

Abstract
Background and Purpose
Angiotensin II (AngII) induces migration and growth of vascular smooth muscle cell (VSMC), which is responsible for vascular remodelling in some cardiovascular diseases. Ang II also activates a Cl− current, but the underlying mechanism is not clear.
Experimental Approach
The A10 cell line and primary cultures of VSMC from control, ClC‐3 channel null mice and WT mice made hypertensive with AngII infusions were used. Techniques employed included whole‐cell patch clamp, co‐immunoprecipitation, site‐specific mutagenesis and Western blotting,
Key Results
In VSMC, AngII induced Cl− currents was carried by the chloride ion channel ClC‐3. This current was absent in VSMC from ClC‐3 channel null mice. The AngII‐induced Cl− current involved interactions between ClC‐3 channels and Rho‐kinase 2 (ROCK2), shown by N‐ or C‐terminal truncation of ClC‐3 protein, ROCK2 siRNA and co‐immunoprecipitation assays. Phosphorylation of ClC‐3 channels at Thr532 by ROCK2 was critical for AngII‐induced Cl− current and VSMC migration. The ClC‐3 T532D mutant (mutation of Thr532 to aspartate), mimicking phosphorylated ClC‐3 protein, significantly potentiated AngII‐induced Cl− current and VSMC migration, while ClC‐3 T532A (mutation of Thr532 to alanine) had the opposite effects. AngII‐induced cell migration was markedly decreased in VSMC from ClC‐3 channel null mice that was insensitive to Y27632, an inhibitor of ROCK2. In addition, AngII‐induced cerebrovascular remodelling was decreased in ClC‐3 null mice, possibly by the ROCK2 pathway.
Conclusions And Implications
ClC‐3 protein phosphorylation at Thr532 by ROCK2 is required for AngII‐induced Cl− current and VSMC migration that are involved in AngII‐induced vascular remodelling in hypertension.
Abbreviations
- ΔCT
C‐terminal truncated ClC‐3
- ΔNT
N‐terminal truncated ClC‐3
- BASMC
basilar artery smooth muscle cells
- caROCK2
constitutively active ROCK2
- dnROCK2
dominant negative ROCK2
- N1
EGFP‐N1 plasmid
- VRCC
volume‐regulated chloride channel
- VSMC
vascular smooth muscle cells
Tables of Links
| TARGETS |
|---|
| Ion channels a |
| ClC‐3 channels |
| Enzymes b |
| Src kinase |
| ROCK2, Rho kinase 2 |
| LIGANDS |
| AngII, angiotensin II |
| SU6656 |
| Tamoxifen |
| Y27632 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a bAlexander et al., 2013a, 2013b).
Introduction
Angiotensin II (AngII) is one of the most potent regulators of blood pressure and has been implicated in cardiovascular remodelling during hypertension, atherosclerosis, and restenosis (Pacurari et al., 2014). The stimulation of the hypertrophy, proliferation and migration of vascular smooth muscle cells (VSMC) has been suggested as the major mechanism underlying AngII‐induced cardiovascular remodelling (Savoia and Volpe, 2011). It is well‐established that AngII stimulates VSMC migration and growth through ion channels and many intracellular signalling molecules. However, the precise molecular mechanism is unclear.
AngII is known to activate Cl− currents in cardiac myocytes (Ren et al., 2008). AngII‐induced Cl− currents were also observed following angiotensin AT1 receptor stimulation, through the downstream pathway of PI3K and NADPH oxidase (Browe and Baumgarten, 2004; Browe and Baumgarten, 2006; Ren et al., 2008). Moreover, continuous perfusion of AngII elicited a Cl− current in follicular cells (Montiel‐Herrera et al., 2011). Overall, these reports suggested a functional association of AngII with Cl− channels. However, it is unknown whether there is an activation of AngII‐induced Cl− current during the development of vascular remodelling. The underlying molecular mechanism also remains to be elucidated.
The anion channel ClC‐3 is a member of the ClC voltage‐gated Cl− channel gene superfamily. ClC‐3 channels are abundantly expressed in almost all species and function as anion channels when expressed on plasma membrane or as a Cl−/H+ antiporter when expressed on lysosomal membranes (Duan et al., 1997; Zhou et al., 2005; Duran et al., 2010; Stauber and Jentsch, 2013). The ClC‐3 channels play a vital role in various cellular functions, including cell volume regulation, vascular remodelling, endothelial inflammation, insulin secretion and neuron excitability (Duan et al., 1997; Dickerson et al., 2002; Deriy et al., 2009; Liu et al., 2010; Yang et al., 2012). We have shown that these channels mediated the cerebrovascular remodelling in hypertension through accelerating cell proliferation and inhibiting apoptosis in VSMC, suggesting that the activation of ClC‐3 channels was related to AngII‐induced Cl− current and vascular remodelling (Liu et al., 2010; Qian et al., 2011).
The phosphorylation or dephosphorylation of ClC‐3 channels is a common, rapid and reversible event in signal transduction. Activation of a serine/threonine kinase, PKC, inhibited ClC‐3 channel activity (Rossow et al., 2006). Mutation of Ser109 within the N‐terminal of ClC‐3 channels abolished the CaMKII‐dependent Cl− current (Robinson et al., 2004). We have also demonstrated that the phosphorylation of Tyr284 in ClC‐3 channels, by Src kinase was an important mechanism for activation of these channels (Wang et al., 2013; Zeng et al., 2014). Moreover, the Rho/Rho‐kinase (ROCK) pathway was involved in the regulation of ClC‐3 channel activity (Liu et al., 2010). Rho‐kinase is a serine/threonine kinases, which is known to play a crucial role in AngII‐induced VSMC migration and vascular remodelling (Loirand et al., 2006). We, therefore, hypothesized that phosphorylation of ClC‐3 channels by Rho/Rho‐kinase might be involved in the Cl− current and migration of VSMC, induced by AngII.
Our experiments showed that AngII induced a Cl− current in VSMC dependent on ClC‐3 channels and involving the phosphorylation of ClC‐3 at Thr532 .by ROCK2 We also investigated the role of ClC‐3 channels in AngII‐induced vascular remodelling and the underlying mechanisms in terms of Ser/Thr phosphorylation and the ROCK2 pathway.
Methods
Plasmids and construction of ClC‐3 protein mutants
The plasmid of ClC‐3 protein was kindly donated by Dr. Hume (University of Nevada, USA). The plasmids of constitutively active ROCK2 (caROCK2) and dominantly negative ROCK2 (dnROCK2) were kindly donated by Dr. Anming Meng (Tsinghua University, China).
NetPhos 2.0 (URL: http://www.cbs.dtu.dk/services/NetPhos/) was used for the prediction of phosphorylation sites in ClC‐3 protein. The NetPhos 2.0 server produces neural network predictions for Ser, Thr and Tyr phosphorylation sites in eukaryotic proteins. The scientific rationale for its use was discussed previously by others (Blom et al., 1999).
Sequential N‐terminal deficient or C‐terminal deficient ClC‐3 protein was created by PCR and, then, was inserted into EGFP‐N1 vector respectively. In the PCR reaction of creating ClC‐3 ∆NT (∆1–56), the sense and antisense primers were 5′‐CCCAAGCTTATGGCATGGGAAATGACAAAAAGT‐3′ and 5′‐CGCGGATCCGCGTTGAACATTATTGAAGCGGG‐3′ respectively. In the PCR reaction of creating ClC‐3 ∆CT (∆642–710), the sense and antisense primers were 5′‐CCCAAGCTTCAATGACAAATGGAGGCAGC‐3′ and 5′‐CGCGGATCCGCTTCTTTTGACATTATGACAGG‐3′ respectively.
The site‐directed mutation of ClC‐3 protein was carried out with Quikchange® Lightning Site‐Direct Mutagenesis Kit (Agilent Technologies, Santa Clara, USA) as previously described (Wang et al., 2013). The primers were designed as follows: T363A, sense, 5′‐GTCGTCGACGCAAGTCCGCCAAATTTGGAAAGTATC‐3′, antisense, 5′‐GATACTTTCCAAATTTGGCGGACTTGCGTCGACGAC‐3′; T532A, sense, 5′‐GCGTGACAAGGATGGCTGTCTCCCTGGTG‐3′, antisense, 5′‐CACCAGGGAGACAGCCATCCTTGTCACGC‐3′; T532D, sense, 5′‐GTGGCGTGACAAGGATGGATGTCTCCCTGGTGGTTA‐3′, antisense, 5′‐TAACCACCAGGGAGACATCCATCCTTGTCACGCCAC‐3′.
Electrophysiological experiments
Membrane whole‐cell Cl− currents were recorded with an Axopatch 200B Amplifier (Axon Instrument, Foster City, USA) as described previously (Liu et al., 2010). The extracellular isotonic solution contained (mmol·L−1): 107 N‐methyl‐d‐glucamine chloride (NMDG‐Cl), 1.5 MgCl2, 2.5 MnCl2, 0.5 CdCl2, 0.05 GdCl3, 10 glucose, 10 HEPES and 70 D‐mannitol, pH 7.4 with NMDG. The osmolarity was 300 mOsm·L−1. The hypertonic solution was made by adding the d‐manitol (mmol·L−1) from 70 to 140 with an osmolarity of 370 mOsm·L−1. Internal pipette solution (300 mOsm·L−1) contained (mmol·L−1): 95 CsCl, 20 TEA chloride, 5 ATP‐Mg, 5 HEPES, 5 EGTA and 80 D‐mannitol, pH 7.2 with CsOH. Patch pipettes were made with borosilicate capillary tubes by means of a Sutter P‐97 horizontal puller (Sutter Instrument, Novato, CA) and had a resistance of 3–5 MΩ when filled with the internal solution. Amphotericin B (40 μmol·L−1) was included in the pipette solution before the recording to perforate the membrane. The currents were elicited with voltage steps from −100 mV to +120 mV in +20 mV increment for 400 ms, and with an interval of 5 s from a holding potential of −40 mV. Currents were sampled at 5 kHz using pCLAMP8.0 software (Axon Instruments) and filtered at 2 kHz.
Cell culture
The A10 cell line of VSMC were purchased from the American Type Culture Collection (Manassas, USA). Basilar artery smooth muscle cells (BASMC) from mice were cultured as previously described (Liu et al., 2010). Briefly, mice were anaesthetized with pentobarbital sodium and (50 mg·kg−1) and decapitated. Basilar arteries were rapidly excised and immersed in cold Krebs buffer. The basilar arteries were then cut into small pieces of about 0.5 mm and incubated in DMEM/F12 supplemented with 20% fetal calf serum at 37 °C, 5% CO2. 7 to 10 days were allowed for the BASMC to migrate from the tissue pieces and the cells were passaged using 0.2% trypsin.
siRNA transfection and plasmid transfection
ClC‐3 siRNA (GeneBank Accession No. NM_053363, 5′‐GGAUGACUGACCUGAAAGATT‐3′) and ROCK2 siRNA (GeneBank Accession No. NM_013022, 5′‐GAGCAACAUGGAAAUAGAUAUGACA‐3′) were designed and synthesized by Invitrogen (Carlsbad, USA). Scrambled RNA was used as negative control. ClC‐3 siRNA, ROCK2 siRNA and negative RNA was transfected into A10 cells by using HiPerfect transfection reagent according to the protocol previously described (Liu et al., 2010).
Plasmids were transfected into the cells with LipofectAMINE2000 reagent in OPTI‐MEMRI reduced serum medium according to the protocol previously described (Liu et al., 2010).
Co‐immunoprecipitation (co‐IP) and Western blot
The cell lysates were incubated with protein G beads for 2 h at 4°C and centrifuged. The supernatants were incubated with pre‐coupled antibodies bound to protein G beads overnight at 4°C. Samples were resolved on 8% SDS‐PAGE gels and transferred onto PVDF membranes. The bound proteins were determined by immunoblotting with the indicated antibodies. The original images of all Western blotting experiments are shown in Figure S8 (Supporting Information).
Migration assays
For the wound‐healing assay, a scratch lesion was created on confluent VSMC cultures, with a pipette tip and three randomly selected fields at the lesion border were examined, using living cells workstations (Zeiss, Oberkochen, Germany). Boyden chamber assays were carried out in transwell filters with 8 μm·L−1 pores (Millipore Billerica, MA, USA) according to the manufacturer's instructions.
Animal models
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China and were approved by the Sun Yat‐Sen University Animal Care and Use Committee. Studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010).
ClC‐3 null and littermate control wild type (WT) mice were kindly provided by Dr. Dean Burkin from the Nevada Transgenic Center (Nevada Transgenic Center, University of Nevada School of Medicine, Nevada, USA) (Huang et al., 2014). Heterozygous 129/SvJ‐C57BL/6 offspring were used to establish breeding colonies.
Male ClC‐3 null mice and littermate control were randomly divided into three groups (con‐treatment, AngII‐treatment, and Y27632‐treatment), and the total number of animal in each group was five. Animals (18–22 g) were implanted with osmotic minipumps set to deliver solution at a rate of 0.25 μL·h−1 (Alzet, USA) and filled with AngII in a buffer consisting of 0.9% saline and 1% acetic acid. AngII was given at a rate of 1.5 mg·kg−1·d for 2 weeks. When Y27632 was used, it was infused at 3 mg·kg−1·d, together with AngII. The control mice underwent the same surgical procedure except for the omission of AngII in the osmotic minipumps. Before the operation, mice were anaesthetized with 60 mg·ml−1 pentobarbital sodium. Then the minipump was implanted subcutaneously between the scapulae as the instruction indicated. The animal recovered within 2 hours after the operation. All of the mice implanted with AngII‐infused pump developed hypertension within 2 weeks and there was no associated mortality.
Tail‐cuff plethysmography (PowerLab 4/30, ADInstruments, Sydney, Australia) was used to measure systolic blood pressure in conscious mice. Care was taken to ensure that the mice had adjusted to the environment and that they were not disturbed before any measurements were taken. Three consecutive measurements were taken for each mouse, and the average was used in the analyses.
Immunohistochemistry
Mice were anaesthetized with 60 mg·ml−1 pentobarbital sodium and were perfused with Krebs buffer containing heparin (100 U·kg−1) and nitroglycerol (0.3 μg·kg−1), followed by 4% freshly depolymerized paraformaldehyde for 20 min. The brain was carefully removed, and sections (8 μm) were prepared as previously described (Liu et al., 2010). Briefly, five sections selected randomly in each group were incubated in the primary antibody for α‐actin at room temperature. After washing with PBS, the sections were incubated with corresponding secondary antibodies of peroxidase‐labelled anti‐mouse IgG. Finally, the α‐actin staining was developed by DAB for 30 s and examined using a microscope (Olympus, Tokyo, Japan).
Data analysis
All data are expressed as mean ± SD. Raw data were applied directly in statistical analysis. An unpaired two‐tailed Student's t‐test was used to determine significant differences between two groups. One‐way or two‐way ANOVA followed by Bonferroni multiple comparison tests was used to compare differences among more than two groups. Values of P < 0.05 were considered statistically significant.
Materials
Cell culture medium (DMEM/F12), fetal calf serum, bovine serum albumin (BSA), LipofectAMINE2000 reagent, OPTI‐MEM®I reduced serum medium and cocktail were obtained from GIBCO/Invitrogen (Carlsbad, CA, USA). AngII, tamoxifen, Y27632 and amphotericin B were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Primary antibody against RhoA (1:1000), ROCK1 (1:1000) and secondary antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Primary antibody against ROCK2 (1:1000) was obtained from Upstate (LakePlacid, NY USA). Primary antibody against ClC‐3 channels (1:500) was obtained from Alomone Labs (Jerusalem, Israel). Primary antibody against TMEM16A (1:200) was purchased from Abcam (Cambridge, UK). Primary antibody against the cystic fibrosis transmembrane conductance regulator chloride channel (1:500) was purchased from Novus (New York, NY, USA). Primary antibody against GFP (1:200) and HA (1:200) were obtained from Vazyme (Nanjing, China). Primary antibody against PRK2 (1:200) and MSK1 (1:200), Protein G beads and IgG were purchased from Santa Cruz Technology (Santa Cruz, CA, USA).
Results
AngII‐induced Cl − current in VSMC was dependent on ClC‐3 channels
In isotonic solution (300 mOsm·L−1), AngII (0.2‐2μM) induced an outwardly rectifying Cl− current in a concentration‐dependent manner in A10 VSMC (n = 5, P < 0.05; Figure 1). The Cl− currents displayed a time‐dependent inactivation at positive test potentials, and the reversal potential was −0.6 ± 1.9 mV, which was near the equilibrium potential for Cl− (ECl = 0 mV). When the Cl− concentration ([Cl−]out) in the bath solution was changed from 116 to 39 mmol·L−1, the reversal potential was also changed +26.4 ± 3.3 mV, which was not statistically different from the potential predicted by the Nernst equation (ΔECl = 25.6 mV) for a Cl− selective current, suggesting that Cl− was the main permeation anion (Figure 1). In addition, AngII‐induced Cl− current was completely suppressed by hypertonic solutions (370 mOsm·L−1) or tamoxifen (10 μmol·L−1), a Cl− channel inhibitor. (Figure 1). These results demonstrated that AngII could induce a Cl− current in A10 VSMC.
Figure 1.
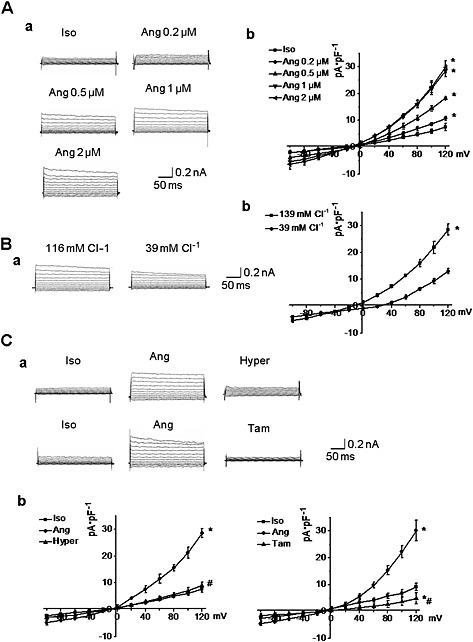
The characteristics of AngII‐induced Cl− current in A10 cells. A. AngII (Ang) activated an outwardly rectifying Cl− current in a concentration‐dependent manner in isotonic solution (Iso) (300 mOsm·L−1). a. Representative recordings of AngII‐induced Cl− current in VSMC treated with different concentrations of AngII. b. I‐V curves of Cl− currents induced by different concentration of AngII (n = 5, *P < 0.05 vs. iso). B. Reduction of extracellular Cl− concentration from 116 mmol·L−1 to 39 mmol·L−1 caused a shift in the reversal potential from −0.6 ± 1.9 to 26.4 ± 3.3 mV. a. Representative recordings of AngII‐induced Cl− current in the presence of different extracellular Cl− concentration. b. I–V curves of AngII‐induced Cl− current in the different extracellular Cl− concentration. (n = 5, *P < 0.05 vs. 139 mmol·L−1 Cl−). C. Infusion with hypertonic solution (Hyper) (370 mOsm·L−1) or tamoxifen (Tam) (10 μmol·L−1) significantly inhibited AngII‐induced Cl− current. a. Representative recordings of AngII‐induced Cl− current after treatment with hypertonic solution or tamoxifen. b. I‐V curves of AngII‐induced Cl− current after treatment with hypertonic solution or tamoxifen. n = 5, *P < 0.05 vs. iso.
Next, the relationship between ClC‐3 channels and AngII‐induced Cl− current was explored. The siRNA for ClC‐3 channel protein was used to reduce the expression of these channels and AngII‐induced Cl− current was recorded. As shown in Figure S1, expression of ClC‐3 channels was significantly decreased after transfection of A10 cells with ClC‐3 siRNA (40 nmol·L−1) for 48 h, while the expression of other Cl− channels, such as TMEM16A (Ano1), and the cystic fibrosis transmembrane conductance regulator were not changed (Figure S1, Supporting Information). Silencing of ClC‐3 protein with ClC‐3 siRNA transfection markedly decreased AngII‐induced Cl− current (Figure 2). Furthermore, this result was confirmed in primary cultures of BASMC from WT and transgenic ClC‐3 null mice. A Cl− current was generated by AngII (1 μmol·L−1) in BASMC from WT mice but not in BASMC from ClC‐3 null mice (Figure 2). These results demonstrated that ClC‐3 was necessary for AngII‐induced Cl− current in VSMC.
Figure 2.
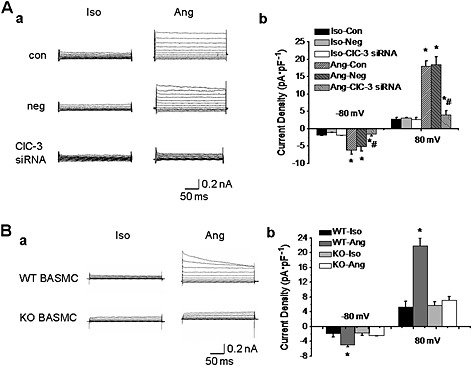
ClC‐3 channels were critically involved in the AngII‐induced Cl− current. A. Downregulation of ClC‐3 protein expression with anti‐ClC‐3 siRNA (40 nmol·L−1) decreased AngII‐induced Cl− current in A10 cells. a. Representative traces of AngII‐induced Cl− current in control (con), negative siRNA (neg) or ClC‐3 siRNA‐transfected A10 cells. b. Bar chart from the experiments measured at −80 mV (downward bars) and +80 mV (upward bars) in control (con), negative siRNA (neg) or ClC‐3 siRNA‐transfected A10 cells (n = 6, *P < 0.05 vs. iso, #P < 0.05 vs. Ang). B. The Cl− current was not induced by Ang II in basilar artery smooth muscle cells (BASMC) from ClC‐3 null mice. a. Representative traces of AngII‐induced Cl− current in BASMC of wild type mice (WT) and ClC‐3 null mice (KO). b. Bar chart from the experiments measured at –80 mV (downward bars) and +80 mV (upward bars) in BASMC of wild type mice (WT) and ClC‐3 null mice (KO). n = 6, *P < 0.05 vs. iso.
AngII‐induced Cl − current was modulated through the interaction between ClC‐3 and ROCK2
RhoA, ROCK1 and ROCK2, the kinases activated downstream of AngII, were individually immunoprecipitated with ClC‐3 channels in A10 cells. ROCK2, but not RhoA or ROCK1, was immunoprecipitated with these channels (n = 4; Figure 3). And then, the interaction between ClC‐3 channels and ROCK2 was determined in A10 cells after Ang II‐treatment for 24 h. As shown in Figure 3, AngII increased the association of ClC‐3 channels with ROCK2 ( Figure 3) although the expression of ClC‐3 channels and ROCK2 was not altered by AngII (n = 4; Figure 3).
Figure 3.
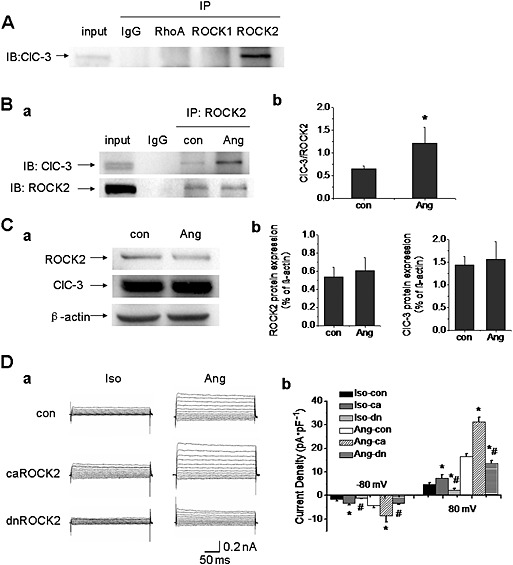
AngII‐induced Cl− current was modulated through the RhoA/Rho‐kinase pathway. A. Co‐immunoprecipitation (co‐IP) showed that ROCK2, but not RhoA or ROCK1, immunoprecipitated with ClC‐3 channels in A10 cells (n = 4). IP, immunoprecipitation; IB, immunoblot. B. A10 cells were incubated with AngII (1 μmol·L−1) for 24 h, and then, co‐IP was carried out to examine the association between ClC‐3 channels and ROCK2. a. The interaction between ClC‐3 channels and ROCK2 was potentiated in the AngII‐pretreated group. b. Bar chart of densitometric analysis from the experiments. n = 4, *P < 0.05 vs. con. C. The effect of AngII on ROCK2 and ClC‐3 protein expression in A10 cells. a. The expression of ROCK2 and ClC‐3 proteins were not altered by the treatment with AngII (1 μmol·L−1) for 24 h. b. Bar chart of densitometric analysis from the experiments (n = 4). D. The effect of ROCK2 activation on AngII(1 μmol·L−1)‐induced Cl− current in A10 cells. a. Cells were transfected with constitutively active ROCK2 (caROCK2) or dominantly negative ROCK2 (dnROCK2) plasmids for 48 h respectively. AngII‐induced Cl− current was increased markedly in caROCK2‐transfected cells, whereas the current was reduced significantly in dnROCK2‐transfected cells. b. Bar chart from the experiments measured at −80 mV (downward bars) and +80 mV (upward bars) in control, caROCK2‐transfected, and dnROCK2‐transfected cells. n = 6, *P < 0.05 vs. con, #P < 0.05 vs. Ang.
To further confirm that ROCK2 mediated AngII‐induced Cl− current, caROCK2 and dnROCK2 plasmids were transfected into A10 cells respectively. AngII‐induced Cl− current was almost doubled in caROCK2 transfected cells, while the current was reduced in dnROCK2 transfected cells (Figure 3). These results suggested that AngII‐induced Cl− current was regulated through ROCK2.
We have already shown that phosphorylation of Tyr284 in ClC‐3 channels by Src kinase is an important mechanism for the activation of these channels by AngII (Wang et al., 2013 ; Zeng et al., 2014). Therefore, we examined the combined effects of ROCK2 and Src on ClC‐3 channel activation and found that either the ROCK2 inhibitor Y27632 or the Src kinase inhibitor SU6656 inhibited but did not abolish the Cl− current carried by ClC‐3 channels. The maximum inhibitory effect was observed when Y27632 or SU6656 was used at 10 μmol·L−1. However, the effect of combining these two inhibitors was greater (Figure S2, Supporting Information).
ROCK2 interacted with the transmembrane region in ClC‐3 channels
NetPhos 2.0 was selected to predict the sites in ClC‐3 protein that might be phosphorylated by ROCK2. A score greater than 0.5 is considered as a possible phosphorylation site. The motif phosphorylated by ROCK2 is R/KXS/T or R/KXXS/T. Combined with all of the selection criteria above, our analysis showed that there were eight serine/threonine sites in ClC‐3 protein that might be phosphorylated by ROCK2 (Table 1). The crystal structure of ClC protein showed that the ClC channels consisted of 16 transmembrane domains; both the N‐terminal and the C‐terminal extend into the cytosol (Dutzler et al., 2002). Among the eight serine/threonine sites in ClC‐3, two sites were located in the N‐terminal, three sites were located in the C‐terminal and three sites were in the transmembrane region of the ClC‐3 protein.
Table 1.
The sites in the ClC‐3 protein predicted to be phosphorylated by ROCK2
| Name | Pos | Context | Score | Pred | Kinases |
|---|---|---|---|---|---|
| Sequence | 51 | RRINSKKKE | 0.948 | *S* | PKG |
| Sequence | 56 | KKKESAWEM | 0.981 | *S* | PKG |
| Sequence | 362 | RRRKSTKFG | 0.998 | *S* | PKG, PKA, PKB |
| Sequence | 466 | IKVPSGLFI | 0.118 | ||
| Sequence | 642 | MSKESQRLV | 0.618 | *S* | |
| Sequence | 695 | LKLRSILDM | 0.865 | *S* | |
| Sequence | 203 | LGKWTLMIK | 0.024 | ||
| Sequence | 210 | IKTITLVLA | 0.165 | ||
| Sequence | 247 | PKYSTNEAK | 0.058 | ||
| Sequence | 363 | RRKSTKFGK | 0.915 | *T* | PKG, PKA |
| Sequence | 394 | TRLNTSELI | 0.137 | ||
| Sequence | 532 | VTRMTVSLV | 0.836 | *T* | PKA |
| Sequence | 655 | RRDLTIAIE | 0.742 | *T* |
To determine which part of the ClC‐3 protein was responsible for the interaction with ROCK2, truncated mutants of this protein, ClC‐3 ΔNT and ClC‐3 ΔCT, were generated (Figure 4), and the interaction between ROCK2 and the truncated proteins was examined using co‐IP. A10 cells were transfected with HA‐tagged ROCK2 and various GFP‐tagged ClC‐3 plasmids, including ClC‐3, ClC‐3 ΔNT and ClC‐3 ΔCT plasmids respectively. Figure S3 (Supporting Information) shows that the co‐transfection efficiency could reach above 10% of the cells transfected with ROCK2 and ClC‐3 or its mutants. (n = 4,100 times). These results demonstrated that the truncation of the N‐terminal or the C‐terminal in the ClC‐3 protein did not change the association between this protein and ROCK2 (n = 4, Figure 4).
Figure 4.
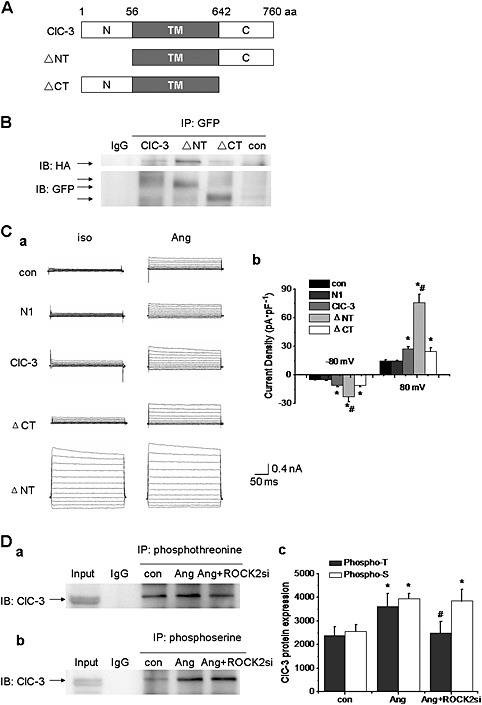
The transmembrane region of ClC‐3 channels was required for the interaction with ROCK2. A. The full length ClC‐3 protein has 760 amino acid (aa), which comprises the N‐terminal (N), transmembrane region (TM), and the C‐terminal. ΔNT represents the N‐terminal truncation and ΔCT the C‐terminal truncation of ClC‐3 protein B. The interaction between ClC‐3 protein and ROCK2 was not affected by N‐terminal or C‐terminal truncation of ClC‐3. A10 cells were transfected with HA‐tagged ROCK2 and various GFP‐tagged ClC‐3 plasmids (ClC‐3, ΔNT, or ΔCT) simultaneously for 48 h, and then cell lysates were immunoprecipitated with anti‐GFP antibody and immunoblotted with anti‐HA antibody. The interaction between ClC‐3 protein and ROCK2 was still present in cells transfected with ΔNT or ΔCT plasmid (n = 4). C. The effects of C‐terminal or N‐terminal in ClC‐3 protein on AngII‐induced Cl− current. a. AngII‐induced Cl− current in A10 cells transfected with plasmids of N1, ClC‐3, ΔNT or ΔCT. b. Bar chart of AngII‐induced Cl− current from the experiments was shown. n = 6, *P < 0.05 vs. con, #P < 0.05 vs. ClC‐3. D. ClC‐3 protein was phosphorylated by ROCK2 on threonine residues, but not on serine residues. a and b. The phosphorylation of threonine and serine in ClC‐3 were both increased in A10 cells pretreated with AngII (1 μmol·L−1) for 24 h. Only the phosphorylation of threonine in ClC‐3 protein was decreased after knocking down the expression of ROCK2 by ROCK2 siRNA transfection (40 nmol·L−1) for 48 h, while the phosphorylation of serine in ClC‐3 protein was not altered by ROCK2 siRNA transfection. c. Bar chart of densitometric analysis from the experiments. n = 5, *P < 0.05 vs. con, #P < 0.05 vs. Ang.
We then determined the effects of different truncated ClC‐3 constructs on AngII‐induced Cl− current. Figure 4 shows that transfection with ClC‐3 ΔCT allowed AngII to facilitate the current as well as in cells with WT ClC‐3, suggesting that the C‐terminal in ClC‐3 channels was not responsible for AngII‐induced Cl− current. By contrast, the ClC‐3 ΔNT construct yielded a constitutively active Cl− current, which was consistent with previous reports (Rossow et al., 2006), indicating that the N‐terminus might act as an inactivation mechanism in the ClC‐3 Cl− channel and might not be involved in the AngII‐induced Cl− current. From these results, we concluded that ClC‐3 channels might interact with ROCK2 through its transmembrane region.
As shown in Table 1, there are three sites in the transmembrane region of ClC‐3 channels that might be phosphorylated by ROCK2, namely, Ser362, Thr363 and Thr532. Therefore, serine or threonine phosphorylation in ClC‐3 channels was further explored, and ROCK2 siRNA was used. As shown in Figure S4 (Supporting Information), the expression of ROCK2 was markedly reduced after transfection with siRNA for ROCK2 (40 nmol·L−1) for 48 h, which did not affect the expression of other related kinases, such as PRK2 and MSK1 (Figure S4, Supporting Information). In addition, knockdown of PRK2 or MSK1 did not inhibit the AngII‐induced Cl− current (n = 6; Figure S5, Supporting Information). Analysis of ClC‐3 protein phosphorylation, in A10 cells pretreated with AngII, showed that both serine and threonine phosphorylation were upregulated but only the threonine phosphorylation was inhibited after knocking down the expression of ROCK2 by ROCK2 siRNA transfection (n = 5, P < 0.05; Figure 4). These results indicated that the ClC‐3 protein was phosphorylated by ROCK2 on a threonine site, but not on a serine site, and these sites were lekyl to be Thr363, Thr532 or both.
Phosphorylation at Thr 532 was required for AngII‐induced Cl − current
In order to identify which threonine site contributed to ROCK2‐mediated ClC‐3 channel activation, the threonine residues (T) were mutated to alanine (A). Therefore, T363A ClC‐3 and T532A ClC‐3 were generated and transfected into A10 cells respectively. Figure 5 shows that AngII evoked a steady increase in current densities in A10 cells (con) and EGFP‐N1 (N1) transfected cells. In A10 cells transfected with WT ClC‐3 protein, AngII further increased the Cl‐ current. There was no significant difference in AngII‐induced Cl− currents between cells transfected with the mutant T363A ClC‐3 and those transfected with WT ClC‐3. These results suggested that the mutation of Thr363 to alanine did not affect ClC‐3 channel activation induced by AngII. However, cells with the T532A ClC‐3 mutant, AngII‐induced Cl− current was reduced to the basal level, suggesting that Thr532 phosphorylation was required for ClC‐3 channel activation induced by AngII (Figure 5).
Figure 5.
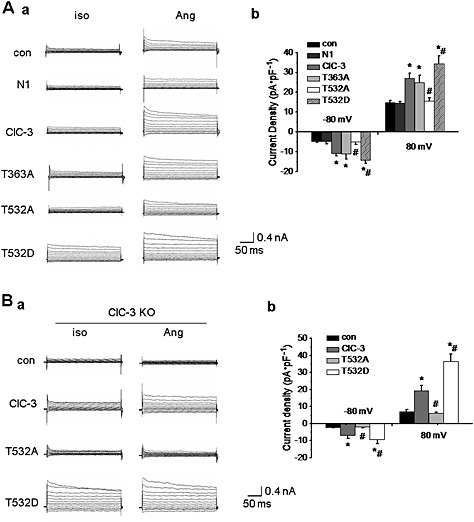
Phosphorylation of Thr532 in ClC‐3 protein was required for AngII‐induced Cl− current. A. The effect of ClC‐3 site mutations on AngII(1 μmol·L−1)‐induced Cl− current in A10 cells. a. Representative traces of Cl− current in A10 cells transfected with EGFP‐N1 (N1), WT ClC‐3, ClC‐3 T363A (T363A), ClC‐3 T532A (T532A) and ClC‐3 T532D (T532D) plasmids for 48 h. Compared with WT ClC‐3 channels, the current density was not altered by the T362A mutation, while the T532A mutation significantly reduced the current density. Moreover, the T532D mutation markedly increased the current density compared with WT ClC‐3 channels. b. Bar chart from the experiments at −80 mV (downward bars) and +80 mV (upward bars) in con, N1‐transfected, ClC‐3‐transfected, T363A‐transfected, T532A‐transfected, and T532D‐ transfected cells (n = 6, *P < 0.05 vs. con, #P < 0.05 vs. ClC‐3). B. The effect of Thr532 phosphorylation on AngII(1 μmol L−1)‐induced Cl− current in ClC‐3 null BASMC (KO). a. Representative traces of Cl− current in ClC‐3 null BASMC transfected with WT ClC‐3, T532A and T532D plasmids for 48 h. AngII could not induce a Cl− current in cells transfected with T532A plasmid, while AngII‐induced Cl− current was observed in cells transfected with WT ClC‐3 plasmid, which was further potentiated in cells transfected with T532D plasmid. b. Bar chart from the experiments at −80 mV (downward bars) and +80 mV (upward bars) in ClC‐3 null BASMC transfected with ClC‐3, T532A and T532D plasmids. n = 6, *P < 0.05 vs. con, #P < 0.05 vs. ClC‐3.
Next, we mutated Thr532 to aspartate (D), a negatively charged residue, which mimicks the phosphorylation of ClC‐3 protein (Wang et al., 2013). In cells transfected with T532D ClC‐3, the AngII‐induced Cl− current was further potentiated (Figure 5). Note that Moreover, the ClC‐3 mutants were expressed at a level, similar to that of the WT protein in both whole cell lysis and membrane protein extracts (Figure S6, Supporting Information). These results demonstrated that Thr532 was the key residue for ClC‐3 channel activation.
The results from ClC‐3 null BASMC were shown as Figure 5. In BASMC from ClC‐3 null mice, AngII produced a Cl− current in cells transfected with WT ClC‐3, but not in cells transfected with the T532A mutant channel. The AngII‐induced Cl− current was potentiated in transfected with T532D channel (n = 6, P < 0.05; Figure 5). This result further demonstrated that phosphorylation of Thr532 in ClC‐3 channels was required for AngII‐induced Cl− current in VSMC.
Phosphorylation of Thr 532 in ClC‐3 channels was critical for AngII‐induced migration in VSMC and AngII‐induced cerebrovascular remodelling was decreased in ClC‐3 null mice
To confirm that activation of ClC‐3 channels was involved in the effect of AngII on vascular function, we explored the influence of ClC‐3 knockdown on VSMC migration induced by AngII. As shown in Figure 6, using the wound‐healing assays, AngII increased the migration of WT BASMC an effect inhibited by treatment with Y27632, an inhibitor of ROCK. However, migration was not increased by AngII in BASMC from ClC‐3 null mice (Figure 6).
Figure 6.
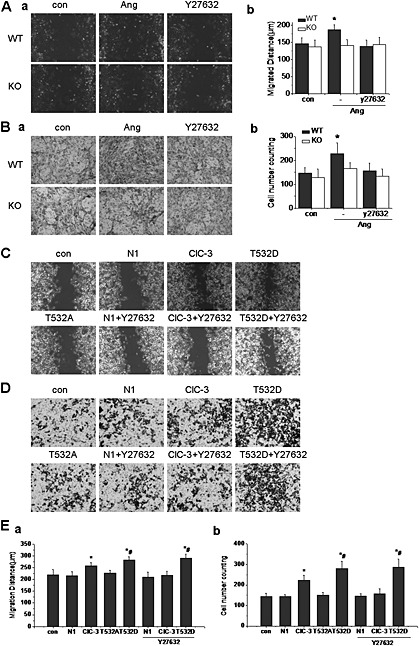
Phosphorylation of Thr532 in ClC‐3 was involved in AngII‐induced migration in A10 cells. A. The effect of ClC‐3 knockdown or ROCK inhibition on AngII (1 μmol·L−1)‐induced wound healing in BASMC from wild type (WT) and ClC‐3 null (KO) mice. a. Representative images of wound healing assay carried out in control, AngII‐treated and Y27632‐ treated groups. AngII increased the wound healing (distance cells migrated), which was inhibited by Y27632 (10 μmol·L−1) or ClC‐3 channel knockdown. b. Bar charts of relative wound healing from the experiments. n = 6, *P < 0.05 vs. WT con. B. The effect of ClC‐3 channel knockdown or ROCK inhibition on AngII(1 μmol·L−1)‐induced transwell migration of BASMC from WT or ClC‐3 KO mice. a. Representative images of transwell assay carried out in control, AngII‐treated and Y27632‐ treated groups. AngII significantly increased the transwell rate, which was inhibited by Y27632 or ClC‐3 knockdown. b. Bar charts of relative migration rate from the experiments. n = 6, *P < 0.05 vs. WT con. C. The effect of Thr532 phosphorylation on AngII(1 μmol·L−1)‐induced wound healing in A10 cells. Representative images of wound healing assay in cells transfected with plasmids of N1,WT ClC‐3, T532A and T532D mutants. Cells were pretreated with or without Y27632 in N1‐transfected, WT ClC‐3‐transfected and T532D‐transfected groups. AngII‐induced wound healing was increased in cells transfected with WT ClC‐3 plasmid, which was inhibited in cells transfected with the T532A mutant and further potentiated in cells transfected with the T532D mutant. Y27632 markedly reduced wound healing in WT ClC‐3‐transfected cells, but had no effect on T532D‐transfected cells. D. The effect of Thr532 phosphorylation on AngII(1 μmol L−1)‐induced transwell migration in A10 cells. Representative images of transwell assays in cells transfected with plasmids of N1, WT ClC‐3, T532A and T532D mutants. Cells were pretreated with or without Y27632 in N1, ClC‐3 and T532D transfected groups. AngII‐induced transwell migration was increased in cells transfected with ClC‐3 plasmid, which was inhibited in cells transfected with T532A, and further potentiated in cells transfected with the T532D mutant. Y27632 markedly reduced the transwell migration in WT ClC‐3 transfected cells, but had no effect on T532D transfected cells. E. Bar charts of relative wound healing and relative migration from the experiments . n = 5, *P < 0.05 vs. con, #P < 0.05 vs. Ang.
Three‐dimensional migration assays were performed simultaneously. Here also, AngII significantly increased migration of WT BASMC and Y27632 inhibited this AngII‐induced cell migration. As in the wound‐healing assays, migration was not increased by AngII in ClC‐3 null BASMC (Figure 6). These results suggested that AngII‐induced BASMC migration was related to the activation of ClC‐3 via the ROCK pathway.
Transwell assays was performed with A10 cells transfected with ClC‐3 siRNA. As shown in Figure S7 (Supporting Information), knocking down the expression of ClC‐3 protein did not affect migration directly but did clearly decrease AngII‐induced migration (Figure S7, Supporting Information).
To determine the specific role of Thr532 in VSMC migration, the three channels, WT ClC‐3, T532A ClC‐3 and T532D ClC‐3, were transfected into A10 cells and AngII‐induced migration was assessed. In the wound‐healing assay, migration in cells transfected with the mutant channel,T532A ClC‐3 was reduced, compared with that in WT ClC‐3 transfected cells, whereas transfection with T532D ClC‐3 further increased the migration. Pretreatment with Y27632 (10 μM) reduced the migration to the basal level in WT ClC‐3 transfected cells, but not in cells transfected with T532D ClC‐3 channels (Figure 6 and 6Ea).
In transwell assays the same pattern of results emerged. Thus, cells transfected with WT and the T532D mutant ClC‐3 channels increased migration in response to AngII, whereas cells with the T532A mutant did not respond. After Y27632 (10 μM), the migration of WT ClC‐3 transfected cells was reduced to the basal level, but not in T532D transfected cells (Figure 6 and 6Eb). These results suggested that phosphorylation of Thr532 in ClC‐3 channels by ROCK was critical for AngII‐induced VSMC migration.
Our previous studies had demonstrated that ClC‐3 channels were involved in the process of cerebrovascular remodelling through the regulation of proliferation and apoptosis of VSMC (Liu et al., 2010; Qian et al., 2011). Furthermore, the activation of vascular cell migration was also an important factor in the process of vascular remodelling. Therefore, the role of ClC‐3 channels in AngII‐induced cerebrovascular remodelling was examined. We found that the basilar arteries of the WT mice made hypertensive by infusion of AngII did show signs of remodelling, as indicated by the vascular remodelling parameters (media/lumen ratio, media thickness and cross‐sectional area). These signs of remodelling were decreased by treatment with Y27632. However, the basilar artery remodelling in ClC‐3 null hypertensive mice was also decreased, although blood pressure was not reduced in these mice, compared with WT hypertensive mice (Figure 7; Table S1, Supporting Information). It appeared that AngII‐induced migration was involved in the process of vascular remodelling through phosphorylation of Thr532 in ClC‐3 channels, by ROCK2.
Figure 7.
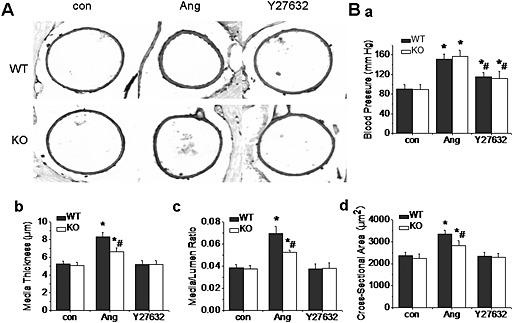
ClC‐3 channels were involved in AngII‐induced cerebrovascular remodelling via ROCK. A. AngII‐induced cerebrovascular remodelling was inhibited in ClC‐3 null and Y27632‐treated mice. Representative pictures showed the morphological characteristics of basilar arteries stained with anti‐α actin. Photographs were taken from control, hypertensive mice, with or without Y27632 treatment, in WT and ClC‐3 null mice. AngII‐induced cerebrovascular remodelling was significantly decreased in ClC‐3 null mice and in Y27632‐treated mice. B. a–d. Bar charts of blood pressure, media thickness, media/lumen ratios and cross‐sectional areas from the experiments. n = 5, *P < 0.05 vs. WT con, #P < 0.05 vs. WT Ang.
Discussion and conclusions
The novel findings in the present study are that AngII, one of the most potent regulators of arterial blood pressure, elicits a Cl− current in VSMC carried by ClC‐3 channels, and that phosphorylation of Thr532 in the ClC‐3 protein, by Rho/Rho‐kinase, is crucial for AngII‐induced Cl− current and VSMC migration.
The ClC‐3 channels are ubiquitously expressed in almost all eukaryotic cells with a variety of physiological functions. ClC‐3 channels could act as anion channels at the cell plasma membrane, related to the volume‐regulated Cl− channels (VRCC) at least in cardiac cells, VSMC and endothelial cells (Duan et al., 1997; Zhou et al., 2005; Yang et al., 2012) or as a Cl−/H+ antiporter in intracellular vesicles in other cells, such as hepatocytes, pancreatic acinar cells, neuronal cells (Stobrawa et al., 2001) and salivary acinar cells (Arreola et al., 2002). It was also suggested that the localization of ClC‐3 channels was different in various cell types. In hepatocytes, the channels were primarily localized in the intracellular vesicles where they were proposed to function primarily in vesicular acidification, whereas in many different kinds of cells, including VSMC, the ClC‐3 channels were located on the plasma membrane (Isnard‐Bagnis et al., 2003; Olsen et al., 2003; Zhou et al., 2005; Wang et al., 2013). In our previous study, we suggested that the ClC‐3 channel was one of molecular components involved in the activation or regulation of VRCC (Zhou et al., 2005; Guan et al., 2006). Although two studies had identified a poorly characterized protein, named LRRC8A as an essential component of VRCC (Qiu et al., 2014; Voss et al., 2014), more recently, it has been demonstrated that bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells (Milenkovic et al., 2015), suggesting that the VRCC is a complex, rather than a single ubiquitous channel, which could be formed by cell type‐ or tissue‐specific subunits. In this study, we found that ClC‐3 channels might serve to carry Cl− ions and was a crucial component of the AngII‐induced Cl− current in VSMC.
Many signalling molecules mediate the activation of ClC‐3 channels. The serine/threonine kinase, PKC, negatively regulated activation of these channels (Rossow et al., 2006). Calcium‐calmodulin kinase II, another serine/threonine kinase, was reported to regulate activation of these channels by interacting with ClC‐3 protein directly and phosphorylating Ser109 within the N‐terminal of the channel, which was intrinsically associated with intracellular calcium concentration, migration, apoptosis and the magnitude of long term potentiation (Robinson et al., 2004; Claud et al., 2008; Cuddapah and Sontheimer, 2010; Farmer et al., 2013). Moreover, the PI3K‐serum and glucocorticoid induced kinase cascade was involved in activation of ClC‐3 channels in PASMCs (Wang et al., 2004). Our previous study has demonstrated that ClC‐3 activation was decreased by inhibitors of protein tyrosine kinase and potentiated by inhibitors of protein tyrosine phosphatase (Zhou et al., 2005). The tyrosine phosphorylation in ClC‐3 channels was upregulated, as vascular cells switched from contractile to proliferative phenotype (Kang et al., 2012). Another study also demonstrated that the phosphorylation of Tyr284 in ClC‐3 channels by Src kinase, was important for activation of these channels (Wang et al., 2013; Zeng et al., 2014). These results suggested that many phosphorylation sites in the ClC‐3 protein were involved in the channel activity. Here, we have further demonstrated that phosphorylation of Thr532 in ClC‐3 channels by ROCK2 was another important molecular mechanism for activation of these channels, and was related to the AngII‐induced Cl− current.
Our results here showed that the truncated ClC‐3 ΔNT construct yielded a constitutively active Cl− current (Figure 4), which was consistent with previous reports (Rossow et al., 2006), indicating the N‐terminus might provide an inactivation mechanism in ClC‐3 Cl− channels. One model to explain such regulation of ClC‐3 channels was to postulate that the N‐terminus acted to block the inner pore vestibule (Rossow et al., 2006). Here, we found that ROCK2 can phosphorylate and activate ClC‐3 channels and that the association between ClC‐3 channels and ROCK2 was increased when the N‐terminal was deleted (Figure 4). Thus, our data indicated that blockade of the interaction between ROCK2 and ClC‐3 protein is a novel mechanism to explain why the N‐terminal negatively regulates theactivation of ClC‐3 channels.
Next, we explored the location of Thr532 in ClC‐3 protein according to the topological structure of ClC family. The crystal structure of a bacterial ClC protein confirms that the ClC channels are homodomeric proteins. Each ClC monomer consists of 18 α‐helical domains designated ‘A‐R’ (Dutzler et al., 2002; Dutzler et al., 2003). Membrane helices D, F, N and R comprise the ClC channel selectivity filter as the channel gate. According to the predicted transmembrane domain, Thr532 is located in the P helix towards the cytoplasmic membrane. The T532A mutation in the ClC‐3 channel may abolish channel activation by affecting the conformation of D, F, N or R helices, which are the crucial domains of the channel gate. Unfortunately, as the crystal structure of ClC‐3 protein is not available now, the precise contribution of Thr532 to the channel activation remains to be is elucidated.
It is noteworthy that phosphorylation of Thr532 in ClC‐3 protein is involved in AngII‐induced vascular cell migration. The role of ClC‐3 channels in cell migration has been shown in several kinds of cells, such as tumour cells (Mao et al., 2008; Cuddapah and Sontheimer, 2010; Lui et al., 2010; Li et al., 2013), neutrophil (Moreland et al., 2006; Volk et al., 2008) and VSMC (Ganapathi et al., 2013). However, exactly how ClC‐3 channels affect cell migration is still not very clear. A previous study showed that the cytosolic C‐terminal of the ClC‐3 protein interacted with subcortical actin filaments and contributed to the hypotonic‐induced shape change (Wang et al., 2005; McCloskey et al., 2007). Lamin A, one of the structural matrix proteins, was regulated by ClC‐3 channels (Qian et al., 2011). Furthermore, [Ca2+]i increase mediated by ClC‐3 channels was one of the mechanisms related to cell migration (Cuddapah et al., 2013; Li et al., 2013). Recently, one study showed that these channels played a role in VSMC migration via calmodulin‐dependent protein kinase II, while the exact mechanism was not fully demonstrated (Ganapathi et al., 2013). We have explored the underlying mechanism in this study and found that the T532A and T532D mutation of the ClC‐3 protein had opposing effects on AngII‐induced migration. This therefore demonstrated that Thr532 phosphorylation in the ClC‐3 protein was required for AngII‐induced vascular cell migration.
The development of vascular remodelling is associated with VSMC proliferation, apoptosis and migration (Hayashi and Naiki, 2009; Savoia and Volpe, 2011). ClC‐3 channels play crucial roles in the regulation of VSMC proliferation (Wang et al., 2002; Tang et al., 2008; Liu et al., 2010; Liang et al., 2014) and apoptosis (Qian et al., 2011; Wang et al., 2013). Deficiency or suppression of ClC‐3 Cl− channel reduced hypertension‐induced cerebrovascular remodelling through inhibition of proliferation and acceleration of apoptosis (Liu et al., 2010; Qian et al., 2011; Zheng et al., 2013). Here, we studied the involvement of ClC‐3 channels in AngII‐induced cerebrovascular remodelling and found that AngII‐induced cerebrovascular remodelling was decreased in ClC‐3 null mice, as well as in Y27632‐treated mice. These results suggested that the activation of ClC‐3 channels may be related to AngII‐induced vascular remodelling through the Rho/Rho‐kinase pathway.
In conclusion, we found that, in VSMC, phosphorylation of Thr532 in the ClC‐3 protein by ROCK2 is required for AngII‐induced Cl− current and cell migration. This suggests that modulation of the activation of ClC‐3 channels could be a novel strategy to prevent AngII‐induced and migration‐related diseases, such as vascular remodelling and atherosclerosis.
Author contributions
Y Y G and G L W proposed this study idea, designed experiments and finally revised the paper. M M M, C X L and C Z L performed the experiments, analysed the data, contributed to design some experiments. M M M wrote the draft manuscript. M G, L S, Y B T and J G Z performed some experiments and analysed the data.
Conflicts of interest
None.
Supporting information
Table S1 The blood pressure of Angiotensin II‐induced hypertensive mice.
Figure S1 A. The expression of ClC‐3 was significantly reduced by ClC‐3 siRNA transfection (40 nmol/L) for 48h in A10 cells, while the expression of TMEM16A and CFTR were not altered by knocking down the expression of ClC‐3. Neg represents cells transfected with scrambled siRNA. B. Bar chart of densitometric analysis from the experiments (n=5, *P<0.05 vs. con).
Figure S2 The effect of Y27632 and SU6656 on AngII‐induced Cl‐ current. A. AngII‐induced Cl‐ current was partially inhibited by the treatment with Y27632 (10 µmol/L) or SU6656 (10 µmol/L) in A10 cells, and this current was further reduced to the basal level by the combination of Y27632 and SU6656. B. Bar chart from the experiments at −80mV (downward bars) and +80mV (upward bars), respectively (n=6, *P<0.05 vs. con, #P<0.05 vs. Ang).
Figure S3 The cotransfection efficiency in A10 cells transfected with ROCK2 and ClC‐3, ΔNT, or ΔCT (n=4, 100×). Green represents cells expressing GFP‐tagged ClC‐3, ΔNT, or ΔCT, respectively. Red represents cells expressing HA‐tagged ROCK2. Blue represents nucleus stained with Hoechst 33258.
Figure S4 A. ROCK2 siRNA transfection (40 nmol/L) for 48h significantly decreased endogenous ROCK2 protein expression in A10 cells, while the expression of PRK2 and MSK1 were not changed by ROCK2 siRNA transfection. Neg represents cells transfected with scrambled siRNA. B. Bar chart of densitometric analysis from the experiments (n=5, *P<0.05 vs. con).
Figure S5 Knockdown of PRK2 or MSK1 had no significant inhibitory effect on AngII‐induced Cl‐ current. A. Western blots showing the expression of PRK2 or MSK1 in A10 cells transfected with PRK2 siRNA (40 nmol/L) or MSK1 siRNA (40 nmol/L) for 48h, respectively (n=5, *P<0.05 vs. con). B. Representative traces of AngII‐induced Cl‐ current in A10 cells transfected with PRK2 siRNA or MSK1 siRNA (n=6).
Figure S6 The expression of ClC‐3 and its mutants were equal in both whole cell lysis and membrane extract. A. The expression of GFP in cells transfected with GFP‐tagged ClC‐3 or its mutants in whole cell lysis (total) or membrane extract (membrane). β‐actin and Na+‐K+‐ATPase were selected as internal reference, respectively. B. Bar chart of densitometric analysis from the experiments (n=5).
Figure S7 AngII (1 µmol/L)‐induced migration was significantly inhibited by ClC‐3 siRNA transfection (40 nmol/L) for 48h in A10 cells. A. Representative images of transwell assay carried out in control, negative, and ClC‐3 siRNA transfection group, with or without the treatment with AngII. B. Bar chart of relative migration rate from the experiments (n=5, *P<0.05 vs. con, #P<0.05 vs. Ang).
Figure S8 The original images of all western blotting experiments.
Supporting info item
Acknowledgements
This work was supported by National Natural Science Foundation of China (Key grants nos. 81230082, 81173055, 81302771, 81370897, 81525025, 81473206, 81273500) and CHINA–CANADA Joint Health Research Program from NSFC‐CIHR (No. 81361128011).
We thank Dr. Robert M K W Lee in McMaster University, Canada, for kindly improving the manuscript.
Ma, M.‐M. , Lin, C.‐X. , Liu, C.‐Z. , Gao, M. , Sun, L. , Tang, Y.‐B. , Zhou, J.‐G. , Wang, G.‐L. , and Guan, Y.‐Y. (2016) Threonine532 phosphorylation in ClC‐3 channels is required for angiotensin II‐induced Cl− current and migration in cultured vascular smooth muscle cells. British Journal of Pharmacology, 173: 529–544. doi: 10.1111/bph.13385.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arreola J, Begenisich T, Nehrke K, Nguyen HV, Park K, Richardson L, et al (2002). Secretion and cell volume regulation by salivary acinar cells from mice lacking expression of the Clcn3 Cl− channel gene. J Physiol 545: 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S (1999). Sequence and structure‐based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294: 1351–1362. [DOI] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM (2004). Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol 124: 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM (2006). EGFR kinase regulates volume‐sensitive chloride current elicited by integrin stretch via PI‐3K and NADPH oxidase in ventricular myocytes. J Gen Physiol 127: 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claud EC, Lu J, Wang XQ, Abe M, Petrof EO, Sun J, et al (2008). Platelet‐activating factor‐induced chloride channel activation is associated with intracellular acidosis and apoptosis of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 294: G1191–G1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah VA, Sontheimer H (2010). Molecular interaction and functional regulation of ClC‐3 by Ca2+/calmodulin‐dependent protein kinase II (CaMKII) in human malignant glioma. J Biol Chem 285: 11188–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah VA, Turner KL, Seifert S, Sontheimer H (2013). Bradykinin‐induced chemotaxis of human gliomas requires the activation of KCa 3.1 and ClC‐3. J Neurosci 33: 1427–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriy LV, Gomez EA, Jacobson DA, Wang X, Hopson JA, Liu XY, et al (2009). The granular chloride channel ClC‐3 is permissive for insulin secretion. Cell Metab 10: 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson LW, Bonthius DJ, Schutte BC, Yang B, Barna TJ, Bailey MC, et al (2002). Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC‐3 voltage‐gated chloride channels. Brain Res 958: 227–250. [DOI] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B (1997). Molecular identification of a volume‐regulated chloride channel. Nature 390: 417–421. [DOI] [PubMed] [Google Scholar]
- Duran C, Thompson CH, Xiao Q, Hartzell HC (2010). Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol 72: 95–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R (2002). X‐ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 415: 287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, MacKinnon R (2003). Gating the selectivity filter in ClC chloride channels. Science 300: 108–112. [DOI] [PubMed] [Google Scholar]
- Farmer LM, Le BN, Nelson DJ (2013). CLC‐3 chloride channels moderate long‐term potentiation at Schaffer collateral‐CA1 synapses. J Physiol 591: 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathi SB, Wei SG, Zaremba A, Lamb FS, Shears SB (2013). Functional regulation of ClC‐3 in the migration of vascular smooth muscle cells. Hypertension 61: 174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan YY, Wang GL, Zhou JG (2006). The ClC‐3 Cl− channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci 27: 290–296. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Naiki T (2009). Adaptation and remodeling of vascular wall; biomechanical response to hypertension. J Mech Behav Biomed Mater 2: 3–19. [DOI] [PubMed] [Google Scholar]
- Huang LY, He Q, Liang SJ, Su YX, Xiong LX, Wu QQ, et al (2014). ClC‐3 chloride channel/antiporter defect contributes to inflammatory bowel disease in humans and mice. Gut 63: 1587–1595. [DOI] [PubMed] [Google Scholar]
- Isnard‐Bagnis C, Da SN, Beaulieu V, Yu AS, Brown D, Breton S (2003). Detection of ClC‐3 and ClC‐5 in epididymal epithelium: immunofluorescence and RT‐PCR after LCM. Am J Physiol Cell Physiol 284: C220–C232. [DOI] [PubMed] [Google Scholar]
- Kang XL, Zhang M, Liu J, Lv XF, Tang YB, Guan YY (2012). Differences between femoral artery and vein smooth muscle cells in volume‐regulated chloride channels. Can J Physiol Pharmacol 90: 1516–1526. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wu DB, Wang J (2013). Effects of volume‐activated chloride channels on the invasion and migration of human endometrial cancer cells. Eur J Gynaecol Oncol 34: 60–64. [PubMed] [Google Scholar]
- Liang W, Huang L, Zhao D, He JZ, Sharma P, Liu J, et al (2014). Swelling‐activated Cl‐ currents and intracellular CLC‐3 are involved in proliferation of human pulmonary artery smooth muscle cells. J Hypertens 32: 318–330. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Wang XG, Tang YB, Chen JH, Lv XF, Zhou JG, et al (2010). Simvastatin ameliorates rat cerebrovascular remodeling during hypertension via inhibition of volume‐regulated chloride channel. Hypertension 56: 445–452. [DOI] [PubMed] [Google Scholar]
- Loirand G, Guerin P, Pacaud P (2006). Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 98: 322–334. [DOI] [PubMed] [Google Scholar]
- Lui VC, Lung SS, Pu JK, Hung KN, Leung GK (2010). Invasion of human glioma cells is regulated by multiple chloride channels including ClC‐3. Anticancer Res 30: 4515–4524. [PubMed] [Google Scholar]
- Mao J, Chen L, Xu B, Wang L, Li H, Guo J, et al (2008). Suppression of ClC‐3 channel expression reduces migration of nasopharyngeal carcinoma cells. Biochem Pharmacol 75: 1706–1716. [DOI] [PubMed] [Google Scholar]
- McCloskey DT, Doherty L, Dai YP, Miller L, Hume JR, Yamboliev IA (2007). Hypotonic activation of short ClC3 isoform is modulated by direct interaction between its cytosolic C‐terminal tail and subcortical actin filaments. J Biol Chem 282: 16871–16877. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic A, Brandl C, Milenkovic VM, Jendryke T, Sirianant L, Wanitchakool P, et al (2015). Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc Natl Acad Sci U S A 112: E2630–E2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel‐Herrera M, Zaske AM, Garcia‐Colunga J, Martinez‐Torres A, Miledi R (2011). Ion currents induced by ATP and angiotensin II in cultured follicular cells of Xenopus laevis . Mol Cells 32: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreland JG, Davis AP, Bailey G, Nauseef WM, Lamb FS (2006). Anion channels, including ClC‐3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. J Biol Chem 281: 12277–12288. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H (2003). Expression of voltage‐gated chloride channels in human glioma cells. J Neurosci 23: 5572–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacurari M, Kafoury R, Tchounwou PB, Ndebele K (2014). The Renin–angiotensin–aldosterone system in vascular inflammation and remodeling. Int J Inflam 2014: 689360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR . (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42: D1098‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Du YH, Tang YB, Lv XF, Liu J, Zhou JG, et al (2011). ClC‐3 chloride channel prevents apoptosis induced by hydrogen peroxide in basilar artery smooth muscle cells through mitochondria dependent pathway. Apoptosis 16: 468–477. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, et al (2014). Swell 1, a plasma membrane protein, is an essential component of volume‐regulated anion channel. Cell 157: 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Raucci FJ, Browe DM, Baumgarten CM (2008). Regulation of swelling‐activated Cl(−) current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res 77: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NC, Huang P, Kaetzel MA, Lamb FS, Nelson DJ (2004). Identification of an N‐terminal amino acid of the CLC‐3 chloride channel critical in phosphorylation‐dependent activation of a CaMKII‐activated chloride current. J Physiol 556: 353–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossow CF, Duan D, Hatton WJ, Britton F, Hume JR, Horowitz B (2006). Functional role of amino terminus in ClC‐3 chloride channel regulation by phosphorylation and cell volume. Acta Physiol (Oxf) 187: 5–19. [DOI] [PubMed] [Google Scholar]
- Savoia C, Volpe M (2011). Angiotensin receptor modulation and cardiovascular remodeling. J Renin Angiotensin Aldosterone Syst 12: 381–384. [DOI] [PubMed] [Google Scholar]
- Stauber T, Jentsch TJ (2013). Chloride in vesicular trafficking and function. Annu Rev Physiol 75: 453–477. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, et al (2001). Disruption of ClC‐3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29: 185–196. [DOI] [PubMed] [Google Scholar]
- Tang YB, Liu YJ, Zhou JG, Wang GL, Qiu QY, Guan YY (2008). Silence of ClC‐3 chloride channel inhibits cell proliferation and the cell cycle via G/S phase arrest in rat basilar arterial smooth muscle cells. Cell Prolif 41: 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk AP, Heise CK, Hougen JL, Artman CM, Volk KA, Wessels D, et al (2008). ClC‐3 and ICls well are required for normal neutrophil chemotaxis and shape change. J Biol Chem 283: 34315–34326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, et al (2014). Identification of LRRC8 heteromers as an essential component of the volume‐regulated anion channel VRAC. Science 344: 634–638. [DOI] [PubMed] [Google Scholar]
- Wang GL, Wang XR, Lin MJ, He H, Lan XJ, Guan YY (2002). Deficiency in ClC‐3 chloride channels prevents rat aortic smooth muscle cell proliferation. Circ Res 91: E28–E32. [DOI] [PubMed] [Google Scholar]
- Wang GX, Dai YP, Bongalon S, Hatton WJ, Murray K, Hume JR, et al (2005). Hypotonic activation of volume‐sensitive outwardly rectifying anion channels (VSOACs) requires coordinated remodeling of subcortical and perinuclear actin filaments. J Membr Biol 208: 15–26. [DOI] [PubMed] [Google Scholar]
- Wang GX, McCrudden C, Dai YP, Horowitz B, Hume JR, Yamboliev IA (2004). Hypotonic activation of volume‐sensitive outwardly rectifying chloride channels in cultured PASMCs is modulated by SGK. Am J Physiol Heart Circ Physiol 287: H533–H544. [DOI] [PubMed] [Google Scholar]
- Wang XG, Tao J, Ma MM, Tang YB, Zhou JG, Guan YY (2013). Tyrosine 284 phosphorylation is required for ClC‐3 chloride channel activation in vascular smooth muscle cells. Cardiovasc Res 98: 469–478. [DOI] [PubMed] [Google Scholar]
- Yang H, Huang LY, Zeng DY, Huang EW, Liang SJ, Tang YB, et al (2012). Decrease of intracellular chloride concentration promotes endothelial cell inflammation by activating nuclear factor‐kappaB pathway. Hypertension 60: 1287–1293. [DOI] [PubMed] [Google Scholar]
- Zeng JW, Wang XG, Ma MM, Lv XF, Liu J, Zhou JG, et al (2014). Integrin beta3 mediates cerebrovascular remodelling through Src/ClC‐3 volume‐regulated Cl(−) channel signalling pathway. Br J Pharmacol 171: 3158–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng LY, Li L, Ma MM, Liu Y, Wang GL, Tang YB, et al (2013). Deficiency of volume‐regulated ClC‐3 chloride channel attenuates cerebrovascular remodelling in DOCA‐salt hypertension. Cardiovasc Res 100: 134–142. [DOI] [PubMed] [Google Scholar]
- Zhou JG, Ren JL, Qiu QY, He H, Guan YY (2005. Regulation of intracellular Cl− concentration through volume‐regulated ClC‐3 chloride channels in A10 vascular smooth muscle cells. J Biol Chem 280: 7301–7308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 The blood pressure of Angiotensin II‐induced hypertensive mice.
Figure S1 A. The expression of ClC‐3 was significantly reduced by ClC‐3 siRNA transfection (40 nmol/L) for 48h in A10 cells, while the expression of TMEM16A and CFTR were not altered by knocking down the expression of ClC‐3. Neg represents cells transfected with scrambled siRNA. B. Bar chart of densitometric analysis from the experiments (n=5, *P<0.05 vs. con).
Figure S2 The effect of Y27632 and SU6656 on AngII‐induced Cl‐ current. A. AngII‐induced Cl‐ current was partially inhibited by the treatment with Y27632 (10 µmol/L) or SU6656 (10 µmol/L) in A10 cells, and this current was further reduced to the basal level by the combination of Y27632 and SU6656. B. Bar chart from the experiments at −80mV (downward bars) and +80mV (upward bars), respectively (n=6, *P<0.05 vs. con, #P<0.05 vs. Ang).
Figure S3 The cotransfection efficiency in A10 cells transfected with ROCK2 and ClC‐3, ΔNT, or ΔCT (n=4, 100×). Green represents cells expressing GFP‐tagged ClC‐3, ΔNT, or ΔCT, respectively. Red represents cells expressing HA‐tagged ROCK2. Blue represents nucleus stained with Hoechst 33258.
Figure S4 A. ROCK2 siRNA transfection (40 nmol/L) for 48h significantly decreased endogenous ROCK2 protein expression in A10 cells, while the expression of PRK2 and MSK1 were not changed by ROCK2 siRNA transfection. Neg represents cells transfected with scrambled siRNA. B. Bar chart of densitometric analysis from the experiments (n=5, *P<0.05 vs. con).
Figure S5 Knockdown of PRK2 or MSK1 had no significant inhibitory effect on AngII‐induced Cl‐ current. A. Western blots showing the expression of PRK2 or MSK1 in A10 cells transfected with PRK2 siRNA (40 nmol/L) or MSK1 siRNA (40 nmol/L) for 48h, respectively (n=5, *P<0.05 vs. con). B. Representative traces of AngII‐induced Cl‐ current in A10 cells transfected with PRK2 siRNA or MSK1 siRNA (n=6).
Figure S6 The expression of ClC‐3 and its mutants were equal in both whole cell lysis and membrane extract. A. The expression of GFP in cells transfected with GFP‐tagged ClC‐3 or its mutants in whole cell lysis (total) or membrane extract (membrane). β‐actin and Na+‐K+‐ATPase were selected as internal reference, respectively. B. Bar chart of densitometric analysis from the experiments (n=5).
Figure S7 AngII (1 µmol/L)‐induced migration was significantly inhibited by ClC‐3 siRNA transfection (40 nmol/L) for 48h in A10 cells. A. Representative images of transwell assay carried out in control, negative, and ClC‐3 siRNA transfection group, with or without the treatment with AngII. B. Bar chart of relative migration rate from the experiments (n=5, *P<0.05 vs. con, #P<0.05 vs. Ang).
Figure S8 The original images of all western blotting experiments.
Supporting info item