

Abstract
Linked Article
This article is commented on by Michel, M. C., pp. 429‐430 of this issue. To view this commentary visit http://dx.doi.org/10.1111/bph.13379.
Background and Purpose
Mirabegron is the first β3‐adrenoceptor agonist approved for treatment of overactive bladder syndrome. This study aimed to investigate the effects of β3‐adrenoceptor agonist mirabegron in mouse urethra. The possibility that mirabegron also exerts α1‐adrenoceptor antagonism was also tested in rat smooth muscle preparations presenting α1A‐ (vas deferens and prostate), α1D‐ (aorta) and α1B‐adrenoceptors (spleen).
Experimental Approach
Functional assays were carried out in mouse and rat isolated tissues. Competition assays for the specific binding of [3H]prazosin to membrane preparations of HEK‐293 cells expressing each of the human α1‐adrenoceptors, as well as β‐adrenoceptor mRNA expression and cyclic AMP measurements in mouse urethra, were performed.
Key Results
Mirabegron produced concentration‐dependent urethral relaxations that were shifted to the right by the selective β3‐adrenoceptor antagonist L‐748,337 but unaffected by β1‐ and β2‐adrenoceptor antagonists (atenolol and ICI‐118,551 respectively). Mirabegron‐induced relaxations were enhanced by the PDE4 inhibitor rolipram, and the agonist stimulated cAMP synthesis. Mirabegron also produced rightward shifts in urethral contractions induced by the α1‐adrenoceptor agonist phenylephrine. Schild regression analysis revealed that mirabegron behaves as a competitive antagonist of α1‐adrenoceptors in urethra, vas deferens and prostate (α1A‐adrenoceptor, pA2 ≅ 5.6) and aorta (α1D‐adrenoceptor, pA2 ≅ 5.4) but not in spleen (α1B‐adrenoceptor). The affinities estimated for mirabegron in functional assays were consistent with those estimated in radioligand binding with human recombinant α1A‐ and α1D‐adrenoceptors (pK i ≅ 6.0).
Conclusion and Implications
The effects of mirabegron in urethral smooth muscle are the result of β3‐adrenoceptor agonism together with α1A and α1D‐adrenoceptor antagonism.
Abbreviations
- CR
concentration ratios
- CRC
concentration–response curve
- KHS
Krebs–Henseleit solution
- LUTS
lower urinary tract symptoms
- OAB
Overactive bladder syndrome
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| α1A-adrenoceptor |
| α1B-adrenoceptor |
| α1D-adrenoceptor |
| β1-adrenoceptor |
| β2-adrenoceptor |
| β3-adrenoceptor |
| Enzymes b |
| PDE4 |
| LIGANDS | |
|---|---|
| 17β-oestradiol | Mirabegron |
| Arginine-vasopressin | Noradrenaline |
| Atenolol | ODQ |
| Desipramine | Propranolol |
| Endothelin-1 | Rolipram |
| IBMX | Yohimbine |
| ICI-118,551 | |
| Isoprenaline | |
| L-748,337 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a b cAlexander et al., 2013a, 2013bc).
Introduction
Lower urinary tract symptoms (LUTS) represent one of the most common clinical complaints in men and women due to structural or functional abnormalities in one or more parts of the lower urinary tract, which comprises the bladder, bladder neck, prostate, distal sphincter mechanism and urethra (Abrams et al., 2002, 2013). Approximately 1.9 billion individuals worldwide are estimated to experience any LUTS, with numbers of affected individuals projected to 2.3 billion (18.4% increase) in 2018 (Irwin et al., 2011). LUTS can be divided into three groups, namely, storage symptoms (increased daytime urinary frequency, nocturia, urgency and incontinence), voiding symptoms (slow stream, splitting or spraying, intermittent stream, hesitancy, straining and terminal dribble) and post‐micturition symptoms (feeling of incomplete emptying and post‐micturition dribble). Overactive bladder syndrome (OAB) is a subset of storage LUTS currently defined as urgency, with or without urge incontinence, usually accompanied by frequency and nocturia (Abrams et al., 2002). In ageing men, LUTS has been attributed to bladder outlet obstruction as a result of benign prostatic enlargement resulting from the histological condition of benign prostatic hyperplasia (Abrams et al., 2013).
The lower urinary tract stores and releases urine via integrated circuits with brain, spinal cord and peripheral ganglia. During the voiding phase, parasympathetic neurons release ACh to contract the bladder smooth muscle through activation of muscarinic M3 cholinoceptors, resulting in an efficient bladder emptying (Hegde et al., 1997). ATP via purinergic P2X1 purinoceptors may act as a co‐transmitter to ACh in parasympathetic nerves, producing bladder contractions in physiological conditions, although this component plays a minor role in human bladder (Burnstock, 2014). The micturition event is followed by the storage phase where activation of sympathetic post‐ganglionic fibers causes the release of noradrenaline that acts on β‐adrenoceptors to promote bladder relaxation (Igawa et al., 1999, 2001). The β2‐ and β3‐adrenoceptors play important roles to induce bladder relaxations in rodent and humans (Fujimura et al., 1999).
The urethra is composed of an inner longitudinal and a middle circular smooth muscle layer innervated by autonomic nerves, along with an outer striated muscle (rhabdosphincter) innervated by somatic nerves (Pradidarcheep et al., 2011). Besides being a conduit for the urine, the urethra contributes to urinary continence by relaxing during the voiding phase and contracting during the urine storage phase (Michel and Vrydag, 2006). The urethral smooth muscle is richly innervated by sympathetic fibers, the activation of which results in the release of noradrenaline that acts on post‐junctional α1‐adrenoceptors to produce contractions during the filing/storage phase of the micturition cycle. Although the role of urethral α1‐adrenoceptors in controlling urinary continence is well documented, the identity of β‐adrenoceptor subtypes and its importance in the urethra smooth muscle are far less studied. An early study showed that β‐adrenoceptor density in rabbit urethra is lower than that in the bladder base (or the detrusor) and these are mostly β2‐adrenoceptors (Latifpour et al., 1990). Later, the urethral relaxations were shown to be predominantly mediated by β2‐ and β3‐adrenoceptors, with a higher level of β2‐adrenoceptors in canine and rat (Takeda et al., 2003) and of β3‐adrenoceptors in pig preparations (Yamanishi et al., 2003). There have been no further attempts to elucidate the importance of β3‐adrenoceptors in urethral tissue.
Mirabegron was recently approved by the Food and Drug Administration for the treatment of OAB and represents the first β3‐adrenoceptor agonist to enter clinical practice. Clinical trials of up to 12 months of mirabegron treatment demonstrated a significant efficacy in treating OAB symptoms, appearing well tolerated with low incidence of dry mouth (Chapple et al., 2014, 2013; Yamaguchi et al., 2014). Taking into consideration that the smooth muscle of the urethra actively contributes to bladder outlet resistance, this study aimed to investigate the mechanisms of mirabegron‐induced relaxations in isolated mouse urethral smooth muscle preparations. In the course of this study, mirabegron showed an unexpected action by competitively antagonizing the urethral contractions induced by the α1‐adrenoceptor agonist phenylephrine. Therefore, this study also aimed to enlarge this initial observation by characterizing the α1‐adrenoceptor blockade by mirabegron, focusing on the α1‐adrenoceptor subtypes in rat vas deferens and prostate (α1A), spleen (α1B) and aorta (α1D). The affinities of mirabegron for each of the human recombinant α1‐adrenoceptors were checked by [3H]prazosin binding assays in membranes from HEK‐293 cells expressing these receptors.
Methods
Animals
All animal care and experimental procedures were in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and were approved by the local Ethics Committee for the Use of Experimental Animals (CEUA #3514–1). Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Male C57BL6/J mice (16–18 weeks old and 30–33 g) were provided by the Central Animal House Services of State University of Campinas (UNICAMP). Male adult Wistar rats (16–20 weeks old and 260–380 g) were provided by the University of São Paulo State (UNESP). The animals were maintained under controlled conditions (23 ± 1 °C, 12 h light/12 h dark cycle and 60 ± 5% relative humidity), housed four per cage on a 12 h light–dark cycle and fed with standard chow (carbohydrate: 70%; protein: 20%; and fat: 10%).
In vitro functional assays and concentration–response curves in mouse isolated urethra
Mice were killed by CO2 asphyxiation and the prostatic urethra was removed, as previously described (Alexandre et al., 2014). Briefly, bladder, urethra and prostate were dissected free as a block and immersed in a Petri dish containing Krebs–Henseleit solution (KHS, mM: 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3 and 11 glucose). Next, the prostate gland was carefully removed, and 2 mm rings of the prostatic urethra were excised from its middle part, equidistant from the bladder neck and the external urethral sphincter. Urethral rings were mounted in 5 mL organ baths containing KHS continuously bubbled with a mixture of 95% O2/5% CO2 at pH 7.4, 37 °C. Changes in isometric force were recorded using a wire myograph for isometric force recording (Danish Myo Technology, Model 610 M, Aarhus, Denmark) coupled with an acquisition system (PowerLab 8/30, LabChart 7, ADInstruments, Sydney, NSW, Australia). The resting tension was adjusted to 2 mN at the beginning of the experiments. The equilibration period was 45 min, and the bathing medium was changed every 15 min until the start of the experiments.
In the first set of experiments, urethral rings were pre‐contracted with the α1‐adrenoceptor agonist phenylephrine (10 μM). Once the contraction had reached plateau, cumulative concentration–response curves (CRCs) to the relaxant agent isoprenaline (non‐selective β‐adrenoceptor agonist; 1 nM–1 mM) and mirabegron (1 nM–100 μM) were obtained, in steps of half a log unit. CRCs to mirabegron and/or isoprenaline were carried out in the absence and in the presence of atenolol (selective β1‐adrenoceptor antagonist; 10 μM), ICI‐118,551 (selective β2‐adrenoceptor antagonist; 10 μM), L‐748,337 (selective β3‐adrenoceptor antagonist; 1–30 μM), ODQ (soluble GC inhibitor; 10 μM) and rolipram (PDE4 inhibitor; 1 and 10 μM). CRCs to mirabegron in urethral rings pre‐contracted with KCl (80 mM), arginine‐vasopressin (60 nM) and endothelin‐1 (100 nM) were also obtained.
In the second set of experiments, cumulative CRCs to the phenylephrine in the absence and in the presence of mirabegron (0.1–100 μM) were evaluated, as described above. Urethral preparations were incubated (30 min) or not with a cocktail of inhibitors containing yohimbine (α2‐adrenoceptor antagonist; 100 nM), propranolol (non‐selective β1/β2‐adrenoceptor antagonist; 100 nM), L‐748,337 (β3‐adrenoceptor antagonist; 10 μM), 17β‐oestradiol (extraneuronal uptake blocker; 10 μM) and desipramine (neuronal monoamine uptake blocker; 100 nM).
In separate urethral rings, contractile curves to the peptide arginine‐vasopressin (0.1 nM to 1 μM) or KCl (80 mM) were carried out in the absence and in the presence of mirabegron (10 μM).
Each urethral ring was used to construct only one CRC for either the relaxant or the contractile protocols.
Rat isolated vas deferens, prostate, spleen and aorta preparations
Male Wistar rats were killed by CO2 asphyxiation and the required tissues were carefully excised and prepared for digital recording of isometric contractions as previously described (Lima et al., 2005; Nojimoto et al., 2010). Briefly, the prostate, vas deferens (epididymal portion), spleen (hemi‐sections) and thoracic aorta (~5 mm rings, endothelium denuded) were cleaned of adherent tissues and mounted in organ baths under 9.8 mN (prostate, vas deferens and spleen) or 14.7 mN (aorta) tension in a solution with the following composition (mM): NaCl 138, KCl 5.7, CaCl2 1.8, NaH2PO4 0.36, NaHCO3 15, dextrose 5.5 (for vas deferens) or KHS (for prostate, spleen and aorta) prepared in glass‐distilled, deionized water, maintained at 30 °C (vas deferens) or 37 °C (prostate, spleen and aorta), pH 7.4, and continuously bubbled with 95%O2/5%CO2. All experiments were performed in the presence of a cocktail of inhibitors containing desipramine (100 nM), corticosterone (10 μM), yohimbine (100 nM) or idazoxan (3 μM, for spleen) and propranolol (0.1 μM), L‐748,337 (300 nM) to block neuronal uptake, extraneuronal uptake, and α2‐ and β‐adrenoceptors respectively. CRCs to noradrenaline (vas deferens, spleen and aorta) or phenylephrine (prostate) were obtained in absence or presence of increasing concentrations of mirabegron (10–100 μM) preincubated for 45 min.
Schild analysis
The pA2 values for mirabegron were calculated by Schild regression analysis (Arunlakshana and Schild, 1959). The ratios between the half‐maximal concentrations of agonists (concentration ratios, CR) were calculated only when the maximal amplitude of the CRCs in the presence of the competitive antagonists was similar to that obtained in its absence. Data were plotted as log antagonist concentrations (M) versus log (CR ‐ 1). For calculation purposes, the slope parameter was constrained to 1.0 when statistically not different from unity, and pA2 was assumed as an estimate of mirabegron affinity. When an insufficient number of concentrations of mirabegron were tested, the pA2 was calculated using the following formula: pA2 = log (CR − 1) – log B, where B is the antagonist concentration and CR the agonist concentration ratio.
Cell culture, transfections and [3H]prazosin binding
HEK‐293 cells were kindly provided by Professor Maria de Fátima Magalhães Lazari (Section of Experimental Endocrinology, Department of Pharmacology, Escola Paulista de Medicina – UNIFESP). These cells were propagated in 100 mm dishes in DMEM with sodium pyruvate, supplemented with 10% heat inactivated FBS, 10 mg·mL−1 streptomycin and 100 U·mL−1 penicillin in a humidified atmosphere with 5% CO2. Cells were transfected with 15 μg of cDNA of the plasmid pDT, containing N‐terminal sequential hexahistidine and FLAG epitope‐tagged human α1A‐, α1B‐ or Δ1–79α1D‐adrenoceptors by Lipofectamin® (Invitrogen, Carlsbad, CA, USA), and stably transfected cells selected with geneticin (400 μg·mL−1). The α1D subtype is improperly folded and primarily found in the intracellular compartment, and due to this limitation, we used a truncated mutant in which the first N‐terminal extracellular 79 amino acids were deleted (Δ1–79α1D‐adrenoceptor). This form of the α1D‐adrenoceptor trafficks more efficiently towards the cell membrane and has pharmacological properties indistinguishable from the full‐length receptor in respect to key α1‐adrenoceptor ligands such as noradrenaline, prazosin and BMY7378 (Pupo et al., 2003; Hague et al., 2004; Nojimoto et al., 2010). The constructs were generously provided by Professor KP Minneman (Emory University, Atlanta, GA) and are described fully elsewhere (Vicentic et al., 2002; Pupo et al., 2003).
For radioligand binding measurements, confluent cultures of HEK‐293 cells in 100 mm plates were washed with PBS (20 mM NaPO4, 154 mM NaCl and pH 7.6) and harvested by scraping. HEK‐293 cells expressing recombinant human α1A‐, α1B‐ or Δ1–79α1D‐adrenoceptors were collected by centrifugation and homogenized with a Polytron. Membranes were collected by centrifugation at 30 000× g for 20 min and resuspended in buffer A (25 mM HEPES, 150 mM NaCl and pH 7.4). The content of the α1A‐, α1B‐ or Δ1–79α1D‐adrenoceptors in these membrane preparations were 1.2, 0.78 and 1.3 pmol·mg−1 protein respectively. The equilibrium dissociation constants (K i) for mirabegron were measured by displacement of specific binding of the α1‐adrenoceptor radioligand [3H]prazosin (350 pM) by mirabegron. Non‐specific binding was defined as binding in the presence of 100 μM phentolamine and ranged from ≅15 to 25% of the [3H]prazosin total binding. The final volume of the assays was 500 μL containing 10 μg of protein and 350 pM [3H]prazosin. Each data point was determined in duplicate and analysed by nonlinear regression analysis using the software package GraphPad Prism (version 5.00).
Quantitative real‐time PCR
For quantitative real‐time PCR and analysis of mRNA encoding β1‐ (Adrb1), β2‐ (Adrb2) and β3‐adrenoceptors (Adrb3), RNA was treated with DNAseI (Fermentas, Life Science, Hanover, MD, USA), and complementary DNA was synthesized from 1 μg RNA, using the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas), according to the manufacturer's protocol. Synthetic PCR primers were designed using the Primer Express software (Life Technologies, Carlsbad, CA, USA) and analysed by blast, to verify specificity, and Gene Runner, to evaluate the formation of structures such as hairpins and dimers. The primer efficiencies for Adrb1, Adrb2 and Adrb3 were 103, 83 and 100%, respectively, and were determined in the heart (Adrb1) and bladder (Adrb2 and Adrb3). Quantitative real‐time PCR was performed using SYBR Green quantification and employing the ABI StepOne Plus equipment (Life Technologies). Amplification specificity was verified using a dissociation curve, and the results were expressed by the difference between Ct values of chosen genes (Adrb1, Adrb2 and Adrb3) and the average of the housekeeping genes actin/GAPDH (deltaCt). The signal strength for actin (Ct: 21.39 ± 0.28) and GAPDH (Ct: 20.87 ± 0.37) did not differ.
Primer sequences employed for quantitative PCR:
| Gene | Primers | Optimal primer concentration (nM) |
|---|---|---|
| Adrb1_F | 5′‐CGA ATC ATC CGA GAC GTA CAG A‐3′ | 70 |
| Adrb1_R | 5′‐AGC CAT ACT AAG CCA CAC TCT CC‐3′ | |
| Adrb2_F | 5′‐AGC TGC AAA CAA GA G AGA GAA ACT‐3′ | 70 |
| Adrb2_R | 5′‐CAG ACA GAC AGA CAG ACT CAG TCC T‐3′ | |
| Adrb3_F | 5′‐ACA GGT TTG ATG GCT ATG AAG G‐3′ | 150 |
| Adrb3_R | 5′‐ATG GGG ATC AAG CAA GCT TC‐3′ | |
| Actb_F | 5′‐ACT GCC GCA TCC TCT TCC T‐3′ | 70 |
| Actb_R | 5′‐GAA CCG CTC GTT GCC AAT A‐3′ | |
| Gapdh_F | 5′‐TGC ACC ACC AAC TGC TTA‐3′ | 70 |
| Gapdh_R | 5′‐GGA TGC AGG GAT GAT GTT C‐3′ |
Determination of cAMP levels in mouse urethra
After animals were killed by CO2 asphyxiation, urethras were immediately excised and equilibrated for 45 min in bubbled KHS. All tissues were then incubated with IBMX (500 μM, 20 min). Urethral rings were stimulated with either mirabegron (10 μM, 3 min) or isoprenaline (10 μM, 3 min) pre‐incubated or not with L‐748,337 (30 μM, 30 min). Tissues were immediately frozen in liquid nitrogen, pulverized and subsequently processed for cAMP measurement using elisa immunoassay kit, according to the manufacturer's protocol (Cayman Chemical Cyclic AMP EIA kit, Ann Arbor, MI, USA). A pool of three urethras was used to constitute each experimental sample.
Data analysis of functional assays and statistical analysis
Data from the functional assays were analyzed by nonlinear regression using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA) to determine the pEC50. Concentration–response data was fitted to a log dose‐response function with variable slope in the form: E = Emax/([1 + (10c/10x)n] + F), where E is the maximum response produced by agonists; c is the logarithm of the EC50, the concentration of drug that produces a half‐maximal response; x is the logarithm of the concentration of the drug; the exponential term, n, is a curve fitting parameter that defines the slope of the concentration–response line, and F is the response observed in the absence of added drug. Concentration‐response data for the relaxation of mouse urethra by isoprenaline and mirabegron were also fitted according a biphasic dose‐response model using GraphPad Prism according the function E = Emax (Frac)/(1 + (10c’/10x’)n’) + Emax(1‐Frac)/(1 + (10c”‐10x”)n”) + F, where Frac is the proportion of maximal response due to the more potent phase, 10c’, 10x’ and n’ are the above described parameters for the first phase and 10c”, 10x” and n” for the second phase. Values of Emax are shown in mN (contraction protocols), whereas relaxing responses were calculated as percentages of the maximal changes from the steady‐state contraction produced by phenylephrine (10 μM) in each tissue (mouse urethra). The n H of the CRCs and slope of the regression line in the Schild plot are expressed as means with 95% confidence intervals. Otherwise, data are expressed as means ± SEM of the number of animals or cell membrane preparations. In the binding assays, each data point was determined in duplicate and analyzed by nonlinear regression analysis using the software package GraphPad Prism (version 5.00, San Diego, California, USA). The program Instat (GraphPad software) was used for statistical analysis. One‐way anova or unpaired Student's t‐test were used to assess the results. P < 0.05 was taken as showing a significant difference. F‐test was used to assess whether log dose‐response or biphasic dose‐response models better described the relaxations of the mouse urethra produced by mirabegron and isoprenaline and the results of the analysis are presented in the form of F value, the associated degrees of freedom (dfn, dfd) and P value.
Materials
(R)‐(−)phenylephrine hydrochloride, L‐(−)‐noradrenaline (+)‐bitartrate salt monohydrate, DL–isoprenaline hydrochloride (isoprenaline), [Arg8]‐vasopressin acetate salt, ICI‐118,551 hydrochloride ((±)‐1‐[2,3‐(dihydro‐7‐methyl‐1 H‐inden‐4‐yl)oxy]‐3‐[(1‐methylethyl)amino]‐2‐butanol hydrochloride), L‐748,337 (N‐(3‐{3‐[2‐(4‐benzenesulfonylamino‐phenyl)‐ethylamino]‐(2S)‐2‐hydroxy‐propoxy}‐benzyl)‐acetamide hydrate; ODQ (1 H‐[1,2,4] oxadiazolo[4,3‐a]quinoxalin‐1‐one), rolipram, yohimbine hydrochloride, (±)‐propranolol hydrochloride, β‐estradiol 3‐benzoate (17β‐estradiol), corticosterone, desipramine hydrochloride, idazoxan hydrochloride, phentolamine mesylate and IBMX were obtained from Sigma‐Aldrich Chem Co. (St. Louis, MO, USA). Mirabegron was purchased from Debye Scientific Co., Ltd. (Hong Kong, China). [3H]prazosin ([7‐methoxy‐3 H]prazosin) was obtained from Amersham Biosciences (GE Healthcare, Buckinghamshire, UK). Stock solutions of ODQ, rolipram, IBMX and mirabegron were prepared in DMSO as vehicle, and the final concentration of DMSO never exceeded 0.53% in the functional or biochemical assays. The other drugs were solubilized in deionized water. Drugs were stored in aliquots at −20 °C, and dilutions were prepared immediately before use.
Results
Mouse urethral relaxations induced by isoprenaline and mirabegron
In mouse urethral rings, phenylephrine (10 μM) induced sustained sub‐maximal contractions with amplitude of 2.1 ± 0.16 mN (n = 4). Cumulative addition of the non‐selective β‐adrenoceptor agonist isoprenaline (1 nM to 1 mM) to phenylephrine‐pre‐contracted tissues produced concentration‐dependent urethral relaxations that were best described by a biphasic dose–response model, rather than a log dose–response with variable slope model [Figure 1; F (dfn, dfd): 40.38 (3, 45); P < 0.0001]. The first relaxation phase was achieved between 1 nM and 1 μM (saturating at 67.5 ± 2.5% of the E max), which was followed by a second relaxation phase achieved between 10 μM and 1 mM (E max: 111.0 ± 1.9%). The estimated pEC50 values for the first and second components of this biphasic CRC were 7.10 ± 0.05 and 4.17 ± 0.06, respectively, whereas the n H were 1.03 (0.79–1.28) and 1.69 (1.02–2.35) for the first and second components respectively. Prior treatment of urethral preparations with the selective β3‐adrenoceptor antagonist L‐748,337 (30 μM, 30 min) converted this biphasic relaxant response to isoprenaline into a CRC best described by a log dose–response with variable slope model [Figure 1, n H = 0.84, 0.75–0.93; F (dfn, dfd): 1.767 (4, 42); P < 0.1536]. In the presence of a cocktail containing atenolol (10 μM), ICI‐118,551 (10 μM) and L‐748,337 (30 μM) preincubated for 30 min, the first relaxation phase was virtually abolished, whereas the second phase was little affected [Figure 1; n H = 1.25, 1.13–1.38; F (dfn, dfd): 2.975 (4, 43); P < 0.1138]. This indicates that mechanisms other than the activation of β‐adrenoceptors are involved in the relaxations of the mouse urethra produced by concentrations of isoprenaline higher than 3 μM.
Figure 1.
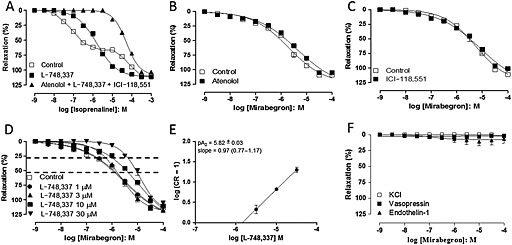
CRCs to isoprenaline and mirabegron in mouse isolated urethra smooth muscle. Responses to isoprenaline (A) were evaluated with or without either L‐748,337 (30 μM) alone or in combination with atenolol (10 μM) and ICI‐118,551 (10 μM). Responses to mirabegron were evaluated with or without atenolol (10 μM; B), ICI‐118,551 (10 μM; C) or L‐748,337 (1–30 μM; D). (D) Dotted lines indicate the urethral relaxations at the levels of 25% and 75%. (E) The Schild plot for L‐748,337 at mirabegron's pEC25. (F) CRCs to mirabegron in urethra pre‐contracted with KCl (80 mM), arginine‐vasopressin (60 nM) and endothelin‐1 (100 nM). Relaxations were calculated relative to the maximal changes from the contraction produced by phenylephrine (10 μM) in each urethral ring, which was taken as 100%. Data are presented as mean ± SEM (n = 3–4 different animals for each curve).
Cumulative addition of mirabegron (1 nM–100 μM) to mouse urethra pre‐contracted with phenylephrine produced concentration‐dependent relaxations. Although less clearly biphasic as the CRC for isoprenaline, the CRC for the relaxation induced by mirabegron was shallow and best described by a biphasic dose–response model rather than a log dose–response with variable slope model [Figure 1 (dfn, dfd): 6.968 (4, 37); P = 0.0003]. The first relaxation phase saturated at 32 ± 14% of the E max with pEC50 = 7.71 ± 0.43, whereas the second phase saturated at 110.9 ± 2.4% with pEC50 = 5.57 ± 0.08; the n H for the first and second components was 0.84 (0.72–2.41) and 1.22 (0.78–1.67) respectively. Prior incubation (30 min) with either the β1‐adrenoceptor antagonist atenolol (10 μM, Figure 1, n = 4) or the β2‐adrenoceptor antagonist ICI‐118,551 (10 μM, Figure 1, n = 5) failed to significantly affect mirabegron‐induced relaxations in mouse urethra. On the other hand, prior incubation with L‐748,337 (3–30 μM, but not 1 μM) produced non‐parallel concentration‐dependent rightward shifts in mirabegron‐induced relaxations, and these CRCs were best described by log dose–response model with variable slope (Figure 1; Table 1). However, it is noticeable that the relaxations produced by the lowest concentrations of mirabegron (1 nM to 0.3 μM, roughly the first phase of the CRC) were more effectively antagonized by L‐748,337 than the relaxations produced by concentrations of mirabegron corresponding to the second phase of the CRC (roughly 3–100 μM, Figure 1). Therefore, the potency of L‐748,337 in antagonizing the relaxations induced by mirabegron was determined at two arbitrarily chosen distinct levels of response, one corresponding to the first phase of the biphasic control CRC (pEC25) and the other corresponding to the midpoint of the second phase of the biphasic CRC (pEC75). In fact, the pA2 values for L‐748,337 derived from the rightward shifts at mirabegron's pEC75 were significantly lower than the pA2 values calculated from the rightward shifts produced at mirabegron's pEC25 (Table 2). The differences in the potencies of L‐748,337 in antagonizing the relaxations induced by ‘low’ and ‘high’ concentrations of mirabegron suggest that different relaxant mechanisms were operating in these two mirabegron concentration ranges.
Table 1.
Parameters of agonism for mirabegron in the mouse urethra (pEC50, E max and n H) in absence (control) and presence of L‐748,337 and fitting for log dose–response versus biphasic dose–response models
| L‐748,337 (μM) | |||||||
|---|---|---|---|---|---|---|---|
| 0 (Control) | 1 | 3 | 10 | 30 | |||
| Mirabegron | First phase | pEC50 | 7.14 ± 1.19 | 6.94 ± 0.97 | 5.66 ± 0.05 | 4.99 ± 0.12 | 4.92 ± 0.06 |
| Saturation (%E max) | 39 ± 31 | 35 ± 0.29 | 121.6 ± 2.6 | 135.4 ± 9.0 | 122.2 ± 4.0 | ||
| n H | 0.955–2.094 | 0.76–1.07 | 0.52–0.70 | 0.85–1.40 | |||
| Second phase | pEC50 | 5.40 ± 0.12 | 5.40 ± 0.14 | ||||
| E max | 118.3 ± 3.3 | 116.7 ± 3.2 | |||||
| n H | 0.44–1.86 | 0.51–1.77 | |||||
| Fitting comparison | F (dfn, dfd) | Biphasic 6.360 (3, 92) | Biphasic 5.034 (3, 37) | Monophasic n.d. | Monophasic n.d. | Monophasic 0.7760 (3, 44) | |
| P‐value | 0.0006 | 0.0050 | 0.5137 | ||||
n.d., not determined, ambiguous fit to the biphasic model.
Table 2.
Potencies of mirabegron (pEC75 and pEC25) in absence (control) and presence of L‐748,337 and the respective pA2 values and n H of the CRCs
| L‐748,337 | |||||
|---|---|---|---|---|---|
| 0 (Control) | 3 μM (pA2) | 10 μM (pA2) | 30 μM (pA2) | ||
| Mirabegron | pEC75 | 5.27 ± 0.10 | 5.37 ± 0.05 (n.d.) | 4.81 ± 0.13 (5.28 ± 0.10) | 4.76 ± 0.07 (5.05 ± 0.07) |
| pEC25 | 6.35 ± 0.07 | 6.18 ± 0.05 (5.83 ± 0.07) | 5.59 ± 0.07 (5.82 ± 0.04 * ) | 5.33 ± 0.07 (5.80 ± 0.04 * ) | |
| n H | 0.54 (0.47–0.60) | 0.79 (0.56–0.94) | 0.61 (0.52–0.70) | 1.12 (0.85–1.40) | |
P < 0.01, significantly different from the respective pA2 value calculated at mirabegron pEC75.
In tissues pre‐contracted with KCl (80 mM, n = 4), arginine‐vasopressin (60 nM, n = 4) or endothelin‐1 (100 nM, n = 3–4), mirabegron (1 nM to 0.3 mM) failed to produce significant urethral relaxations (E max: 1.0 ± 0.9%, 2.3 ± 2.2% and 5.7 ± 5.6%, respectively, Figure 1).
Cyclic AMP production and involvement of the NO/cGMP signalling pathway in mirabegron‐induced relaxations of mouse urethra
Rolipram (10 μM) was incubated with the urethral rings for 30 min, after which preparations were pre‐contracted with phenylephrine. At this concentration, rolipram affected neither the basal tone of the preparations (not shown) nor that induced by phenylephrine (1.70 ± 0.20 and 1.47 ± 0.13 mN, n = 4). However, rolipram significantly increased the mirabegron‐induced relaxations, as observed at the E max level (P < 0.05; Figure 2). The pEC50 did not significantly change between control and rolipram‐treated preparations (5.53 ± 0.11 and 5.45 ± 0.16 for control and rolipram; n = 4). Likewise, co‐incubation of a threshold dose of mirabegron (0.1 and 1 μM) enhanced by 29.7% (P < 0.05) and 75.8% (P < 0.01) the rolipram (1 μM)‐induced urethral relaxations (30.6 ± 3.0, 39.8 ± 2.2 and 53.8 ± 3.5 for rolipram alone, and rolipram plus mirabegron at 0.1 and 1 μM respectively; n = 5–6; Figure 2).
Figure 2.
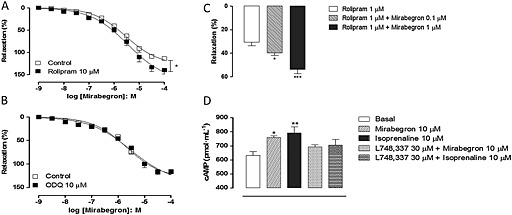
Concentration–response curves to mirabegron in mouse isolated urethra smooth muscle and cyclic AMP production. Relaxant responses were measured with or without either rolipram (10 μM; A) or ODQ (10 μM; C). Responses to rolipram (1 μM) were measured in the presence of threshold concentrations of mirabegron (0.1 and 1 μM; B). Cyclic AMP production in mirabegron and isoprenaline‐stimulated urethra in the presence of L‐748,337 is shown in (D). Relaxations were calculated relative to the maximal changes from the contraction produced by phenylephrine (10 μM) in each urethral ring, which was taken as 100%. Data are presented as mean ± SEM. (n = 4–6). * P < 0.05, ** P < 0.01; significantly different from corresponding respective control/basal; anova followed by Tukey's test.
Stimulation of urethral rings with either mirabegron (10 μM, 3 min) or isoprenaline (10 μM, 3 min) significantly elevated (P < 0.05) the cAMP levels above basal levels (Figure 2; n = 5–6). Pre‐incubation of urethra with L‐748,337 (30 μM, 30 min) fully prevented the increases in cAMP produced by isoprenaline and mirabegron (Figure 2).
Relaxations of vascular smooth muscle in response to β3‐adrenoceptor activation may be coupled to the NO/cGMP pathway (Flacco et al., 2013). Therefore, CRCs for mirabegron were also carried out in the presence of the soluble GC inhibitor ODQ (10 μM, 20 min, n = 4–5). Figure 2 shows that ODQ failed to affect mirabegron‐induced urethral relaxations, as evaluated at the level of pEC50 (5.67 ± 0.10 and 5.52 ± 0.14 for untreated and treated preparations, respectively) and E max values (115 ± 6.4% and 116 ± 4.3% for untreated and treated preparations respectively).
Phenylephrine‐induced contractions of mouse urethra
Cumulative addition of phenylephrine (0.01–300 μM) produced concentration‐dependent contractions in isolated urethra rings, achieving maximal responses at 100 μM (Figure 3). Prior incubation of urethra (30 min) with mirabegron (0.1–10 μM, n = 4) produced concentration‐dependent rightward shifts in the phenylephrine‐induced contractions (pEC50: 5.57 ± 0.03, 5.48 ± 0.07, 5.08 ± 0.03 and 4.74 ± 0.07 for control and mirabegron at 0.1, 1 and 10 μM respectively; P < 0.001 for mirabegron 1 and 10 μM when compared with control group). However, Schild analysis showed that mirabegron presented a complex behaviour, as the slope in the Schild plot was much lower than 1.0 (slope = 0.63, 0.47–0.80, Figure 3).
Figure 3.
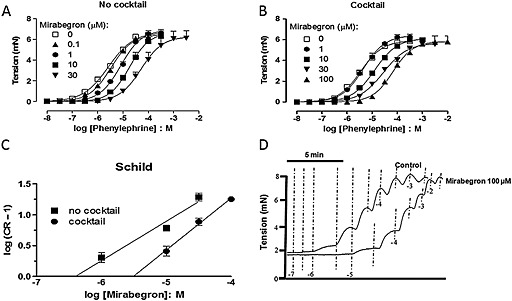
Concentration–response curves to phenylephrine (PE) in mouse isolated urethra with or without different concentrations of mirabegron. Urethral preparations were not pretreated (A) or pretreated (B) with a cocktail of inhibitors containing yohimbine (100 nM), propranolol (100 nM), L‐748,337 (10 μM), 17β‐estradiol (10 μM) and desipramine (100 nM). (C) Schild plots for mirabegron with and without the cocktail of inhibitors. (D) Representative original traces for PE‐induced urethral contractions in a preparation pretreated with the cocktail of inhibitors. Data are presented as mean ± SEM (n = 4–6).
In a separate set of experiments, urethral rings were incubated with a cocktail of inhibitors containing yohimbine (100 nM), propranolol (100 nM), L‐748,337 (10 μM), 17β‐estradiol (10 μM) and desipramine (100 nM). CRCs to phenylephrine did not significantly change in comparison with the preparations in the absence of this inhibitor cocktail (Figure 3). In addition, mirabegron (10–100 μM, n = 4) produced similar concentration‐dependent rightward shifts in the phenylephrine‐induced contractions with no changes in the maximal response (Figure 3; Table 3). The Schild analysis showed that mirabegron behaved as a competitive antagonist of α1‐adrenoceptors with slope in the Schild plot not different from theoretical unity (0.84, 0.65–1.03 and pA2 = 5.34 ± 0.04, Figure 3).
Table 3.
Maximal responses (E max) and potency (pEC50) values derived from concentration–response curves to phenylephrine (0.1 μM to 10 mM) in mouse isolated urethral preparations in the absence and in the presence of mirabegron (1–100 μM)
| Groups | E max (mN) | pEC50 |
|---|---|---|
| Control | 5.8 ± 0.3 | 5.45 ± 0.05 |
| Mirabegron 1 μM | 6.2 ± 0.2 | 5.34 ± 0.03 |
| Mirabegron 10 μM | 5.9 ± 0.1 | 4.90 ± 0.02 *** |
| Mirabegron 30 μM | 5.6 ± 0.1 | 4.55 ± 0.03 *** |
| Mirabegron 100 μM | 5.8 ± 0.5 | 4.17 ± 0.06 *** |
Urethral preparations were pretreated (30 min) with a cocktail of inhibitors containing yohimbine (100 nM), propranolol (100 nM), L‐748,337 (10 μM), 17β‐estradiol (10 μM) and desipramine (100 nM).
P < 0.001, significantly different from control group; anova followed by Tukey's test. Data are presented as mean ± SEM of four experiments.
Mirabegron (10 μM, 30 min) did not affect the urethral contractions induced by arginine‐vasopressin (0.1 nM to 1 μM), as evaluated at the level of both pEC50 (7.66 ± 0.04 and 7.66 ± 0.08 in absence and presence of mirabegron, respectively) and E max (5.64 ± 0.3 and 5.88 ± 0.5 mN in absence and presence of mirabegron, respectively; n = 4). Mirabegron (10 μM, 30 min) also failed to affect the urethral contractions induced by KCl (80 mM; 4.46 ± 0.34 and 4.74 ± 0.09 mN; n = 4–5).
Selectivity of mirabegron for α1‐adrenoceptor subtypes of the rat vas deferens, prostate, spleen and aorta
Contractions induced by noradrenaline and phenylephrine in the rat vas deferens/prostate, spleen and aorta are mediated predominantly by activation of α1A‐, α1B‐ and α1D‐adrenoceptors respectively (Aboud et al., 1993; Marshall et al., 1996; Nojimoto et al., 2010). Therefore, we have used these rat preparations to evaluate the potential antagonism of α1A‐, α1B‐ and α1D‐adrenoceptors by mirabegron.
In the rat vas deferens and prostate (α1A‐adrenoceptor), mirabegron (10–100 μM) produced concentration‐dependent rightward shifts with a slope in the Schild plot not different from theoretical unity (vas deferens 1.01, 0.76–1.25, and prostate 0.91, 0.69–1.13, Figure 4; n = 6). Likewise, in the rat aorta (α1D‐adrenoceptor), mirabegron displaced to the right the CRC to noradrenaline with a slope in the Schild plot not different from theoretical unity (1.08, 0.66–1.50, Figure 4; n = 4–8). The pA2 values estimated for mirabegron in the vas deferens (5.64 ± 0.05), prostate (5.55 ± 0.04) and aorta (5.51 ± 0.07) were not different from the values found in urethra (pA2 = 5.35). On the other hand, the noradrenaline‐induced contractions in the rat spleen (α1B‐adrenoceptor) remained unchanged by mirabegron (up to 100 μM, Figure 4).
Figure 4.

Concentration–response curves to noradrenaline or phenylephrine (PE) in absence and presence of increasing concentrations of mirabegron (10–100 μM) in rat isolated vas deferens (A), prostate (B), aorta (C) and spleen (D) preparations. (E) The Schild plots for mirabegron in vas deferens, prostate and aorta. (F) Competition for the specific binding of [3H]prazosin by mirabegron in membrane preparations from HEK‐293 cells expressing α1A‐, α‐ and Δ1–79α1D‐adrenoceptors. Data are presented as mean ± SEM (n = 4–6).
Selectivity of mirabegron for recombinant α1‐adrenoceptor subtypes in HEK‐293 cells
The competition for the specific binding of [3H]prazosin by mirabegron in membrane preparations from HEK‐293 cells expressing α1A‐, α1B‐ and Δ1‐79α1D‐adrenoceptors is shown in Figure 4. Mirabegron presented much higher affinities for α1A‐ (pK i = 6.36 ± 0.17, n = 3,) and Δ1–79α1D‐adrenoceptors (pK i = 5.74 ± 0.16, n = 3), than for α1B‐adrenoceptors (pK i = 4.59 ± 0.11, n = 3).
MRNA expression for β‐adrenoceptors in mouse urethral smooth muscle
The quantitative real‐time PCR (ΔCt) for β1, β2 and β3‐adrenoceptors was, respectively, 10.6 ± 0.44, 5.7 ± 0.14 and 11.9 ± 0.47.
Discussion
Mirabegron has been successfully introduced in clinical practice as the first β3‐adrenoceptor agonist for OAB treatment (Chapple et al., 2013). In the present study, we have identified β3‐adrenoceptors in the mouse urethral smooth muscle, the activation of which resulted in efficient in vitro urethral relaxations. Mirabegron also unexpectedly behaved as a competitive α1‐adrenoceptor antagonist, presenting higher affinities for rat α1A‐ (vas deferens and prostate) and α1D‐adrenoceptors (aorta), which was confirmed in radioligand binding with human recombinant α1A‐ and α1D‐adrenoceptors.
Activation of the sympathetic nervous system in the urinary bladder is well known to contribute to urine storage by relaxing the detrusor muscle via activation of β2‐ and β3‐adrenoceptors (Yamazaki et al., 1998; Wuest et al., 2009). However, much less information exists on the presence and functional importance of β3‐adrenoceptors in the urethral smooth muscle. Coordinated contraction and relaxation of the urethra smooth muscle are essential for maintaining continence and effective voiding, and an altered function of the bladder neck and urethra may lead to inappropriate or un‐coordinated functions of the bladder outlet (Michel and Vrydag, 2006). The importance of the β3‐adrenoceptors was first shown by Takeda et al. (2003) and Yamanishi et al. (2003) who demonstrated the ability of the β3‐adrenoceptor agonists CL316243 or BRL37344 to produce, in vitro, urethral relaxations in rat, dog and guinea pig preparations, with little information on β‐adrenoceptors in the mouse urethra. In the present study, the non‐selective β‐adrenoceptor agonist isoprenaline clearly produced a biphasic pattern of relaxation in the mouse isolated urethra, which turned to be a monophasic curve in the presence of a high concentration of the selective β3‐adrenoceptor antagonist L‐748,337 (or in the presence of a cocktail of antagonists containing atenolol, ICI‐118,551 and L‐748,337). Apart from implying that a heterogeneous population of β‐adrenoceptor subtypes mediated the mouse urethral relaxations, these results also indicated that mechanisms other than the agonism of β‐adrenoceptors were involved in the relaxation produced by isoprenaline in the mouse urethra pre‐contracted with phenylephrine. It is known that isoprenaline in concentrations higher than 1 μM interacts with α1‐adrenoceptors, usually behaving as a weak partial agonist (Trendelenburg, 1974), and this may explain why the second, low potency phase of the CRC for isoprenaline was little affected by β‐adrenoceptor antagonists. In addition, mirabegron produced a full relaxant response in the mouse urethra that was unaffected by atenolol (selective β1‐adrenoceptor antagonist) and ICI‐118,551 (selective β2‐adrenoceptor antagonist) but was instead displaced to the right by the selective β3‐adrenoceptor antagonist L‐748,337. However, it is important to note that the CRCs for mirabegron in the relaxation of mouse urethra pre‐contracted with phenylephrine were very shallow (n H lower than 1.0) and that L‐748,337 was more effective in antagonizing the effects produced by concentrations of mirabegron up to 1 μM than those produced by higher concentrations. This indicated that different mechanisms might be responsible for the relaxations produced by ‘low’ (<1 μM) and high concentrations of mirabegron (>1 μM).
The potency of L‐748,337 against mirabegron‐induced relaxations of mouse urethra (pA2 ≈ 5.8) was relatively low but consistent with the broad range of reported potency/affinity values at rodent β3‐adrenoceptors determined in functional studies or radioligand binding assays (pK B = 5.4–7.95, Deba et al., 2009; Palea et al., 2012; van Wieringen et al. 2013; pK i = 6.5, Candelore et al., 1999) but was significantly lower than the reported potencies/affinities at the human β3‐adrenoceptor (pK B = 7.6–9.5, Sato et al., 2008; Wuest et al., 2009; D'Agostino et al., 2015; pK i = 8.4–8.6, Candelore et al., 1999; van Wieringen et al., 2013), indicating that there are important pharmacological differences depending both on the species investigated and on the method employed in the quantification of affinity of L‐748,337 (i.e. radioligand binding vs. functional assays). Actually, a recent study revised the usefulness of the pharmacological tools currently available for β3‐adrenoceptors and pointed that although L‐748,337 remains the most suitable among the selective β3‐adrenoceptor antagonists, this ligand presents considerably lower affinities for rodent β3‐adrenoceptors than for the human receptor (Cernecka et al., 2014).
To assess the presence of β‐adrenoceptor in the mouse urethral tissue, we processed real‐time PCR analysis and identified similar levels of mRNA encoding β1‐, β2‐ and β3‐adrenoceptors in this tissue. This finding is consistent with our functional data showing rightward shifts for isoprenaline‐induced relaxations in the presence of selective antagonists for these receptors (atenolol, ICI‐118,551 and L‐748,337 respectively). The classical signalling pathway of β‐adrenoceptor, including the β3‐adrenoceptor subtype, is activation of adenylyl cyclase with consequent generation of cAMP, which is inactivated by hydrolysis to AMP, by PDE4 (Hatanaka et al., 2013). However, no previous study has evaluated the role of cAMP signalling in modulation of urethral relaxations. Therefore, to evaluate the cAMP pathway, we conducted relaxation curves in mouse urethra with the PDE4 inhibitor rolipram and measured the urethral cAMP production. In fact, mirabegron increased cAMP production in mouse urethra, and this effect was prevented by L‐748,337, indicating involvement of β3‐adrenoceptors. However, incubation with the PDE4 inhibitor rolipram produced only a small (but significant) increase of mirabegron‐induced relaxations of mouse urethra, suggesting that cAMP plays a minor role in this effect. Interestingly, in the rat bladder under no pre‐contraction, selective β2‐ and β3‐adrenoceptor activation produced concentration‐dependent relaxations that were accompanied by concomitant increases of cAMP levels; however, in KCl‐pre‐contracted tissues, the bladder relaxations did not correlate linearly with the cAMP production (Uchida et al., 2005). It has been proposed that calcium‐dependent potassium channels account for the β‐adrenoceptor activation in bladders pre‐contracted with KCl (Frazier et al., 2005).
NO released by the nitrergic fibers is the main neurotransmitter involved in urethral relaxations during the voiding phase. The target of NO is the enzyme soluble GC, which catalyses the conversion of GTP to the intracellular second messenger cGMP, mediating NO‐induced relaxations. Given that β3‐adrenoceptors may be coupled to NO release in vascular (Graves and Poston, 1993; Flacco et al., 2013) and bladder smooth muscle (Birder et al., 2002), we next examined the potential involvement of NO in the mirabegron‐induced relaxations. Preincubation with the soluble GC inhibitor ODQ failed to affect mirabegron‐induced relaxations, excluding the involvement of the cGMP pathway. At the concentration used, ODQ markedly reduced the mouse urethral relaxations induced by the NO donor compounds sodium nitrite, S‐nitrosoglutathione and glyceryl trinitrate (Alexandre et al., 2014).
In the lower urinary tract, α1‐adrenoceptor subtypes have been identified in bladder neck, urethra and prostate. In human proximal urethra smooth muscle, the mRNA for α1A‐adrenoceptors accounts for 90–100% and that for α1D‐adrenoceptors for 10%, whereas the mRNA for the α1B‐adrenoceptors was not detectable (Nasu et al., 1998). In the rat urethra, there are all three α1‐adrenoceptor subtypes, with the α1A‐adrenoceptor mRNA as the predominant gene transcript (Yono et al., 2006). Activation of α1‐adrenoceptor during the storage phase contributes to the bladder outlet resistance, a mechanism greatly increased in elderly men with enlarged prostates (Michel and Vrydag, 2006). As discussed above, mirabegron produces mouse urethral relaxations with high efficacy in tissues pre‐contracted with the α1‐adrenoceptor agonist phenylephrine. Our findings that mirabegron failed to promote efficient relaxation of urethral preparation when tissues are pre‐contracted with the peptide arginine‐vasopressin or KCl prompted us to hypothesize that mirabegron‐induced urethral relaxations were due, at least in part, to α1‐adrenoceptor antagonism. Mirabegron concentration dependently produced rightward shifts in the CRC to phenylephrine in mouse urethra with no modifications of the maximal responses, but the behaviour presented by mirabegron was not consistent with simple competitive antagonism, as the slope in the Schild plot was much lower than the theoretical unity. In separate urethral preparations, we repeated the CRCs to phenylephrine in the presence of a cocktail of inhibitors aimed to isolate the α1‐adrenoceptor and to block the extraneuronal and neuronal monoamine uptake systems (yohimbine, propranolol, L‐748,337, 17β‐estradiol and desipramine). Similarly, mirabegron markedly displaced to the right the CRCs to phenylephrine, and the Schild analysis revealed a slope not different from theoretical unity in tissues incubated with the cocktail of inhibitors, supporting competitive antagonism of α1‐adrenoceptor by mirabegron in the mouse urethra. Interestingly, mirabegron was about three times more potent to shift the phenylephrine‐induced CRC to the right in the absence (pA2 = 5.92), compared with its potency on preparations treated with the cocktail of inhibitors (pA2 = 5.40). This finding is likely to reflect the two combined actions of mirabegron, that is, (i) functional antagonism resulting from β3‐adrenoceptor‐induced relaxations counteracting the urethral contractions and (ii) competitive antagonism due to α1‐adrenoceptor blockade. In the presence of the cocktail of inhibitors, which contains the β3‐adrenoceptor antagonist L‐748,337, the rightward displacement by mirabegron would be solely produced by the α1‐adrenoceptor blockade explaining the lower potency for mirabegron in the antagonism of contractions induced by phenylephrine. Consistent with this explanation is the fact that the Schild plot for mirabegron in the absence of the cocktail yields a slope much lower than unity, whereas in the presence of the cocktail, the slope did not differ from the theoretical unity predicted for competitive antagonism.
We next used the rat vas deferens, spleen and aorta to investigate the α1‐adrenoceptor subtypes antagonized by mirabegron. The full α1‐adrenoceptor agonists noradrenaline and phenylephrine contract these three tissues predominantly through α1A‐, α1B and α1D‐adrenoceptors respectively (Minneman et al., 1994; Taniguchi et al., 1999; Lima et al., 2005). The α1A‐adrenoceptor is also the predominant adrenoceptor subtype in the rat and human prostate smooth muscle, which is why we used the prostate preparations in our assays (Nasu et al., 1998; Yono et al., 2006). In our study, mirabegron produced concentration‐dependent rightward shifts in the rat vas deferens, prostate and aorta (but not spleen) with a slope in the Schild plot not different from theoretical unity, confirming that mirabegron selectively antagonizes α1A‐ and α1D‐adrenoceptors but not α1B‐adrenoceptors. In addition, mirabegron displaced [3H]prazosin binding to membrane preparations from HEK‐293 cells transfected with each of the recombinant human α1‐adrenoceptor subtypes presenting affinities consistent with those estimated in functional assays further supporting the competitive nature of the interaction between mirabegron and α1A‐ and α1D‐adrenoceptors. We were surprised by the paucity of information in the literature related to the affinities of mirabegron for secondary targets other than β1‐ and β2‐adrenoceptors, and after extensive search, we could find only preliminary indications that mirabegron interacted with rat recombinant α1A‐adrenoceptors, showing micromolar affinity in evidence submitted to a regulatory agency (pages 33 and 34 of the document available in the Department of Health and Human Services, U.S. Food and Drug Administration, 2012). Interestingly, a similar pharmacological profile was described for nebivolol, which acted as a β3‐adrenoceptor agonist and α1D‐adrenoceptor antagonist in the rat thoracic aorta (Rozec et al., 2006).
The LUTS in women has usually been equated to the OAB and assumed to be caused by detrusor overactivity, whereas in men, it typically occurs in association with bladder outlet obstruction secondary to benign prostatic hyperplasia (Andersson and Chapple, 2014). In ageing men, currently available therapies for LUTS have been focused on reducing the tension of the prostate smooth muscle against the urethra, thus including α1‐adrenoceptor antagonists, 5α‐reductase inhibitors and PDE5 inhibitors (Bechara et al., 2008; McVary et al., 2011; Yan et al., 2014). A combination of mirabegron with the α1‐adrenoceptor antagonist tamsulosin was more effective and safe than tamsulosin monotherapy for patients with benign prostatic obstruction and OAB (Ichihara et al., 2015). Mirabegron has also recently reported to produce potent human and rabbit prostatic relaxations (Calmasini et al., 2015).
Whether this novel property of mirabegron, that is, competitive antagonism of α1A‐ and α1D‐adrenoceptors, is relevant or not for its therapeutic efficacy obviously depends on the doses administered to the patient. Multiple 50 mg doses of mirabegron (approved dose level) to humans result in C max values from 20 to 60 ng·mL−1, leading to plasma concentrations in the range of 60–150 nM, of which ≅70% is bound to plasma proteins (Iitsuka et al., 2014, 2015), leaving a free plasma concentration of ≅20 to 50 nM. Pre‐clinical pharmacokinetic studies in rats after a single oral dose of 14C‐mirabegron showed that the tissue : plasma radioactivity ratios are highest in the alimentary canal and excretory organs and that in some cases, upon repeated doses, the tissue : plasma ratios nearly reached 20‐fold (Department of Health and Human Services, U.S. Food and Drug Administration, 2012; Department of Health Therapeutic Goods Administration, Australian Public Assessment Report, 2014). As mirabegron has a large volume of distribution at steady state (Vdss: 1670 L), it is possible that its levels in the excretory organs such as bladder and urethra exceeds by far that of plasma and that the effective concentrations of mirabegron at the receptor biophase might approximate those required to occupy a significant fraction of α1‐adrenoceptors. Actually, it seems that the accumulation of mirabegron in tissues owing to the highly lipophilic nature of the molecule is a fundamental condition to be assumed; otherwise, it is difficult to reconcile the relatively low potencies of mirabegron in the relaxations of rodent and human bladder (EC50s from ≈780 to 5100 nM; Takasu et al., 2007; Hatanaka et al., 2013; Svalø et al., 2013; Michel, 2014), unless we assume that its therapeutic efficacy results from relaxations produced by concentrations as low as 50 nM, which are nearly ineffective in vitro.
In conclusion, this study shows that in addition to its major β3‐adrenoceptor agonism promoting urethral relaxations, mirabegron exhibits selective α1A‐ and α1D‐adrenoceptor antagonism. Our study may be important not only for human therapeutics by revealing a novel property of mirabegron but also as cautionary findings for pre‐clinical studies by guiding the choice of appropriate agonists to contract the tissues that will be relaxed by mirabegron.
Author contributions
E. C. A., F. B. C., L. R. K., K. P. S., F. H. S., C. A. R. and R. F. performed the research. E. C. A., A. S. P., L. R. K. and E. A. designed research study. A. S. P., F. Z. M. and E. A. contributed essential reagents or tools. E. C. A., A. S. P., L. R. K., F. Z. M. and E. A. analysed the data. E. C. A., A. S. P. and E. A. wrote the paper.
Conflict of interest
The authors state no conflict of interest.
Acknowledgements
Eduardo Costa Alexandre, André S. Pupo and Edson Antunes are grateful to Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2014/02196‐2 to E. C. A and E. A and FAPESP 08/50423‐7 to A. S. P) for financial support.
Alexandre, E. C. , Kiguti, L. R. , Calmasini, F. B. , Silva, F. H. , da Silva, K. P. , Ferreira, R. , Ribeiro, C. A. , Mónica, F. Z. , Pupo, A. S. , and Antunes, E. (2016) Mirabegron relaxes urethral smooth muscle by a dual mechanism involving β3‐adrenoceptor activation and α1‐adrenoceptor blockade. British Journal of Pharmacology, 173: 415–428. doi: 10.1111/bph.13367.
References
- Aboud R, Shafii M, Docherty JR (1993). Investigation of the subtypes of alpha 1‐adrenoceptor mediating contractions of rat aorta, vas deferens and spleen. Br J Pharmacol 109: 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams P, Cardozo L, Fall M, Griffiths D, Rosier P, Ulmsten U et al. (2002). The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub‐committee of the International Continence Society. NeurourolUrodyn 21: 167–178. [DOI] [PubMed] [Google Scholar]
- Abrams P, Chapple C, Khoury S, Roehrborn C, de la Rosette J (2013). Evaluation and treatment of lower urinary tract symptoms in older men. J Urol 189: S93–S101. [DOI] [PubMed] [Google Scholar]
- Alexandre EC, Leiria LO, Silva FH, Mendes‐Silverio CB, Calmasini FB, Davel AP et al. (2014). Soluble guanylyl cyclase (sGC) degradation and impairment of nitric oxide‐mediated responses in urethra from obese mice: reversal by the sGC activator BAY 60–2770. J Pharmacol Exp Ther 349: 2–9. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KE, Chapple CR (2014). Current pharmacotherapy of lower urinary tract symptoms. Surgery (Oxford) 32: 292–296. [Google Scholar]
- Arunlakshana O, Schild HO (1959). Some quantitative uses of drug antagonists. Br J Pharmacol Chemother 14: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A, Romano S, Casabe A, Haime S, Dedola P, Hernandez C et al. (2008). Comparative efficacy assessment of tamsulosin vs. tamsulosin plus tadalafil in the treatment of LUTS/BPH. Pilot study. J Sex Med 5: 2170–2178. [DOI] [PubMed] [Google Scholar]
- Birder LA, Nealen ML, Kiss S, de Groat WC, Caterina MJ, Wang E et al. (2002). Beta‐adrenoceptor agonists stimulate endothelial nitric oxide synthase in rat urinary bladder urothelial cells. J Neurosci 22: 8063–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G (2014). Purinergic signaling in the urinary tract in health and disease. Purinergic Signal 10: 103–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmasini FB, Candido TZ, Alexandre EC, D'Ancona CA, Silva D, de Oliveira MA et al. (2015). The beta‐3 adrenoceptor agonist, mirabegron relaxes isolated prostate from human and rabbit: new therapeutic indication? Prostate 75: 440–447. [DOI] [PubMed] [Google Scholar]
- Candelore MR, Deng L, Tota L, Guan XM, Amend A, Liu Y et al. (1999). Potent and selective human beta(3)‐adrenergic receptor antagonists. J Pharmacol Exp Ther 290: 649–655. [PubMed] [Google Scholar]
- Cernecka H, Sand C, Michel MC (2014). The odd sibling: features of β3‐adrenoceptor pharmacology. Mol Pharm 86: 479–484. [DOI] [PubMed] [Google Scholar]
- Chapple CR, Cardozo L, Nitti VW, Siddiqui E, Michel MC (2014). Mirabegron in overactive bladder: a review of efficacy, safety, and tolerability. NeurourolUrodyn 33: 17–30. [DOI] [PubMed] [Google Scholar]
- Chapple CR, Kaplan SA, Mitcheson D, Klecka J, Cummings J, Drogendijk T et al. (2013). Randomized double‐blind, active‐controlled phase 3 study to assess 12‐month safety and efficacy of mirabegron, a beta(3)‐adrenoceptor agonist, in overactive bladder. Eur Urol 63: 296–305. [DOI] [PubMed] [Google Scholar]
- D'Agostino G, Condino MA, Calvi P (2015). Involvement of β3‐adrenoceptors in the inhibitory control of cholinergic activity in human bladder: direct evidence by [(3)H]‐acetylcholine release experiments in the isolated detrusor. Eur J Pharmacol 758: 115–122. [DOI] [PubMed] [Google Scholar]
- Deba A, Palea S, Rouget C, Westfall TD, Lluel P (2009). Involvement of beta (3)‐adrenoceptors in mouse urinary bladder function: role in detrusor muscle relaxation and micturition reflex. Eur J Pharmacol 618: 76–83. [DOI] [PubMed] [Google Scholar]
- Department of Health and Human Services, U.S. Food and Drug Administration . 2012. Pharmacology/Toxicology NDA/BLA Review and Evaluation (NDA 202–611). Available at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202611Orig1s000PharmR.pdf (accessed 6/8/2015).
- Department of Health Therapeutic Goods Administration, Australian Public Assessment Report . 2014. (199664 and 199668). Available at: https://www.tga.gov.au/file/1307/download (accessed 6/8/2015).
- Flacco N, Segura V, Perez‐Aso M, Estrada S, Seller JF, Jiménez‐Altayó F et al. (2013). Different β‐adrenoceptor subtypes coupling to cAMP or NO/cGMP pathways: implications in the relaxant response of rat conductance and resistance vessels. Br J Pharmacol 169: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier EP, Mathy MJ, Peters SL, Michel MC (2005). Does cyclic AMP mediate rat urinary bladder relaxation by isoproterenol? J Pharmacol Exp Ther 313: 260–267. [DOI] [PubMed] [Google Scholar]
- Fujimura T, Tamura K, Tsutsumi T, Yamamoto T, Nakamura K, Koibuchi Y et al. (1999). Expression and possible functional role of the beta3‐adrenoceptor in human and rat detrusor muscle. J Urol 161: 680–685. [PubMed] [Google Scholar]
- Graves J, Poston L (1993). Beta‐adrenoceptor agonist mediated relaxation of rat isolated resistance arteries: a role for the endothelium and nitric oxide. Br J Pharmacol 108: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hague C, Chen Z, Pupo AS, Schulte NA, Toews ML, Minneman KP (2004). The N terminus of the human alpha1D‐adrenergic receptor prevents cell surface expression. J Pharmacol Exp Ther 309: 388–397. [DOI] [PubMed] [Google Scholar]
- Hatanaka T, Ukai M, Watanabe M, Someya A, Ohtake A, Suzuki M et al. (2013). In vitro and in vivo pharmacological profile of the selective beta3‐adrenoceptor agonist mirabegron in rats. Naunyn Schmiedebergs Arch Pharmacol 386: 247–253. [DOI] [PubMed] [Google Scholar]
- Hegde SS, Choppin A, Bonhaus D, Briaud S, Loeb M, Moy TM et al. (1997). Functional role of M2 and M3 muscarinic receptors in the urinary bladder of rats in vitro and in vivo . Br J Pharmacol 120: 1409–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara K, Masumori N, Fukuta F, Tsukamoto T, Iwasawa A, Tanaka Y (2015). A randomized controlled study to evaluate the efficacy of tamsulosin monotherapy and its combination with mirabegron in patients with overactive bladder induced by benign prostatic obstruction. J Urol 193: 921–926. [DOI] [PubMed] [Google Scholar]
- Igawa Y, Yamazaki Y, Takeda H, Hayakawa K, Akahane M, Ajisawa Y et al. (1999). Functional and molecular biological evidence for a possible beta3‐adrenoceptor in the human detrusor muscle. Br J Pharmacol 126: 819–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igawa Y, Yamazaki Y, Takeda H, Kaidoh K, Akahane M, Ajisawa Y et al. (2001). Relaxant effects of isoproterenol and selective beta3‐adrenoceptor agonists on normal, low compliant and hyperreflexic human bladders. J Urol 165: 240–244. [DOI] [PubMed] [Google Scholar]
- Irwin DE, Kopp ZS, Agatep B, Milsom I, Abrams P (2011). Worldwide prevalence estimates of lower urinary tract symptoms, overactive bladder, urinary incontinence and bladder outlet obstruction. BJU Int 108: 1132–1138. [DOI] [PubMed] [Google Scholar]
- Iitsuka H, Tokuno T, Amada Y, Matsushima H, Katashima M, Sawamoto T et al. (2014). Pharmacokinetics of mirabegron, a β3‐adrenoceptor agonist for treatment of overactive bladder, in healthy Japanese male subjects: results from single‐ and multiple‐dose studies. Clin Drug Investig 34: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iitsuka H, van Gelderen M, Katashima M, Takusagawa S, Sawamoto T (2015). Pharmacokinetics of mirabegron, a β3‐adrenoceptor agonist for treatment of overactive bladder, in healthy East Asian subjects. Clin Ther 37: 1031–1044. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifpour J, Kondo S, O'Hollaren B, Morita T, Weiss RM (1990). Autonomic receptors in urinary tract: sex and age differences. J Pharmacol Exp Ther 253: 661–667. [PubMed] [Google Scholar]
- Lima V, Mueller A, Kamikihara SY, Raymundi V, Alewood D, Lewis RJ et al. (2005). Differential antagonism by conotoxin rho‐TIA of contractions mediated by distinct alpha1‐adrenoceptor subtypes in rat vas deferens, spleen and aorta. Eur J Pharmacol 508: 183–192. [DOI] [PubMed] [Google Scholar]
- Marshall I, Burt RP, Green GM, Hussain MB, Chapple CR (1996). Different subtypes of alpha 1A‐adrenoceptor mediating contraction of rat epididymal vas deferens, rat hepatic portal vein and human prostate distinguished by the antagonist RS 17053. Br J Pharmacol 119: 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVary KT, Roehrborn CG, Avins AL, Barry MJ, Bruskewitz RC, Donnell RF et al. (2011). Update on AUA guideline on the management of benign prostatic hyperplasia. J Urol 185: 1793–1803. [DOI] [PubMed] [Google Scholar]
- Michel MC (2014). Do β‐adrenoceptor agonists induce homologous or heterologous desensitization in rat urinary bladder? Naunyn Schmiedebergs Arch Pharmacol 387: 215–224. [DOI] [PubMed] [Google Scholar]
- Michel MC, Vrydag W (2006). Alpha1‐, alpha2‐ and beta‐adrenoceptors in the urinary bladder, urethra and prostate. Br J Pharmacol 147: S88–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minneman KP, Theroux TL, Hollinger S, Han C, Esbenshade TA (1994). Selectivity of agonists for cloned alpha 1‐adrenergic receptor subtypes. Mol Pharmacol 46: 929–936. [PubMed] [Google Scholar]
- Nasu K, Moriyama N, Fukasawa R, Tsujimoto G, Tanaka T, Yano J et al. (1998). Quantification and distribution of alpha1‐adrenoceptor subtype mRNAs in human proximal urethra. Br J Pharmacol 123: 1289–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojimoto FD, Mueller A, Hebeler‐Barbosa F, Akinaga J, Lima V, Kiguti LR et al. (2010). The tricyclic antidepressants amitriptyline, nortriptyline and imipramine are weak antagonists of human and rat alpha1B‐adrenoceptors. Neuropharmacology 59: 49–57. [DOI] [PubMed] [Google Scholar]
- Palea S, Rekik M, Rouget C, Camparo P, Botto H, Rischmann P et al. (2012). Fenoterol functionally activates the β3‐adrenoceptor in human urinary bladder, comparison with rat and mouse: implications for drug discovery. Eur J Pharmacol 690: 202–206. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradidarcheep W, Wallner C, Dabhoiwala NF, Lamers WH (2011). Anatomy and histology of the lower urinary tract. Handb Exp Pharmacol 202: 117–148. [DOI] [PubMed] [Google Scholar]
- Pupo AS, Uberti MA, Minneman KP (2003). N‐terminal truncation of human alpha 1D‐adrenoceptors increases expression of binding sites but not protein. Eur J Pharmacol 462: 1–8. [DOI] [PubMed] [Google Scholar]
- Rozec B, Quang TT, Noireaud J, Gauthier C (2006). Mixed beta3‐adrenoceptor agonist and alpha1‐adrenoceptor antagonist properties of nebivolol in rat thoracic aorta. Br J Pharmacol 147: 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Hutchinson DS, Evans BA, Summers RJ (2008). The beta3‐adrenoceptor agonist 4‐[[(hexylamino)carbonyl]amino]‐N‐[4‐[2‐[[(2S)‐2‐hydroxy‐3‐(4‐hydroxyphenoxy) propyl]amino]ethyl]‐phenyl]‐benzenesulfonamide (L755507) and antagonist(S)‐N‐[4‐[2‐[[3‐[3‐(acetamidomethyl)phenoxy]‐2‐hydroxypropyl]amino]‐ethyl]phenyl] benzenesulfonamide (L748337) activate different signaling pathways in Chinese hamster ovary‐K1 cells stably expressing the human beta3‐adrenoceptor. Mol Pharmacol 74: 1417–1428. [DOI] [PubMed] [Google Scholar]
- Svalø J, Nordling J, Bouchelouche K, Andersson KE, Korstanje C, Bouchelouche P (2013). The novel β3‐adrenoceptor agonist mirabegron reduces carbachol‐induced contractile activity in detrusor tissue from patients with bladder outflow obstruction with or without detrusor overactivity. Eur J Pharmacol 15: 101–105. [DOI] [PubMed] [Google Scholar]
- Takasu T, Ukai M, Sato S, Matsui T, Nagase I, Maruyama T et al. (2007). Effect of (R)‐2‐(2‐aminothiazol‐4‐yl)‐4′‐{2‐[(2‐hydroxy‐2‐phenylethyl)amino]ethyl} acetanilide (YM178), a novel selective beta3‐adrenoceptor agonist, on bladder function. J Pharmacol Exp Ther 321: 642–647. [DOI] [PubMed] [Google Scholar]
- Takeda H, Matsuzawa A, Igawa Y, Yamazaki Y, Kaidoh K, Akahane S et al. (2003). Functional characterization of beta‐adrenoceptor subtypes in the canine and rat lower urinary tract. J Urol 170: 654–658. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Inagaki R, Murata S, Akiba I, Muramatsu I (1999). Microphysiometric analysis of human alpha1a‐adrenoceptor expressed in Chinese hamster ovary cells. Br J Pharmacol 127: 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg U (1974). An analysis of the alpha‐ and beta‐effects of isoprenaline on the isolated nictitating membrane. Naunyn Schmiedebergs Arch Pharmacol 285: 375–393. [DOI] [PubMed] [Google Scholar]
- Uchida H, Shishido K, Nomiya M, Yamaguchi O (2005). Involvement of cyclic AMP‐dependent and ‐independent mechanisms in the relaxation of rat detrusor muscle via beta‐adrenoceptors. Eur J Pharmacol 518: 195–202. [DOI] [PubMed] [Google Scholar]
- Vicentic A, Robeva A, Rogge G, Uberti M, Minneman KP (2002). Biochemistry and pharmacology of epitope‐tagged alpha(1)‐adrenergic receptor subtypes. J Pharmacol Exp Ther 302: 58–65. [DOI] [PubMed] [Google Scholar]
- van Wieringen JP, Michel‐Reher MB, Hatanaka T, Ueshima K, Michel MC (2013). The new radioligand [(3)H]‐L 748,337 differentially labels human and rat β3‐adrenoceptors. Eur J Pharmacol 720: 124–130. [DOI] [PubMed] [Google Scholar]
- Wuest M, Eichhorn B, Grimm MO, Wirth MP, Ravens U, Kaumann AJ (2009). Catecholamines relax detrusor through beta 2‐adrenoceptors in mouse and beta 3‐adrenoceptors in man. J Pharmacol Exp Ther 328: 213–222. [DOI] [PubMed] [Google Scholar]
- Yamaguchi O, Marui E, Kakizaki H, Homma Y, Igawa Y, Takeda M et al. (2014). Phase III, randomised, double‐blind, placebo‐controlled study of the beta3‐adrenoceptor agonist mirabegron, 50 mg once daily, in Japanese patients with overactive bladder. BJU Int 113: 951–960. [DOI] [PubMed] [Google Scholar]
- Yamanishi T, Chapple CR, Yasuda K, Yoshida K, Chess‐Williams R (2003). The functional role of beta‐adrenoceptor subtypes in mediating relaxation of pig urethral smooth muscle. J Urol 170: 2508–2511. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Takeda H, Akahane M, Igawa Y, Nishizawa O, Ajisawa Y (1998). Species differences in the distribution of beta‐adrenoceptor subtypes in bladder smooth muscle. Br J Pharmacol 124: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Zong H, Cui Y, Li N, Zhang Y (2014). The efficacy of PDE5 inhibitors alone or in combination with alpha‐blockers for the treatment of erectile dysfunction and lower urinary tract symptoms due to benign prostatic hyperplasia: a systematic review and meta‐analysis. J Sex Med 11: 1539–1545. [DOI] [PubMed] [Google Scholar]
- Yono M, Foster HE Jr, Weiss RM, Latifpour J (2006). Age related changes in the functional, biochemical and molecular properties of alpha1‐adrenoceptors in the rat genitourinary tract. J Urol 176: 1214–1219. [DOI] [PubMed] [Google Scholar]