, allowing for the added volume of EIA buffer. Raw data were used for statistical comparison.
Figure 3.
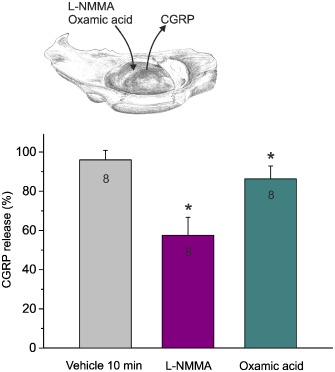
CGRP release from the dura mater in the hemisected rat head (inset) determined as the concentration of CGRP collected in the superfusate within 5 min after treatment with vehicle, L‐NMMA (2 mM) or oxamic acid (2 mM) for 10 min, normalized to the first release value prior to treatment. *P < 0.05, significantly different from vehicle.
Histochemistry
Rats deeply anaesthetized by breathing 5% isoflurane and i.p. injection of thiopentone (Trapanal®, 150–200 mg·kg−1) were thoracotomized and perfused transcardially with physiological saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The skin and muscles of the skull were removed and the skull was divided into halves along the midline. After removal of the brain, samples of the dura mater were harvested and processed for microscopic examination.
For indirect immunofluorescence, whole mounts of the dura mater were rinsed in PBS (0.01 M, pH 7.4), pre‐incubated for 2 h at room temperature with a solution of 5% goat serum (Dianova, Hamburg, Germany), 0.5% Triton X‐100 and 1% BSA containing PBS, rinsed in PBS, and incubated overnight with rabbit polyclonal antisera raised against rat CGRP 1:1000 (Peninsula T 4032; Bachem, Weil/Rhein, Germany) or rat nNOS 1:1000 (Calbiochem ST 1995; Merck Millipore, Darmstadt, Germany). For TRPA1 immunostaining, unfixed whole mounts of rat dura mater were treated accordingly and incubated with rabbit polyclonal antiserum raised against rat TRPA1 1:500 (ab58844; Abcam, Cambridge, UK). Then whole mounts were washed with PBS, incubated with goat anti‐rabbit IgG conjugated with indocarbocyanine 1:200 (Cy3; Dianova 111‐165‐144) for 2 h at room temperature, rinsed again and coverslipped in Fluoromount G (Science Services, München, Germany). Some samples were mounted with Roti®‐Mount Fluor Care (Roth, Karlsruhe, Germany) containing DAPI for nuclear DNA staining.
For NADPH‐diaphorase histochemistry, the dura mater was pre‐incubated in 0.1 M PBS containing 0.3% Triton X‐100 for 60 min and then incubated in reaction solutions for NADPH‐diaphorase activity (1 mg·mL−1 nitroblue tetrazolium, 0.4 mg·mL−1 ß‐NADPH, 0.3% Triton X‐100 in 0.1 M PBS) at 37°C for 15 min. Dura samples were mounted on glass slides, air dried and coverslipped.
For detection of NO, H2S and HNO by fluorescence sensors, isolated (unfixed) dura mater was incubated for 15 min at 37°C with DAF (4‐amino‐5‐methylamino‐2′,7′‐difluoresceine diacetate) for NO, with WSP‐1 (Cayman, distributed by Alexis, Grünberg, Germany) for H2S, or with the HNO‐selective fluorescent dye CuBOT1, synthesized as previously described by Rosenthal and Lippard (2010), then washed with SIF and further incubated in SIF for additional 30 min at 37°C. In other experiments, the isolated dura mater was pre‐incubated with L‐NMMA (2 mM) or with oxamic acid (2 mM) for 45 min before the above sensors were applied. The green fluorescence was visualized with the FITC filter setting using the LSM 780 confocal microscope.
In order to examine the production of NO and H2S from the dura, isolated rat dura was incubated with the NO sensor DAF for 30 min following pretreatment with oxamic acid (2 mM) for 30 min at 37°C or, in the same way, with the H2S sensor WSP‐1 following L‐NMMA (2 mM). The fluorescence of the supernatant was analysed with Jasco spectrofluorometer (Jasco Analytical Instruments, Gross‐Umstadt, Germany) with λex 485 and λem 538 nm for DAF‐FM‐DA and λex 465 and λem 515 nm for WSP‐1. The intensities (arbitrary units per wet weight of tissue) compared with the value of the contralateral dura half, incubated without pretreatment.
Microscopy
The stained dura mater was analysed using an LSM 780 light and confocal microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany) mounted on an inverted Axio Observer Z1. Two dry objective lenses (10× and 20× with numerical apertures of 0.3 and 0.8) were used. Fluorescent structures were observed in the light path mode using red and green filters. Confocal images were taken using filter settings for Cy3 (excitation 514 nm, emission 530–566 nm) and for FITC (excitation 488 nm, emission 505–520 nm). The number of image pixels was 1024 × 1024 or 512 × 512. Pictures were converted to a 12‐bit RGB tiff file using confocal assistant software ZEN 2010. The final layout was made with CorelDraw (Corel, Dublin, Ireland).
Data analysis
All values are expressed as means ± SEM. Statistical analysis of the data was performed using Statistica 7 (StatSoft, Tulsa, OK, USA). The Student's t‐test was used for group sizes of n ≥ 10. The Wilcoxon matched pairs test was used for dependent measurements and the Mann–Whitney U‐test for independent measurements of group sizes <10. anova was applied for analysing the time course of the flow increase after application of Na2S. A probability level of P < 0.05 was regarded as statistically significant.
Materials
The NO donor DEANONOate purchased from Sigma (St. Louis, MO, USA) was prepared in 10 mM KOH. The H2S source, Na2S, was purchased from Sigma as the anhydrous form and stored in an atmosphere with <1 p.p.m. O2 and H2O. A stock solution of 100 mM was prepared using argon‐bubbled nanopure water and stored in glass vials with PTFE septa at 4°C until use, for a maximum of 1 week. Adequate volumes taken from the vial with a small syringe were diluted in SIF immediately before use. The TRPA1 channel antagonist HC030031 (Tocris, Bristol, UK) was stored as a 10 mM stock solution in DMSO. The CGRP receptor antagonist CGRP8‐37 (Tocris), the NOS inhibitor L‐NG‐monomethylarginine acetate (L‐NMMA; Sigma) and oxamic acid (Sigma) were dissolved in saline, the sGC inhibitor 1H‐[1,2,4]oxadiazole[4,3‐a]quinoxalin‐1‐one (ODQ, Calbiochem, distributed by Merck, Schwalbach, Germany) as a 25 mM stock solution in ethanol. All stock solutions were diluted with SIF immediately before use, except for L‐NMMA which was administered i.v., in saline.
Results
Role of NO in meningeal blood flow
Topical administration of the NO donor DEANONOate at a concentration of 200 μM onto the exposed dura mater for 10 min caused an increase in meningeal blood flow in each experiment, on average by about 30% above baseline (Figure 1A and 1C), consistent with previous findings (Strecker et al., 2002b). In addition, the increase in flow has already been shown to be reduced (albeit not totally abolished) by blockade of CGRP receptors (Strecker et al., 2002a). This suggests that besides the classical NO‐cGMP signalling pathway, a CGRP receptor‐mediated arterial vasodilatation is involved in the blood flow elevating effect of DEANONOate. Because there is no mechanism known by which NO can directly activate CGRP receptors (Miranda et al., 2003), we assumed that the NO derivative, HNO, may have been responsible for this effect (Eberhardt et al., 2014). Knowing that HNO can be formed by the reaction of NO with H2S, and the formation of H2S is accelerated by endogenous CBS and CSE, we examined if inhibition of these enzymes by oxamic acid has an influence on the DEANONOate‐evoked blood flow.
Pretreatment of the dura mater with oxamic acid (2 mM), a non‐selective inhibitor of both CBS and CSE (Asimakopoulou et al., 2013), reduced the blood flow increase evoked by DEANONOate (200 μM) in a time‐dependent manner (Figure 1A and C). When oxamic acid (10 mg·kg−1) was given i.v. a comparable inhibition of the increased blood was observed, though with a faster onset. The basal meningeal blood flow was not significantly changed by oxamic acid alone (5.2 ± 2.0% increase after topical, n = 14; 4.5 ± 2.9% increase after i.v. administration; n = 12).
The remaining blood flow increase induced by administration of DEANONOate was presumably due to the direct sGC activating effect of NO. However, in experiments with additional topical or systemic administration of the sGC inhibitor ODQ following i.v. oxamic acid treatment, the effect of DEANONOate was not significantly different compared with the DEANONOate effect after oxamic acid alone (Wilcoxon, n = 8, P > 0.5), although it tended to be further reduced (Figure 1A and C). The basal flow was not changed by adding ODQ after i.v. oxamic acid. Topical administration of oxamic acid or ODQ had no significant effect on the mean systemic blood pressure, which was 85.4 ± 5.4 mmHg throughout the measurements.
Taken together, these results suggested that DEANONOate was fully effective in increasing meningeal blood flow only if there was endogenous production of H2S, suggesting that the blood flow increase was due to the formation of HNO. In contrast, sGC activation linked to cGMP production seemed to play a minor role in the DEANONOate‐induced effect in rat meningeal blood vessels. We therefore wanted to explore if supply of exogenous H2S could further activate the synthesis of HNO from endogenously produced NO.
Role of H2S in meningeal blood flow
Topical application of the H2S source Na2S (300 μM) to the dura mater induced a rapid increase in meningeal blood flow, within a few minutes and outlasting the application period of 10 min (Figure 2A and B). The mean flow increase within the 10 min period (about 19%) is shown in Figure 2C. Repeated application of H2S at intervals of 30 min after washout of the substance caused similar blood flow increases (data not shown).
Assuming that the effect of H2S was mediated through the reduction of the endogenous NO to HNO, followed by HNO‐induced activation of TRPA1 channels to release CGRP, which eventually caused the increased blood flow, as suggested by Eberhardt et al. (2014), we inhibited each step of this signal cascade. Pretreatment of the dura mater with the CGRP receptor antagonist CGRP8‐37 (100 μM) or the TRPA1 channel antagonist HC030031 (50 μM) abolished the blood flow increase following application of Na2S (Figure 2A, B and C). CGRP8‐37 or HC030031 given alone had no significant effects on the basal meningeal blood flow.
The contribution of endogenous NO production in the Na2S‐induced blood flow was tested by topical and systemic administration of the non‐specific NOS inhibitor L‐NMMA (Figure 2C right side and 2D). Topical application of L‐NMMA (1 mM) significantly decreased the basal meningeal flow by 21.2 ± 4.4% (t‐test, n = 10, P < 0.01) but had no effect on the arterial pressure. At the end of the 10 min application period of L‐NMMA, the new baseline was stable, but the effect of topical Na2S was markedly less (Figure 2C). Intravenous administration of L‐NMMA (12 mg·kg−1) increased the systemic blood pressure by 31.2 ± 7.2% and the basal meningeal blood flow by 22.6 ± 7.1% (t‐test, n = 10, P < 0.01), which was stable after some minutes. Under these conditions, the blood flow increase following topical Na2S was abolished.
Role of NO and H2S in CGRP release from the dura mater
In the hemisected rat head preparation with adhering dura mater in vitro, the basal release of CGRP in the incubation fluid (0.5 mL SIF), which was applied for 5 min at 37°C, was around 15 pg·mL−1 with small variations depending on the batch of the elisa. After application of the non‐specific NOS inhibitor L‐NMMA (2 mM) twice for 5 min each, the CGRP release was about 50% of basal release, which is close to the detection level of the assay (Figure 3, see also Figure 4B). After application of the CBS/CSE inhibitor oxamic acid (2 mM), for two periods of 5 min each, the basal CGRP release was only slightly decreased , while vehicle applied in the same way had no significant effects (Figure 3).
Figure 4.
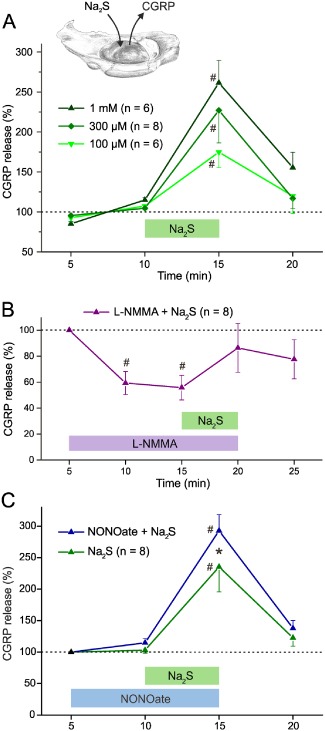
CGRP release from the dura mater, measured as the concentration of CGRP collected in the superfusate of the hemisected head (inset) at periods of 5 min. #P < 0.05, significantly different from baseline at 5–10 or 5 min. (A) Effect of different concentrations of Na2S applied for 5 min. (B) CGRP release decreased during administration of L‐NMMA (2 mM). Na2S (300 μM) after L‐NMMA administration did not increase CGRP release above baseline. (C) Application of NONOate (200 μM) tends to increase CGRP release (at 10 min) while co‐administration of Na2S (300 μM) causes a synergistic effect significantly different from Na2S alone. *P < 0.05, significant effect of NONOate.
Administration of Na2S (0.1–1 mM) significantly increased CGRP release over mean baseline values of 15.0–17.8 pg·mL−1 (Figure 4A) but the Na2S‐induced increases in CGRP release were not significantly different between the concentrations (Mann–Whitney U‐test, P = 0.7; Figure 4A). For the subsequent experiments, we used Na2S at the intermediate concentration of 300 μM.
To test if the release of CGRP by Na2S depended on the presence of NO, we inhibited endogenous NO production by pre‐treatment with the non‐specific NOS inhibitor, L‐NMMA (2 mM). After this pre‐treatment, the CGRP release evoked by Na2S (300 μM) was not different from baseline (Figure 4B). Conversely, to examine if the Na2S‐evoked CGRP release was influenced by a synergistic action of NO, Na2S (300 μM) was co‐administered with DEANONOate (200 μM) and compared with the effect of Na2S alone, applied to the contralateral skull half. Mean basal CGRP levels were 15–15.3 pg·mL−1 and Na2S alone raised CGRP release to about double this level. Although DEANONOate did not affect basal levels of CGRP release, it did potentiate the increase induced by Na2S (Figure 4C).
Histochemical evidence for HNO‐TRPA1‐CGRP signalling in the dura mater
In the whole mount preparations of the dura mater, a dense network of CGRP‐immunofluorescent nerve fibres was observed surrounding the MMA and its branches, indicating that CGRP can be released close to the arterial vessel wall (Figure 5A). We indirectly visualized different sources for NO; NADPH‐diaphorase, a specific marker for NO‐producing cells, was localized by its activity mostly in the wall of the MMA and arterioles up to the transition into capillaries and at a low intensity in some nerve fibre bundles accompanying the artery (Figure 5B). Some nerve fibres immunoreactive for the neuronal subtype of NOS were identified accompanying branches of the MMA (Figure 5C). TRPA1 channel immunoreactivity was detected in perivascular nerves of the dura mater staining several axons in a patchy manner (Figure 5D).
Figure 5.
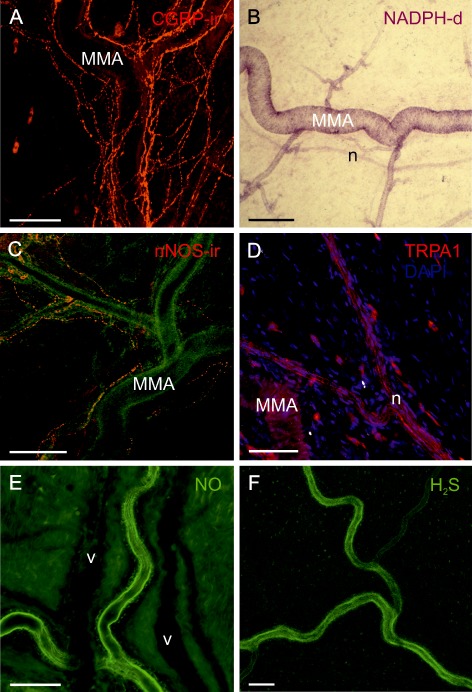
Histochemical demonstration of components of HNO‐CGRP signalling in the dura mater; size bars 100 μm. (A) Confocal immunofluorescence image of CGRP‐immunoreactive nerve fibres densely surrounding the MMA and its branches. (B) Transmission light microscopic image showing NADPH‐diaphorase staining of the MMA and some arterioles up to the transition into capillaries, indicative for the production of NO. An adjacent nerve bundle (n) is weakly stained. (C) Confocal image of nerve fibres immunoreactive for nNOS surrounding the MMA, which is visible in the green background channel. (D) Confocal image showing TRPA1‐immunoreactive nerve fibres in a nerve bundle (n) adjacent to the MMA. (E) Confocal image showing arterial endothelium of the MMA stained by DAF indicating NO. Two unstained accompanying veins are visible in the background as dark shadows. (F) Confocal image showing endothelial staining of the MMA by WSP1 demonstrating the presence of H2S.
The sensors for NO and H2S stained the endothelium of the MMA and its branches but not venous vessels or other structures of the dura mater (Figure 5E and F). This may be due to the low sensitivity of the used sensors coupled with relatively low steady‐state levels of both NO and H2S in the neuronal cells. HNO staining was visible in the arterial vessel walls and particularly in nerve fibre bundles adjacent to the arterial vessels (Figure 6A). Pretreatment of the dura mater with oxamic acid significantly reduced the fluorescence after incubation with the HNO sensor (Figure 6B). Pretreatment with L‐NMMA or a combination of oxamic acid and L‐NMMA abolished the staining (Figure 6C and D).
Figure 6.

Confocal images of dura mater stained by CuBOT1 indicative of HNO (green colour). Branches of the MMA (A) are accompanied by a venous vessel (V) and bundles of nerve fibres (n). Nuclei are stained with DAPI (blue); size bars 100 μm. (A) Control dura. Nerve bundles and smooth muscle cells (m) of the artery as well as other perivascular cells appear HNO positive. (B) The HNO staining was significantly reduced after pretreatment with oxamic for 45 min. After pretreatment with L‐NMMA (C) or the combination of oxamic acid and L‐NMMA (D), the staining was lost indicative of blocked HNO production.
In addition, we assessed the intensity of fluorescence in the supernatant after incubating the dura with the NO sensor or H2S sensor as a measure of NO and H2S generation from this tissue. Following pretreatment with oxamic acid, the fluorescence of the supernatant created by the NO fluorescence sensor was increased by 51.4 ± 9.2% compared with the contralateral control dura, indicating that less NO was consumed in the tissue when the H2S production was inhibited. Similarly, after pretreatment with L‐NMMA, the fluorescence of the H2S sensor in the supernatant was increased by 32.9 ± 10.8% compared with the control side indicative of less H2S consumption in the dura upon inhibition of the NO synthesis.
Discussion and conclusions
CGRP release upon formation of HNO inducing meningeal blood flow
The present results reveal an important role for NO and H2S crosstalk in controlling the release of CGRP from afferent endings, which is involved in the regulation of meningeal blood flow. We have recently described the cellular mechanism underlying the interaction of these two gasotransmitters (Filipovic et al., 2012; 2013; Miljkovic et al., 2013; Eberhardt et al., 2014). NO and H2S generate HNO, which is an activator of TRPA1 cation channels by modifying cysteine residues of the receptor protein to form disulfide bonds, which opens the receptor channels and the consequent influx of Ca2+ activates the exocytosis of CGRP (Eberhardt et al., 2014). In the histochemical preparations shown here (Figures 5 and 6), we localized endothelial and neuronal sources for NO in the dura mater, while H2S production was predominantly visible in arterial endothelium. CSE, CBS and MST are the key enzymes of H2S production. CSE has been found in endothelium and tunica media cells of extra‐ and intracranial arteries (Chertok and Kotsyuba, 2012) and CBS in the brain (Kimura, 2002). In addition, our previous results showed that CBS immunoreactivity in small trigeminal neurons partly co‐localizes with TRPA1 channel immunoreactivity (Eberhardt et al., 2014). The clearly demonstrated co‐dependence of these two gasotransmitters is reflected in the data presented here showing that higher amounts of NO leave the dura mater when H2S production is inhibited and that higher concentrations of H2S can be detected when NO production is inhibited. This confirms that these two gasotransmitters react to form HNO, which has been previously shown to increase intracellular Ca2+ levels by opening TRPA1 channels in primary afferent neurons and to thus activate CGRP exocytosis (Eberhardt et al., 2014). Indeed, for the first time, the ongoing basal HNO production could also be visualized in a tissue using the HNO sensitive dye CuBOT1. The strong staining, which is due to the high sensitivity of the HNO sensor, is clearly reduced after treatment of the dura mater with L‐NMMA or oxamic acid, which inhibit the production of NO or H2S respectively (see Figure 6). In the dura mater, the close apposition of HNO sources and meningeal arteries surrounded by CGRP expressing trigeminal afferents provide the morphological substrate for this putative HNO‐TRPA1‐CGRP signalling path, which was substantiated by our observation that blockade of either TRPA1 channels or CGRP receptors abolished the blood flow increasing effect of Na2S (Figure 2).
Our experimental results indicate that the modest basal release of CGRP in the dura mater measured in the hemisected skull preparation is partly the result of constitutive HNO production, as both inhibition of NO production by L‐NMMA and inhibition of the H2S synthesis by oxamic acid, reduced the (unstimulated) basal level of CGRP release (Figures 3 and 4B). Application of additional Na2S, i.e,.exogenous H2S, induced CGRP release from the dura mater, which was strongly dependent on the NO/HNO production, as it was abolished by the NOS inhibitor L‐NMMA. Conversely, providing additional NO through the NO donor DEANONOate further increased the Na2S‐induced CGRP release.
The amount of CGRP released under physiological conditions is probably below the level needed to maintain vasodilatation, because application of the CGRP receptor antagonist CGRP8‐37 did not change the basal meningeal blood flow. On the other hand, the flow increase induced by H2S in the presence of endogenous NO was fully antagonized by CGRP8‐37, suggesting that HNO formed by H2S and NO exerts its vasodilator effects largely through CGRP release. Furthermore, the CGRP release evoked by HNO depends on the activation of TRPA1 channels, because the H2S‐induced flow increase was abolished by inhibition of these channels with HC030031 (Figure 2B).
The proposed pathway of NO‐TRPA1‐CGRP signalling in the dura mater with the experimental interventions is summarized in Figure 7. We assume that under natural conditions, most of the NO is produced in the endothelial cells of meningeal arteries by endothelial NOS. NO diffuses into perivascular trigeminal nerve fibres and is converted to HNO in the presence of H2S, which is formed by CBS, an enzyme that co‐localizes with TRPA1 channels, HNO then activates TRPA1 channels inducing Ca2+ influx and consequent CGRP release . CGRP activates CGRP receptors of smooth arterial muscle cells causing vasorelaxation through up‐regulation of cyclic monophosphates followed by increased perfusion of the meninges. This fits nicely with the data presented here showing that H2S‐induced increase of meningeal blood flow was inhibited by TRPA1 channel and CGRP receptor antagonists, as well as by the NOS inhibitors (Figures 1 and 2).
Figure 7.
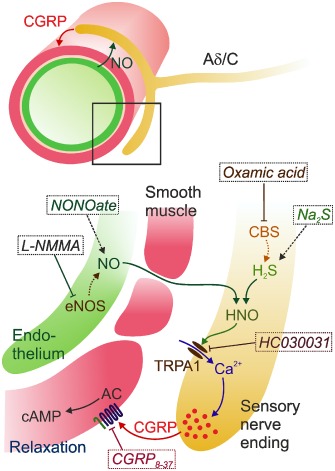
Scheme illustrating the proposed HNO‐TRPA1‐CGRP signalling pathway at meningeal arteries innervated by trigeminal afferent nerve fibres (Aδ/C). NO mainly produced by the endothelial NOS (eNOS) diffuses into perivascular nerve fibres, where HNO is formed together with H2S produced in the presence of neuronal CBS. HNO activates TRPA1 channels leading to Ca2+‐induced CGRP release. CGRP activates its receptors in arterial smooth muscle cells causing vasodilatation upon up‐regulation of the AC‐cAMP and other pathways. Experimentally used NO and H2S donors (NONOate, Na2S) and blockers of the NO and H2S generation (L‐NMMA, oxamic acid), as well as blockers of TRPA1 channels or CGRP receptors (HC030031 and CGRP 8‐37) are indicated in italics.
Other factors mediating NO‐induced blood flow increase
Following inhibition of the endogenous H2S production by oxamic acid and thereby of HNO, DEANONOate still induced an increase in meningeal blood flow by about 10% above baseline (Figure 1A and C). This component was obviously not mediated by CGRP but may have resulted partly from the direct cGMP regulating effect of NO. Indeed, some further reduction of the blood flow increase was seen after blockade of cGMP production with ODQ. Nevertheless, a 6–8% increase in blood flow was maintained. The maintained increase in flow could have been due to a low, persistent production of H2S. Activation of KATP channels by H2S mediating relaxation of smooth muscle cells has previously been shown (Mustafa et al., 2011; Eberhardt et al., 2014). Furthermore, Na2S relaxed isolated aortic rings by maintaining a tonic inhibitory effect on the phosphodiesterase type 5 in endothelial cells, thereby inhibting the degradation of cGMP and permitting physiological cGMP signalling (Coletta et al., 2012). However, this mechanism of vasorelaxation seems to play a minor role in the meningeal vasodilatation, as blocking the TRPA1‐CGRP pathway almost completely abolished the blood flow increase induced by Na2S and inhibition of NO production significantly reduced the effect. This differs from recently examined functions of HNO in the heart, where Angeli's salt increased coronary blood flow and left ventricular inotropy (Chin et al., 2014). Scavenging of HNO and inhibition of sGC but not of CGRP receptors reduced these effects.
Role of CGRP in meningeal nociception and headache
According to vascular and neuroinflammatory theories of migraine, the dilatation of meningeal arteries has been considered to account for the generation of headaches (Moskowitz et al., 1989). Neurogenic vasodilatation in the dura mater induced by the release of potent vasodilators like CGRP is still considered as an important process in the pathophysiology of primary headaches such as migraine (Geppetti et al., 2012). Although these hypotheses have widely been modified and amended by central migraine theories (May and Goadsby, 1999), it is evident that nociceptive events in the trigeminovascular system causing the sensation of headaches are linked to vascular processes (Olesen et al., 2009). The concept of the neurovascular generation of headaches results from the observation that infusion of vasodilatory mediators can induce headaches which resemble spontaneously occurring primary headaches, such as migraine (Olesen, 2008). Most intriguingly, infusion of CGRP induced acute and delayed migraine‐like headaches in migraineurs (Lassen et al., 2002), an effect which was prevented by the CGRP receptor antagonist olcegepant, concomitant with an inhibition of the dilatation of extracranial arteries (Petersen et al., 2005). Contrariwise, the infusion of agents such as vasoactive intestinal polypeptide (Rahmann et al., 2008) and adrenomedullin (Petersen et al., 2009), which dilate blood vessels but do not induce headaches, has been used to argue against vascular migraine theories (Panconesi et al., 2009).
Finally, we have to state that currently no theory based on acute peripheral CGRP effects in migraine generation is satisfying for the following reasons. Nociceptive afferents in the meninges, the activation of which is regarded as the primary source of headache generation, are neither activated directly by CGRP nor by vasodilatation induced by CGRP (Levy et al., 2005; Bhatt et al., 2015). Injection of CGRP or the CGRP receptor antagonist olcegepant into the ganglion did not change the activity of spinal trigeminal neurons with meningeal afferent input (Covasala et al., 2012). In this respect, acute CGRP release and vasodilatation during experimental activation of meningeal afferents (Zagami et al., 1990) or during attacks of primary headaches (Edvinsson and Goadsby, 1995) may rather be consequences than causes of meningeal afferent activation. On the other hand, the application of CGRP antagonists or antibodies seems to be very efficient in preventing migraines (Edvinsson, 2001; Elvidge, 2014; Bigal et al., 2015). It appears possible that long‐term effects of CGRP can trigger trigeminal sensitization and the generation of primary headaches. CGRP receptors have immunohistochemically been located to a major proportion of trigeminal ganglion neurons and satellite cells, noticeably not co‐localized with CGRP‐immunoreactive neurons (Lennerz et al., 2005; Eftekhari et al., 2010). In trigeminal ganglion cell cultures, CGRP has been shown to up‐regulate the expression of iNOS and NO production in glial cells (Li et al., 2008), involving MAPK pathways (Vause and Durham, 2009). NO donors in turn increased CGRP promoter activity and CGRP release, which surprisingly was not dependent on the cGMP signalling pathway (Bellamy et al., 2006), possibly indicating a vicious cycle of CGRP and NO production in the trigeminal ganglion. The mechanism of CGRP release may be the same as in the dura mater, namely conversion of NO to HNO catalysed by CBS in the presence of H2S followed by HNO‐induced TRPA1 channel activation and Ca2+ influx. Apart from CGRP release, Ca2+‐activated gene expression could thus increase neuronal sensitivity.
Possible role of HNO‐TRPA1‐CGRP signalling in meningeal nociception and headache
Activation of TRPA1 channels expressed in nociceptive afferents is likely not only to release CGRP but also to contribute to the activation of the nociceptive system, as shown by recent patch‐clamp recordings of rat trigeminal afferent projection to the dura mater and by behavioural studies (Edelmayer et al., 2012). Regarding meningeal afferents, we have currently no final results but there is evidence that TRPA1 channel agonists cause at least transient activation in rodents. Activation of TRPA1 channels of the trigeminal afferents innervating the nasal mucosa is considered as a pathophysiological mechanism involved in the generation of headache induced by inhaled irritants (Kunkler et al., 2011). TRPA1 channels are present in 36% of trigeminal neurons and they are frequently co‐localized with CGRP (Story et al., 2003; Huang et al., 2012). Thus, the described activation of TRPA1 channels by HNO as a result of the interaction of the two gasotransmitters NO and H2S may be highly relevant as a headache‐provoking mechanism. Indeed, increased plasma levels not only of CGRP but also of NO metabolites have been found during migraine attacks (Sarchielli et al., 2000). Furthermore, some researchers have described higher plasma levels of homocysteine in patients suffering from migraine with aura, which could be indicative of an increased metabolism of H2S for HNO formation (Moschiano et al., 2008). It would be worthwhile to look further for metabolites of the NO‐H2S‐HNO pathway in primary headache patients.
These putative processes are probably not restricted to the periphery but are present also in the spinal trigeminal nucleus, where CGRP release from the central terminals of sensory neurons seems to facilitate glutamatergic neurotransmission (Storer et al., 2004) and the activation and/or sensitization of the nociceptive pathway, a matter which is currently under investigation in our laboratory. In the spinal system, the contribution to central sensitization of CGRP has long been shown in a variety of animal experiments (Sun et al., 2009). The release of CGRP and other neurotransmitters from presynaptic neuronal elements is likely to be controlled by HNO. Nitroglycerin is able to activate and sensitize spinal trigeminal neurons (Lambert et al., 2000; Jones et al., 2001), and this could be reversed and prevented by inhibition of CGRP receptors (Koulchitsky et al., 2009; Feistel et al., 2013), which is probably, at least in part, a central effect. We do not yet know to which extent the central CGRP release depends on the proposed HNO‐TRPA1 signalling but all elements of this pathway, NOS, CBS, TRPA1 channels, CGRP and its receptor, are present in the spinal trigeminal nucleus (Lennerz et al., 2005; Schlechtweg et al., 2009; Eberhardt et al., 2014). It is tempting to speculate that CBS‐produced H2S modifies nitroglycerin to form HNO and activates the TRPA1‐CGRP process, thus stimulating a migraine attack.
Therapeutic considerations
Consistent with above‐mentioned findings in animal experiments, drugs that block the release (triptans) or the receptor binding of CGRP (antagonists, antibodies) are all therapeutic in migraine and partly in other primary headaches (see Bigal et al., 2013). However, there is a great deal of scientific discussion around the question, on which level of the trigeminal system these inhibitory effects take place. In particular, the newly approved CGRP antibodies and also triptans and CGRP receptor antagonists will not readily pass the blood brain barrier, so that a peripheral action (i.e. in the meninges or the trigeminal ganglion) appears likely. Because the HNO‐TRPA1‐CGRP signalling may take place on all levels of the trigeminal system, the trigeminal ganglion seems to be the most likely site of interaction. In line with the discussed signal cascade, inhibition of NO generation by L‐NMMA has also proven to be therapeutic in migraine, while specific inhibitors of iNOS were ineffective (see Olesen, 2008; Hoffmann and Goadsby, 2012). The role of H2S production has never been addressed before in the context of migraine, so this might be the missing link between NO, CGRP and migraine attacks. Regarding possible therapeutic options by targeting the generation of H2S and HNO, further preclinical investigations are necessary to elucidate the role of the HNO‐TRPA1‐CGRP signalling cascade in meningeal nociception and headache generation.
Author contributions
M. D., C. W., B. V., M. R. F. and K. M. performed the research. M. R. F. and K. M. designed the research study. M. D., C. W. and B. V. analysed the data. M. D., M. R. F. and K. M. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
We like to thank Jana Schramm, Annette Kuhn and Susanne Haux‐Oertel for excellent technical assistance. The project was supported by the Friedrich‐Alexander‐University Erlangen‐Nürnberg (Emerging Fields Project: Medicinal Redox Inorganic Chemistry), the Humboldt Foundation (travel grant to M. D.) and the FP7 project EUROHEADPAIN (Grant Agreement No. 602633).
Dux, M. , Will, C. , Vogler, B. , Filipovic, M. R. , and Messlinger, K. (2016) Meningeal blood flow is controlled by H2S‐NO crosstalk activating a HNO‐TRPA1‐CGRP signalling pathway. Br J Pharmacol, 173: 431–445. doi: 10.1111/bph.13164.
References
- Akerman S, Williamson DJ, Kaube H, Goadsby PJ (2002). Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene‐related peptide induced dilation of dural meningeal vessels. Br J Pharmacol 137: 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G et al (2013). Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br J Pharmacol 169: 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy J, Bowen EJ, Russo AF, Durham PL (2006). Nitric oxide regulation of calcitonin gene‐related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci 23: 2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DK, Ramachandran R, Christensen SL, Gupta S, Jansen‐Olesen I, Olesen J (2015). CGRP infusion in unanesthetized rats increases expression of c‐Fos in the nucleus tractus solitarius and caudal ventrolateral medulla, but not in the trigeminal nucleus caudalis. Cephalalgia 35: 220–233. [DOI] [PubMed] [Google Scholar]
- Bigal ME, Walter S, Rapoport AM (2013). Calcitonin gene‐related peptide (CGRP) and migraine current understanding and state of development. Headache 53: 1230–1244. [DOI] [PubMed] [Google Scholar]
- Bigal ME, Walter S, Rapoport AM (2015). Therapeutic antibodies against CGRP or its receptor. Br J Clin Pharmacol doi: 10.1111/bcp.12591; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain SD, Grant AD (2004). Vascular actions of calcitonin gene‐related peptide and adrenomedullin. Physiol Rev 84: 903–934. [DOI] [PubMed] [Google Scholar]
- Brian JE, Faraci FM, Heistad DD (1996). Recent insights into the regulation of cerebral circulation. Clin Exp Pharmacol Physiol 23: 449–457. [DOI] [PubMed] [Google Scholar]
- Chertok VM, Kotsyuba AE (2012). Distribution of H2S synthesis enzymes in the walls of cerebral arteries in rats. Bull Exp Biol Med 154: 104–107. [DOI] [PubMed] [Google Scholar]
- Chin KY, Qin C, Cao N, Kemp‐Harper BK, Woodman OL, Ritchie RH (2014). The concomitant coronary vasodilator and positive inotropic actions of the nitroxyl donor Angeli's salt in the intact rat heart: contribution of soluble guanylyl cyclase‐dependent and ‐independent mechanisms. Br J Pharmacol 171: 1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P et al (2012). Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium‐dependent vasorelaxation. Proc Natl Acad Sci U S A 109: 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covasala O, Stirn SL, Albrecht S, De Col R, Messlinger K (2012). Calcitonin gene‐related peptide receptors in rat trigeminal ganglion do not control spinal trigeminal activity. J Neurophysiol 108: 431–440. [DOI] [PubMed] [Google Scholar]
- Dux M, Schwenger N, Messlinger K (2002). Possible role of histamine (H1‐ and H2‐) receptors in the regulation of meningeal blood flow. Br J Pharmacol 137: 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dux M, Sántha P, Jancsó G (2003). Capsaicin‐sensitive neurogenic sensory vasodilatation in the dura mater of the rat. J Physiol 552: 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt M, Dux M, Namer B, Miljkovic J, Cordasic N, Will C et al (2014). H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO‐TRPA1‐CGRP signalling pathway. Nat Commun 5: 4381–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger A, Averbeck B, Messlinger K, Reeh PW (1999). Release of substance P, calcitonin gene‐related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neuroscience 89: 901–907. [DOI] [PubMed] [Google Scholar]
- Edelmayer RM, Le LN, Yan J, Wei X, Nassini R, Materazzi S et al (2012). Activation of TRPA1 on dural afferents: a potential mechanism of headache pain. Pain 153: 1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L (2001). Calcitonin gene‐related peptide (CGRP) and the pathophysiology of headache: therapeutic implications. CNS Drugs 15: 745–753. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Goadsby PJ (1995). Neuropeptides in the cerebral circulation: relevance to headache. Cephalalgia 15: 272–276. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Uddman R (1981). Adrenergic, cholinergic and peptidergic nerve fibres in dura mater–involvement in headache? Cephalalgia 1: 175–179. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Ekman R, Jansen I, McCulloch J, Uddman R (1987). Calcitonin gene‐related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab 7: 720–728. [DOI] [PubMed] [Google Scholar]
- Eftekhari S, Salvatore CA, Calamari A, Kane SA, Tajti J, Edvinsson L (2010). Differential distribution of calcitonin gene‐related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 169: 683–696. [DOI] [PubMed] [Google Scholar]
- Ellis A, Li CG, Rand MJ (2000). Differential actions of L‐cysteine on responses to nitric oxide, nitroxyl anions and EDRF in the rat aorta. Br J Pharmacol 129: 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvidge S (2014). Anti‐CGRP antibodies for migraine turn industry heads. Nat Biotechnol 32: 707. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Heistad DD (1991). Regulation of cerebral blood vessels by humoral and endothelium‐dependent mechanisms. Update on humoral regulation of vascular tone. Hypertension 17: 917–922. [DOI] [PubMed] [Google Scholar]
- Feistel S, Albrecht S, Messlinger K (2013). The calcitonin gene‐related peptide receptor antagonist MK‐8825 decreases spinal trigeminal activity during nitroglycerin infusion. J Headache Pain 14: 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic MR, Miljkovic JL, Nauser T, Royzen M, Klos K, Shubina T et al (2012). Chemical characterization of the smallest S‐nitrosothiol, HSNO; cellular cross‐talk of H2S and S‐nitrosothiols. J Am Chem Soc 134: 12016–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic MR, Eberhardt M, Prokopovic V, Mijuskovic A, Orescanin‐Dusic Z, Reeh P et al (2013). Beyond H2S and NO interplay: hydrogen sulfide and nitroprusside react directly to give nitroxyl (HNO). A new pharmacological source of HNO. J Med Chem 56: 1499–1508. [DOI] [PubMed] [Google Scholar]
- Flores‐Santana W, Salmon DJ, Donzelli S, Switzer CH, Basudhar D, Ridnour L et al (2011). The specificity of nitroxyl chemistry is unique among nitrogen oxides in biological systems. Antioxid Redox Signal 14: 1659–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuto JM, Carrington SJ (2011). HNO signalling mechanisms. Antioxid Redox Signal 14: 1649–1657. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Switzer CH, Miranda KM, Wink DA (2005). Nitroxyl (HNO): chemistry, biochemistry, and pharmacology. Annu Rev Pharmacol Toxicol 45: 335–355. [DOI] [PubMed] [Google Scholar]
- Geppetti P, Rossi E, Chiarugi A, Benemei S (2012). Antidromic vasodilatation and the migraine mechanism. J Headache Pain 13: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray DW, Marshall I (1992). Human alpha‐calcitonin gene‐related peptide stimulates adenylate cyclase and guanylate cyclase and relaxes rat thoracic aorta by releasing nitric oxide. Br J Pharmacol 107: 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Mehrotra S, Avezaat CJJ, Villalón CM, Saxena PR, Maassenvandenbrink A (2006). Characterisation of CGRP receptors in the human isolated middle meningeal artery. Life Sci 79: 265–271. [DOI] [PubMed] [Google Scholar]
- Hoffmann J, Goadsby PJ (2012). New agents for acute treatment of migraine: CGRP receptor antagonists, iNOS inhibitors. Curr Treat Options Neurol 14: 50–59. [DOI] [PubMed] [Google Scholar]
- Hope BT, Michael GJ, Knigge KM, Vincent SR (1991). Neuronal NADPH diaphorase is a nitric oxide synthase. Proc Natl Acad Sci U S A 88: 2811–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Li S, Dhaka A, Story GM, Cao Y‐Q (2012). Expression of the transient receptor potential channels TRPV1, TRPA1 and TRPM8 in mouse trigeminal primary afferent neurons innervating the dura. Mol Pain 8: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MG, Lever I, Bingham S, Read S, McMahon SB, Parsons A (2001). Nitric oxide potentiates response of trigeminal neurones to dural or facial stimulation in the rat. Cephalalgia 21: 643–655. [DOI] [PubMed] [Google Scholar]
- Kabil O, Banerjee R (2014). Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal 20: 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Motl N, Banerjee R (2014). H2S and its role in redox signaling. Biochim Biophys Acta 1844: 1355–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JT, Marfurt CF (1991). Peptidergic and serotoninergic innervation of the rat dura mater. J Comp Neurol 309: 515–534. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H (2002). Hydrogen sulfide as a neuromodulator. Mol Neurobiol 26: 13–19. [DOI] [PubMed] [Google Scholar]
- Koulchitsky S, Fischer MJ, Messlinger K (2009). Calcitonin gene‐related peptide receptor inhibition reduces neuronal activity induced by prolonged increase in nitric oxide in the rat spinal trigeminal nucleus. Cephalalgia 29: 408–417. [DOI] [PubMed] [Google Scholar]
- Kunkler PE, Ballard CJ, Oxford GS, Hurley JH (2011). TRPA1 receptors mediate environmental irritant‐induced meningeal vasodilatation. Pain 152: 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosawa M, Messlinger K, Pawlak M, Schmidt RF (1995). Increase of meningeal blood flow after electrical stimulation of rat dura mater encephali: mediation by calcitonin gene‐related peptide. Br J Pharmacol 114: 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert GA, Donaldson C, Boers PM, Zagami AS (2000). Activation of trigeminovascular neurons by glyceryl trinitrate. Brain Res 887: 203–210. [DOI] [PubMed] [Google Scholar]
- Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J (2002). CGRP may play a causative role in migraine. Cephalalgia 22: 54–61. [DOI] [PubMed] [Google Scholar]
- Lennerz JK, Rühle V, Ceppa EP, Neuhuber WL, Bunnett NW, Grady EF et al (2005). Calcitonin receptor‐like receptor (CLR), receptor activity‐modifying protein 1 (RAMP1), and calcitonin gene‐related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol 507: 1277–1299. [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R, Strassman AM (2005). Calcitonin gene‐related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol 58: 698–705. [DOI] [PubMed] [Google Scholar]
- Li J, Vause CV, Durham PL (2008). Calcitonin gene‐related peptide stimulation of nitric oxide synthesis and release from trigeminal ganglion glial cells. Brain Res 1196: 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May A, Goadsby PJ (1999). The trigeminovascular system in humans: pathophysiologic implications for primary headache syndromes of the neural influences on the cerebral circulation. J Cereb Blood Flow Metab 19: 115–127. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messlinger K, Hanesch U, Baumgärtel M, Trost B, Schmidt RF (1993). Innervation of the dura mater encephali of cat and rat: ultrastructure and calcitonin gene‐related peptide‐like and substance P‐like immunoreactivity. Anat Embryol (Berl) 188: 219–237. [DOI] [PubMed] [Google Scholar]
- Messlinger K, Suzuki A, Pawlak M, Zehnter A, Schmidt RF (2000). Involvement of nitric oxide in the modulation of dural arterial blood flow in the rat. Br J Pharmacol 129: 1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messlinger K, Fischer MJM, Lennerz JK (2011). Neuropeptide effects in the trigeminal system: pathophysiology and clinical relevance in migraine. Keio J Med 60: 82–89. [DOI] [PubMed] [Google Scholar]
- Miljkovic JL, Kenkel I, Ivanović‐Burmazović I, Filipovic MR (2013). Generation of HNO and HSNO from nitrite by heme‐iron‐catalyzed metabolism with H2S. Angew Chem Int Ed Engl 52: 12061–12064. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD et al (2003). A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc Natl Acad Sci U S A 100: 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschiano F, D'Amico D, Usai S, Grazzi L, Di Stefano M, Ciusani E et al (2008). Homocysteine plasma levels in patients with migraine with aura. Neurol Sci 29 (Suppl. 1): S173–S175. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Buzzi MG, Sakas DE, Linnik MD (1989). Pain mechanisms underlying vascular headaches. Progress report 1989. Rev Neurol (Paris) 145: 181–193. [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK et al (2011). Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K, Moskowitz MA, Maynard KI, Koketsu N, Dawson TM, Bredt DS et al (1993). Possible origins and distribution of immunoreactive nitric oxide synthase‐containing nerve fibers in cerebral arteries. J Cereb Blood Flow Metab 13: 70–79. [DOI] [PubMed] [Google Scholar]
- Olesen J (2008). The role of nitric oxide (NO) in migraine, tension‐type headache and cluster headache. Pharmacol Ther 120: 157–171. [DOI] [PubMed] [Google Scholar]
- Olesen J, Burstein R, Ashina M, Tfelt‐Hansen P (2009). Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol 8: 679–690. [DOI] [PubMed] [Google Scholar]
- Owman C (1990). Peptidergic vasodilator nerves in the peripheral circulation and in the vascular beds of the heart and brain. Blood Vessels 27: 73–93. [DOI] [PubMed] [Google Scholar]
- Panconesi A, Bartolozzi ML, Guidi L (2009). Migraine pain: reflections against vasodilatation. J Headache Pain 10: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM et al (2001). Nitroxyl anion exerts redox‐sensitive positive cardiac inotropy in vivo by calcitonin gene‐related peptide signaling. Proc Natl Acad Sci U S A 98: 10463–10468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolocci N, Katori T, Champion HC, St John ME, Miranda KM, Fukuto JM et al (2003). Positive inotropic and lusitropic effects of HNO/NO‐ in failing hearts: independence from beta‐adrenergic signaling. Proc Natl Acad Sci U S A 100: 5537–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KA, Lassen LH, Birk S, Lesko L, Olesen J (2005). BIBN4096BS antagonizes human alpha‐calcitonin gene related peptide‐induced headache and extracerebral artery dilatation. Clin Pharmacol Ther 77: 202–213. [DOI] [PubMed] [Google Scholar]
- Petersen KA, Birk S, Kitamura K, Olesen J (2009). Effect of adrenomedullin on the cerebral circulation: relevance to primary headache disorders. Cephalalgia 29: 23–30. [DOI] [PubMed] [Google Scholar]
- Pozsgai G, Bodkin JV, Graepel R, Bevan S, Andersson DA, Brain SD (2010). Evidence for the pathophysiological relevance of TRPA1 receptors in the cardiovascular system in vivo. Cardiovasc Res 87: 760–768. [DOI] [PubMed] [Google Scholar]
- Pozsgai G, Hajna Z, Bagoly T, Boros M, Kemény Á, Materazzi S et al (2012). The role of transient receptor potential ankyrin 1 (TRPA1) receptor activation in hydrogen‐sulphide‐induced CGRP‐release and vasodilation. Eur J Pharmacol 689: 56–64. [DOI] [PubMed] [Google Scholar]
- Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M (2008). Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia 28: 226–236. [DOI] [PubMed] [Google Scholar]
- Reuter U, Bolay H, Jansen‐Olesen I, Chiarugi A, Sanchez del Rio M, Letourneau R et al (2001). Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain 124: 2490–2502. [DOI] [PubMed] [Google Scholar]
- Rosenthal J, Lippard SJ (2010). Direct detection of nitroxyl in aqueous solution using a tripodal copper(II) BODIPY complex. J Am Chem Soc 132: 5536–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarchielli P, Alberti A, Codini M, Floridi A, Gallai V (2000). Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia 20: 907–918. [DOI] [PubMed] [Google Scholar]
- Schlechtweg PM, Röder J, Fischer MJ, Neuhuber W, Messlinger K (2009). Increase in NADPH‐diaphorase‐positive and neuronal NO synthase immunoreactive neurons in the rat spinal trigeminal nucleus following infusion of a NO donor–evidence for a feed‐forward process in NO production involved in trigeminal nociception. Cephalalgia 29: 566–579. [DOI] [PubMed] [Google Scholar]
- Storer RJ, Akerman S, Goadsby PJ (2004). Calcitonin gene‐related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br J Pharmacol 142: 1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR et al (2003). ANKTM1, a TRP‐like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112: 819–829. [DOI] [PubMed] [Google Scholar]
- Strecker T, Dux M, Messlinger K (2002a). Increase in meningeal blood flow by nitric oxide–interaction with calcitonin gene‐related peptide receptor and prostaglandin synthesis inhibition. Cephalalgia 22: 233–241. [DOI] [PubMed] [Google Scholar]
- Strecker T, Dux M, Messlinger K (2002b). Nitric oxide releases calcitonin‐gene‐related peptide from rat dura mater encephali promoting increases in meningeal blood flow. J Vasc Res 39: 489–496. [DOI] [PubMed] [Google Scholar]
- Sun RQ, Tu YJ, Lawand NB, Yan JY, Lin Q, Willis WD (2009). Calcitonin gene‐related peptide receptor activation produces PKA‐ and PKC‐dependent mechanical hyperalgesia and central sensitization. J Neurophysiol 92: 2859–2866. [DOI] [PubMed] [Google Scholar]
- Vause CV, Durham PL (2009). CGRP stimulation of iNOS and NO release from trigeminal ganglion glial cells involves mitogen‐activated protein kinase pathways. J Neurochem 110: 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DJ, Hargreaves RJ, Hill RG, Shepheard SL (1997). Intravital microscope studies on the effects of neurokinin agonists and calcitonin gene‐related peptide on dural vessel diameter in the anaesthetized rat. Cephalalgia 17: 518–524. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K et al (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma‐lyase. Science 322: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong QC, Hu LF, Wang S, Huang D, Bian JS (2010). Hydrogen sulfide interacts with nitric oxide in the heart: possible involvement of nitroxyl. Cardiovasc Res 88: 482–491. [DOI] [PubMed] [Google Scholar]
- Yong QC, Cheong JL, Hua F, Deng LW, Khoo YM, Lee HS et al (2011). Regulation of heart function by endogenous gaseous mediators‐crosstalk between nitric oxide and hydrogen sulfide. Antioxid Redox Signal 14: 2081–2091. [DOI] [PubMed] [Google Scholar]
- Zagami AS, Goadsby PJ, Edvinsson L (1990). Stimulation of the superior sagittal sinus in the cat causes release of vasoactive peptides. Neuropeptides 16: 69–75. [DOI] [PubMed] [Google Scholar]