

Abstract
Background and Purpose
In the phase III clinical trial, RELAX‐AHF, serelaxin caused rapid and long‐lasting haemodynamic changes. However, the cellular mechanisms involved are unclear in humans.
Experimental Approach
This study examined the effects of serelaxin in co‐cultures of human primary endothelial cells (ECs) and smooth muscle cells (SMCs) on cAMP and cGMP signalling.
Key Results
Stimulation of HUVECs or human coronary artery endothelial cells (HCAECs) with serelaxin, concentration‐dependently increased cGMP accumulation in co‐cultured SMCs to a greater extent than in monocultures of either cell type. This was not observed in human umbilical artery endothelial cells (HUAECs) that do not express the relaxin receptor, RXFP1. Treatment of ECs with l‐NG‐nitro arginine (NOARG; 30 μM, 30 min) inhibited serelaxin‐mediated (30 nM) cGMP accumulation in HUVECs, HCAECs and co‐cultured SMCs. In HCAECs, but not HUVECs, pre‐incubation with indomethacin (30 μM, 30 min) also inhibited cGMP accumulation in SMCs. Pre‐incubation of SMCs with the guanylate cyclase inhibitor ODQ (1 μM, 30 min) had no effect on serelaxin‐mediated (30 nM) cGMP accumulation in HUVECs and HCAECs but inhibited cGMP accumulation in SMCs. Serelaxin stimulation of HCAECs, but not HUVECs, increased cAMP accumulation concentration‐dependently in SMCs. Pre‐incubation of HCAECs with indomethacin, but not l‐NOARG, abolished cAMP accumulation in co‐cultured SMCs, suggesting involvement of prostanoids.
Conclusions and Implications
In co‐cultures, treatment of ECs with serelaxin caused marked cGMP accumulation in SMCs and with HCAEC also cAMP accumulation. Responses involved EC‐derived NO and with HCAEC prostanoid production. Thus, serelaxin differentially modulates vascular tone in different vascular beds.
Abbreviations
- AHF
acute heart failure
- DEA
diethylamine NONOate
- ECs
endothelial cells
- HCAEC
human coronary artery endothelial cell
- HUAEC
human umbilical artery endothelial cell
- HUASMC
human umbilical artery smooth muscle cell
- HUVSMC
human umbilical vein smooth muscle cell
- l‐NOARG
l‐NG‐nitro arginine
- SMCs
smooth muscle cells
Tables of Links
| TARGETS |
|---|
| GPCRsa |
| RXFP1, relaxin family peptide receptor 1, |
| Enzymesb |
| COX |
| GC, guanylate cyclase |
| NOS |
| LIGANDS |
|---|
| Indomethacin |
| cAMP |
| cGMP |
| NO |
| ODQ |
| Relaxin, human H2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a cAlexander et al., 2013b, 2013).
Introduction
Vasodilators are a cornerstone of therapy for acute heart failure (AHF). Standard therapies such as loop diuretics, nitrates, β‐blockers and ACE inhibitors cause vasodilation and/or prevent vasoconstriction (Hollenberg, 2007). However, most vasodilators exhibit side effects with hypotension being the most commonly reported example in patients with heart failure (Hollenberg, 2007). Serelaxin, the recombinant form of the human hormone relaxin, presents as a novel treatment option for AHF and in the phase III clinical trial, RELAX‐AHF, serelaxin relieved dyspnoea and congestion in patients with AHF but also significantly reduced patient mortality at day 180 without notable side effects (Teerlink et al., 2013). Serelaxin treatment was also associated with rapid and long‐lasting haemodynamic changes including reductions in pulmonary capillary wedge pressure, pulmonary artery pressure (systolic and diastolic), pulmonary vascular resistance, right atrial pressure and systemic vascular resistance (Ponikowski et al., 2013). These could be attributed to the vasodilatory effects of relaxin that have been reported in vitro (Bani et al., 1998; McGuane et al., 2011b; Sarwar et al., 2014; Boccalini et al., 2015), in vivo (Masini et al., 1997; Danielson et al., 1999; Masini et al., 2002; Conrad et al., 2004; Debrah et al., 2005, 2006; Conrad and Shroff, 2011; McGuane et al., 2011a; Segal et al., 2012) and in patients with AHF (Voors et al., 2011; Ponikowski et al., 2013; Voors et al., 2014).
Relaxin acts at the cognate relaxin receptor, RXFP1,that is expressed in endothelial cells (ECs) and smooth muscle cells (SMCs) of arteries and veins, although the expression pattern does not always necessarily correlate with function (Jelinic et al., 2013). Studies on human isolated vessels are rare, but relaxin does cause vasodilation in human isolated s.c. and small systemic resistance arteries (McGuane et al., 2011b). Although the precise cellular mechanisms of the haemodynamic effects of relaxin in humans are poorly understood, two distinct mechanisms have been described. Rapid relaxin‐mediated vasodilation occurs via a Gαi/PI3K/cAMP/NO‐dependent mechanism (McGuane et al., 2011b), whereas sustained relaxin‐mediated responses are associated with changes in activity or expression of gelatinases, endothelin receptor B (ETB), vascular endothelial growth factor (VEGF) and nitric oxide synthase (NOS) (Dschietzig et al., 2003; Jeyabalan et al., 2003; McGuane et al., 2011a).
We have previously shown that these signalling mechanisms occur in primary ECs, SMCs and fibroblasts from the human vasculature (Sarwar et al., 2014), thereby identifying blood vessels as an important potential target for serelaxin in humans. We also showed that serelaxin had a variety of effects in cells from arteries and veins. However, in vivo, the vascular cells are organized as layers in blood vessels, and crosstalk between these cells has an important role to play in regulating the function of the vessel. Monocultures in vitro fail to integrate this natural physiological organization of blood vessels and on their own do not reflect the impact of cellular crosstalk on signal transduction.
The endothelium is known to release vasoactive substances that act on smooth muscle cells to regulate vessel tone. Acetylcholine and bradykinin cause endothelium‐dependent vasorelaxation via their respective G protein‐coupled receptors (Furchgott and Zawadzki, 1980) and the EC/SMC interactions involve NO (Palmer et al., 1987), prostacyclin (Radomski et al., 1987) and endothelium‐derived hyperpolarizing factor (EDHF) (Bolton et al., 1984). These interactions have been shown to affect cGMP and cAMP signalling, second messengers that are known to regulate cardiovascular function and are altered in disease (Ganz et al., 1986; Majed and Khalil, 2012). Indeed, relaxin‐mediated relaxation is abolished in human gluteal arteries that are endothelium denuded (Fisher, 2009), suggesting that relaxin signalling is endothelium‐dependent. Because there is a lack of information on the signal transduction mechanisms activated by relaxin in a physiologically relevant environment, we have investigated signalling in a cell co‐culture model of ECs and SMCs from human arteries and veins in order to better understand serelaxin‐mediated signal transduction in human blood vessels.
Methods
Human primary cells
Primary cultures of human umbilical artery endothelial cells (HUAEC), HUVEC, human coronary artery endothelial cells (HCAEC), human umbilical artery smooth muscle cells (HUASMC) and human umbilical vein smooth muscle cells (HUVSMC) were obtained from ScienCell Research Laboratories (San Diego, CA, USA). These cells were characterized as detailed previously (Sarwar et al., 2014). All cells were maintained in Medium 199 containing 5% FBS, penicillin (100 units per mL), streptomycin (100 μg⋅mL−1) and the relevant growth supplements for optimal growth of each cell type. As such, ECs were grown in EC growth supplement, smooth muscle cells in SMGS and fibroblasts in FGS‐2 (ScienCell) as detailed previously (Sarwar et al., 2014). Early culture passages (2–5) were used for each cell type.
Cell culture
For monoculture assays, both ECs and SMCs were plated in standard 24‐well CELLSTAR® multiwell plates (Greiner Bio‐One) at a density of 2 × 105 cells per well in a volume of 500 μL of growth medium per well. The cells were allowed to adhere and grow overnight. For co‐culture assays, ECs were plated on 24‐well ThinCerts (Greiner Bio‐One), comprising translucent membranes with 0.4 μm pores, at a density of 1 × 105 cells per insert in a volume of 400 μL of growth medium per insert. Smooth muscle cells were plated in standard 24‐well CELLSTAR multiwell plates (Greiner Bio‐One) at a density of 2 × 105 cells per well in a volume of 500 μL of growth medium per well. The cells were allowed to adhere and grow overnight and just prior to the experiment, ThinCerts were placed in wells containing smooth muscle cells.
cAMP and cGMP accumulation
cAMP accumulation was determined as previously described (Sarwar et al., 2014). Briefly, cells grown in monocultures, were pre‐incubated with stimulation buffer and treated with serelaxin at the given concentrations for 30 min. Forskolin (50 μM, 30 min) and diethylamine NONOate (DEA) (1 μM, 5 min) were used as positive controls for stimulating cAMP and cGMP synthesis respectively. Where appropriate, cells were pre‐incubated with the NOS inhibitor, l‐NG‐nitro arginine (l‐NOARG; 30 μM, 30 min), the non‐specific COX inhibitor, indomethacin; (30 μM, 30 min) or ODQ, the guanylate cyclase (GC) inhibitor (1 μM, 30 min). Following stimulation with serelaxin (30 min), the cells were rapidly lysed, and cAMP and cGMP levels were measured with AlphaScreen cAMP and cGMP kits (Perkin‐Elmer, Australia). For co‐culture studies, cells on the ThinCerts were stimulated with serelaxin (30 min), and/or cells were treated with the relevant inhibitors. Before stimulation with serelaxin, ThinCerts were placed directly on top of the wells containing the smooth muscle cells, and after completion of the assay, cells were separated and lysed. cAMP and cGMP levels were detected in each cell type using the AlphaScreen cAMP and cGMP kits (Perkin‐Elmer).
Data analysis
All data represent the means ± SEM of at least five individual experiments unless otherwise indicated in the text. Data was analysed using graphpad prism v6.0. Replicates were averaged before entry as a single data point. Concentration–response curves were fitted using a sigmoidal or Gaussian distribution function. Statistical significance was determined using one‐way anova with significance accepted at P < 0.05. If F reached significance, the Dunnett's post hoc test was used to compare groups.
Materials
Serelaxin (the recombinant form of human gene 2 relaxin) was kindly provided by Corthera, Inc. (a subsidiary of Novartis AG, Switzerland). 1H‐[1,2,4]Oxadiazolo[4,3‐a]quinoxalin‐1‐one (ODQ), l‐NG‐nitro arginine (l‐NOARG) and indomethacin were purchased from Sigma (Australia). Cell co‐culture ThinCerts™ were purchased from Greiner Bio‐One (Germany).
Results
Serelaxin stimulation of HUVEC and HCAEC but not HUAEC enhances cGMP accumulation in co‐cultures of HUASMC and HUVSMC
The addition of serelaxin (30 min) to HUAEC co‐cultured with HUASMC (Figure 1A) or HUVSMC (Figure 1B) failed to produce a cGMP response in HUAEC (Figure 1C,D) or in HUASMC (Figure 1C) or HUVSMC (Figure 1D). This can be explained by the lack of cell surface RXFP1 expression in HUAEC (Sarwar et al., 2014) because direct stimulation of either HUASMC (Figure 1C, dashed line; pEC50: 9.5 ± 0.5) or HUVSMC (Figure 1D, dashed line; pEC50: 9.3 ± 0.3) with serelaxin (30 min) produced concentration‐dependent increases in cGMP accumulation of 30% and 32% of the DEA response respectively. The absence of a cGMP response in SMCs co‐cultured with HUAEC demonstrates that after addition of serelaxin, although the peptide may penetrate the 0.4 μm pores within the insert, it fails to reach a concentration in the SMC chamber sufficient to cause a response.
Figure 1.
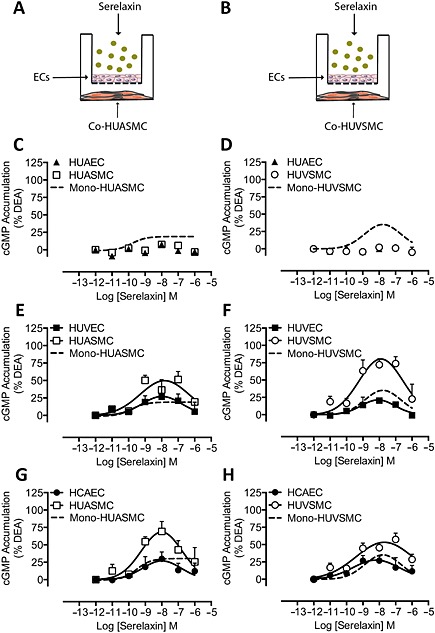
cGMP accumulation in co‐cultures of human primary vascular smooth muscle cells following addition of serelaxin to endothelium. HUAEC , HUVEC or HCAEC were co‐cultured with (A) HUASMC or (B) HUVSMC (all n = 5), and the ECs were treated with serelaxin for 30 min. Serelaxin addition to HUAEC did not cause cGMP accumulation in HUAEC (▲) (C) HUASMC (□) or (D) HUVSMC (◯) co‐cultured with HUAEC, whereas direct stimulation of either (C) HUASMC (n = 5) or (D) HUVSMC with serelaxin caused a concentration‐dependent increase in cGMP accumulation (dashed lines). In contrast, serelaxin addition to HUVEC concentration‐dependently increased cGMP accumulation not only in HUVEC (■) but also in (E) HUASMC (□) or (F) HUVSMC (◯) co‐cultured with HUVEC with the responses in smooth muscle cells being greater or in the case of HUVSMC much greater than cGMP responses to direct stimulation of (E) HUASMC or (F) HUVSMC (dashed lines). A similar pattern of cGMP accumulation was observed with (G, H) HCAEC (●) and (G) HUASMC (□) or (H) HUVSMC (◯) co‐cultured with HCAEC.
In contrast, addition of serelaxin (30 min) to HUVEC, which do express RXFP1 (Sarwar et al., 2014), when co‐cultured with HUASMC, not only increased cGMP accumulation to 27% of the DEA response in HUVEC (Figure 1E; pEC50: 9.8 ± 1.2) but also caused a large, concentration‐dependent increase in cGMP accumulation in HUASMC (Figure 1E; pEC50: 9.8 ± 0.5) to 50% of the DEA response or 1.7‐fold higher than the maximal response observed when HUASMC were directly stimulated with serelaxin (Figure 1E, dashed line). Similarly, when HUVEC were co‐cultured with HUVSMC (Figure 1F), serelaxin treatment (30 min) increased cGMP accumulation to 21% of the DEA response in HUVEC (Figure 1F, pEC50: 9.7 ± 0.6) but also caused a robust increase in cGMP accumulation in the co‐cultured HUVSMC reaching 80% of DEA response (Figure 1F, pEC50: 9.5 ± 0.3), or 2.5 times higher than the maximal response obtained with HUVSMC directly stimulated with serelaxin (Figure 1F, dashed line). It was noted that whereas the concentration–response relationship in HUASMC in monocultures was sigmoidal, it became bell‐shaped in co‐cultures with HUVEC.
To examine whether the difference between co‐cultures involving HUAEC and HUVEC represented a difference between arterial and venous ECs or a regional difference between ECs, we also utilized co‐cultures involving HCAEC. In co‐cultures of HCAEC/HUASMC, treatment of HCAEC with serelaxin (30 min) produced a modest increase in cGMP accumulation to 27% of the DEA response (Figure 1G, pEC50: 9.8 ± 0.9) but also robustly increased cGMP accumulation in the co‐cultured HUASMC reaching 68% of DEA response (Figure 1G, pEC50: 9.7 ± 0.6), or 2.1 times higher than cGMP responses observed in HUASMC directly stimulated with serelaxin (Figure 1G, dashed line). In co‐cultures of HCAEC/HUVSMC, treatment of HCAEC with serelaxin (30 min) produced a modest increase in cGMP accumulation to about 28% of DEA response (Figure 1H, pEC50: 9.9 ± 0.7) but also increased cGMP accumulation in HUVSMC to 53% of the DEA response (Figure 1H, pEC50: 9.5 ± 0.4), about 1.8 times that of cGMP responses observed in HUVSMC directly stimulated with serelaxin (Figure 1H, dashed line).
Serelaxin‐mediated NO generation in HUVEC and HCAEC is responsible for cGMP accumulation in HUASMC and HUVSMC
To determine how serelaxin treatment of HUVEC and HCAEC caused cGMP accumulation in arterial and venous SMCs, we used pharmacological inhibitors to disrupt key signalling pathways. Because in intact blood vessels NO is known to be generated by ECs to stimulate cGMP in smooth muscle cells (Furchgott and Vanhoutte, 1989), we incubated ECs with the NOS inhibitor l‐NOARG and stimulated with serelaxin (30 nM, 30 min). In monocultures, pre‐treatment with the general NOS inhibitor, l‐NOARG (30 μM, 30 min) significantly inhibited serelaxin‐mediated cGMP responses (% DEA) in HUVEC (Figure 2A), HCAEC (Figure 2B), HUASMC (Figure 2C) and HUVSMC (Figure 2D, Table S1) suggesting that serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in these cells is NO dependent.
Figure 2.
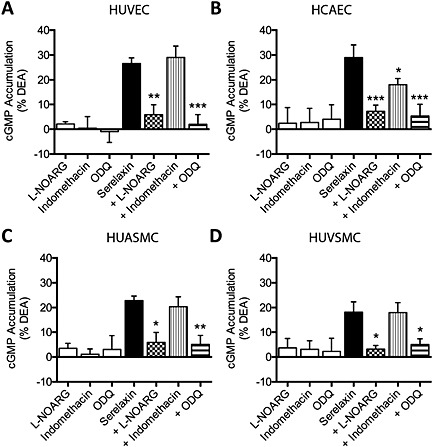
Serelaxin‐mediated cGMP accumulation in monocultures of human primary vascular cells (all n = 5). Serelaxin (30 nM, 30 min) increased cGMP accumulation in (A) HUVEC, (B) HCAEC, (C) HUASMC and (D) HUVSMC. Pre‐incubation with l‐NOARG (30 μM, 30 min) or ODQ (1 μM, 30 min) almost abolished serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in all cell types. Pre‐treatment with indomethacin (30 μM, 30 min) significantly inhibited serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in (B) HCAEC but had no effect in (A) HUVEC, (C) HUASMC or (D) HUVSMC. *P < 0.05, **P < 0.02, ***P < 0.005; significantly different from serelaxin alone; one‐way anova with Dunnett's post hoc test.
We next determined whether NO mediates crosstalk between ECs and SMCs by incubating ECs with l‐NOARG (30 μM, 30 min) and serelaxin (30 nM, 30 min) (Figure 3A). In co‐cultures (Figure 3A), pre‐treatment of HUVEC with l‐NOARG (30 μM, 30 min) abolished serelaxin‐mediated (30 nM, 30 min) cGMP responses (% DEA) not only in HUVEC (Figure 3B) but also in both HUASMC (Figure 3D) and HUVSMC (Figure 3E, Table S2). In co‐cultures with HCAEC, pre‐treatment with l‐NOARG (30 μM, 30 min) almost abolished serelaxin‐mediated (30 nM, 30 min) cGMP accumulation not only in HCAEC (Figure 3C) but also in HUASMC (Figure 3F) and HUVSMC (Figure 3G, Table S1). These results suggest that endothelial NO production is essential for cGMP responses in co‐cultured arterial and venous SMCs.
Figure 3.
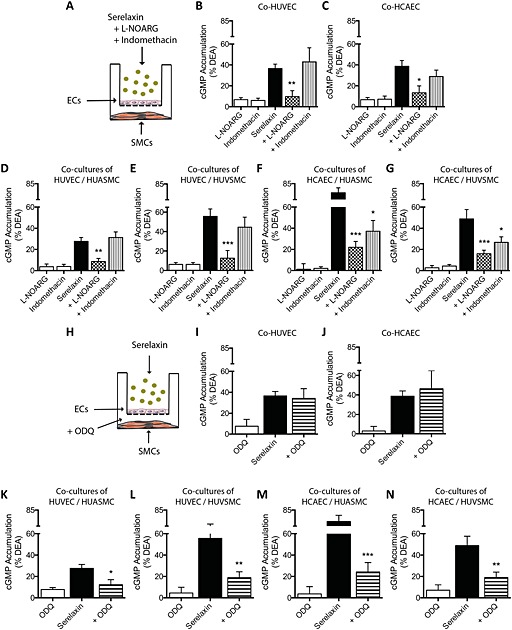
Serelaxin‐mediated cGMP accumulation in human primary vascular smooth muscle cells co‐cultured with HUVEC or (A) HCAEC (all n = 6 except where otherwise indicated). Stimulation of HUVEC or HCAEC with serelaxin (30 nM, 30 min) increased cGMP accumulation not only in (B) HUVEC and (C) HCAEC but also in co‐cultures of (D, F) HUASMC or (E, G) HUVSMC. Pre‐incubation of HUVEC or HCAEC with l‐NOARG (30 μM, 30 min) before addition of serelaxin (30 nM, 30 min) significantly inhibited cGMP accumulation not only in HUVEC and (C) HCAEC but also in (D, F) HUASMC and (E, G) HUVSMC. Pre‐incubation of HUVEC with indomethacin (30 μM, 30 min) did not affect serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in (B) HUVEC or in co‐incubated (D) HUASMC or (E) HUVSMC (n = 5). Pre‐incubation of HCAEC with indomethacin (30 μM, 30 min) had no significant effect on serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in (C) HCAEC but produced marked and significant reductions in cGMP accumulation in co‐incubated (F) HUASMC or (G) HUVSMC (n = 5). Pre‐treatment of HUASMC or HUVSMC with ODQ (1 μM, 30 min) had no significant effect on serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in (I) HUVEC or (J) HCAEC but reduced or abolished cGMP accumulation in (K, M) HUASMC or (L, N) HUVSMC (n = 5). *P < 0.05, **P < 0.02, ***P < 0.005 significantly different from serelaxin alone; one‐way anova with Dunnett's post hoc test.
Serelaxin‐mediated prostanoid production in HCAEC but not HUVEC influences cGMP accumulation in HUASMC and HUVSMC
We next determined whether prostanoids had a role in endothelium‐dependent responses in co‐cultures because previous studies have shown that endothelial prostanoids can act on smooth muscle cells to affect cAMP signalling (Furchgott and Vanhoutte, 1989; Majed and Khalil, 2012). While little is known of the role of prostanoids in vasodilator responses to serelaxin, indomethacin treatment is known to affect responses in some blood vessels (Fisher, 2009).
Indomethacin pre‐treatment (30 μM, 30 min) did not influence serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in monocultures of HUVEC (Figure 2A), HUASMC (Figure 2C) and HUVSMC (Figure 2D) but significantly inhibited serelaxin‐mediated cGMP accumulation in HCAEC (Figure 2B, Table S1). In co‐cultures, pre‐treatment with indomethacin (30 μM, 30 min) had no effect on serelaxin‐mediated cGMP accumulation in HUVEC (Figure 3D) or on cGMP responses in HUASMC (Figure 3D) or HUVSMC (Figure 3E, Table S2), showing that serelaxin does not stimulate prostanoid production in HUVEC. However, indomethacin pre‐treatment did (as in the monocultures) appear to reduce cGMP accumulation in HCAEC (Figure 3C) and significantly reduced cGMP accumulation in the co‐cultures of both HUASMC (Figure 3F) and HUVSMC (Figure 3G, Table S2). This suggests that in HCAEC, endothelial prostanoid production has a significant influence on cGMP signalling in arterial and venous smooth muscle cells.
GC activation and cGMP accumulation in HUASMC and HUVSMC is dependent on HUVEC and HCAEC
Because previous studies showed that NO activates GC in SMCs (Martin et al., 2005), we pre‐treated SMCs with the GC inhibitor ODQ and stimulated ECs with serelaxin. In monocultures, pre‐treatment with ODQ (1 μM, 30 min), significantly inhibited serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in HUVEC (Figure 2A), HCAEC (Figure 2B), HUASMC (Figure 2C) and HUVSMC (Figure 2D, Table S1). In co‐cultures with HUVEC, pre‐treatment of HUASMC or HUVSMC with ODQ (1 μM, 30 min) had no significant effect on serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in HUVEC (Figure 3I) but markedly reduced cGMP accumulation in both HUASMC (Figure 3K) and HUVSMC (Figure 3L, Table S2). Likewise in co‐cultures with HCAEC, pre‐treatment of HUASMC or HUVSMC with ODQ (1 μM, 30 min) had no significant effect on serelaxin‐mediated (30 nM, 30 min) cGMP accumulation in HCAEC (Figure 3J), but significantly reduced cGMP accumulation in both HUASMC (Figure 3M) and HUVSMC (Figure 3N, Table S2).
Treatment of HCAEC but not HUAEC or HUVEC with serelaxin enhances cAMP accumulation in HUASMC and HUVSMC
In order to examine whether cAMP was another mediator involved in the vasodilator response in SMCs in response to serelaxin treatment, we investigated the effect of the peptide in EC/SMC co‐culture (Figure 4A,B) on cAMP accumulation. In co‐cultures with HUAECs, serelaxin (30 min) treatment failed to produce a cAMP response in HUAECs (Figure 4C,D), HUASMC (Figure 4C) or HUVSMC (Figure 4D). In monocultures, treatment with serelaxin (30 min) increased cAMP accumulation in HUASMC (Figure 4C, dashed line, pEC50: 9.6 ± 0.7) and HUVSMC (Figure 4D, dashed line, pEC50: 9.4 ± 0.4), with maximal responses 5% and 6% of the forskolin response respectively.
Figure 4.
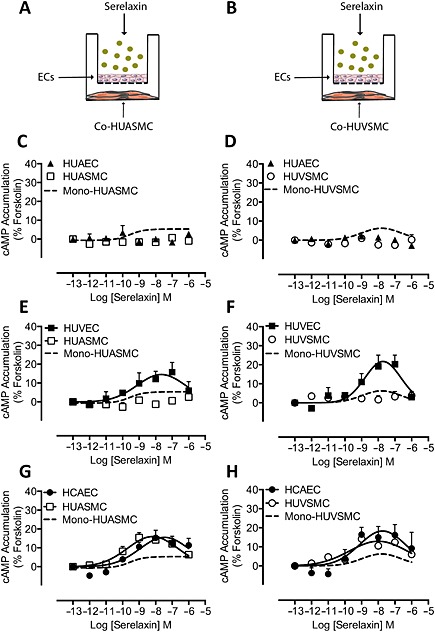
cAMP accumulation in co‐cultures of human primary vascular smooth muscle cells following addition of serelaxin to endothelium (all n = 5). HUAEC, HUVEC or HCAEC were co‐cultured with (A) HUASMC or (B) HUVSMC, and the endothelial cells were treated with serelaxin for 30 min. Serelaxin added to HUAEC did not cause cAMP accumulation either in (C, D) HUAEC (▲), (C) HUASMC (□) or (D) HUVSMC (◯), whereas direct stimulation of (C) HUASMC or (D) HUVSMC with serelaxin caused a concentration‐dependent increase in cAMP accumulation (dashed lines). Although direct addition of serelaxin to HUVEC concentration‐dependently increased cAMP accumulation in (E, F) HUVEC (■), there was no significant effect on cAMP accumulation in (E) HUASMC (□) or (F) HUVSMC (◯). Direct addition of serelaxin to (E) HUASMC or (F) HUVSMC stimulated cAMP accumulation (dashed lines). Serelaxin concentration‐dependently increased cAMP accumulation in (G, H) HCAEC (●) but also caused a robust concentration‐dependent increase in cAMP accumulation in both (G) HUASMC (□) and (H) HUVSMC (◯).
In co‐cultures of HUVEC (that express RXFP1) with HUASMC, serelaxin treatment (30 min) increased cAMP accumulation to 15% of the forskolin response in HUVEC (Figure 4E, pEC50: 9.9 ± 0.6), but there was no increase in cAMP accumulation in HUASMC (Figure 4E), whereas direct stimulation of HUASMC with serelaxin (30 min) increased cAMP accumulation concentration‐dependently (Figure 4E, dashed line: pEC50: 9.6 ± 0.7). In co‐cultures of HUVEC and HUVSMC (Figure 4B), serelaxin treatment (30 min) increased cAMP accumulation to 22% of forskolin response in HUVEC (Figure 4F, pEC50: 9.1 ± 0.4), with no significant effect on cAMP accumulation in HUVSMC (Figure 4F), even though direct stimulation of HUVSMC with serelaxin (30 min) increased cAMP accumulation (Figure 4F, dashed line: pEC50: 9.4 ± 0.4).
In co‐cultures of HCAEC/HUASMC, treatment of HCAEC with serelaxin increased cAMP accumulation to 16% of the forskolin response (Figure 4G, pEC50: 9.8 ± 0.3). However, treatment of HCAECs with serelaxin (30 min) also increased cAMP accumulation in HUASMC to 16% of the forskolin response (Figure 4G, pEC50: 9.30 ± 0.3), or 3.2 times higher than cAMP responses observed in HUASMC directly stimulated with serelaxin (Figure 4G, dashed line). In co‐cultures of HCAEC/HUVSMC, treatment of HCAEC with serelaxin (30 min) increased cAMP accumulation to 18% of the forskolin response (Figure 4H, pEC50: 9.8 ± 0.4). Stimulation of HCAEC with serelaxin (30 min) also increased cAMP accumulation in HUVSMC to 13% of the forskolin response (Figure 4H, pEC50: 9.6 ± 0.3), or 2.2 times higher than cAMP responses observed in HUVSMC directly stimulated with serelaxin (Figure 4H, dashed line; pEC50: 9.4 ± 0.4). Thus, in HCAEC, not only did serelaxin promote NO release, it also increased the release of another mediator that increased cAMP levels in co‐cultured SMCs.
The effects of serelaxin on cAMP signalling in HCAEC co‐cultures is dependent on prostanoid secretion from ECs
To provide information on the mediator released from HCAEC by serelaxin treatment to influence cAMP signalling in SMCs, we used pharmacological inhibitors on ECs and SMCs to disrupt key signalling pathways (Figure 5A). In monocultures, pre‐treatment with indomethacin (30 μM, 30 min) significantly inhibited serelaxin‐mediated (30 nM, 30 min) cAMP accumulation in HCAEC (Figure 5B) but not in HUVEC (Figure 5A), HUASMC (Figure 5C) or HUVSMC (Figure 5D, Table S1) suggesting that cellular background determines whether serelaxin causes prostanoid production in human primary vascular cells. However, pre‐treatment with l‐NOARG (30 μM, 30 min) had no significant effect on serelaxin‐mediated (30 nM, 30 min) cAMP accumulation in HUVEC (Figure 5A), HCAEC (Figure 5B), HUASMC (Figure 5C) or HUVSMC (Figure 5D, Table S1). Similarly, pre‐treatment with ODQ (1 μM, 30 min) had no effect on serelaxin‐mediated (30 nM, 30 min) cAMP accumulation in HUVEC (Figure 5A), HCAEC (Figure 5B), HUASMC (Figure 5C) or HUVSMC (Figure 5D, Table S1) suggesting that NOS and GC do not influence cAMP accumulation in human primary vascular cells.
Figure 5.
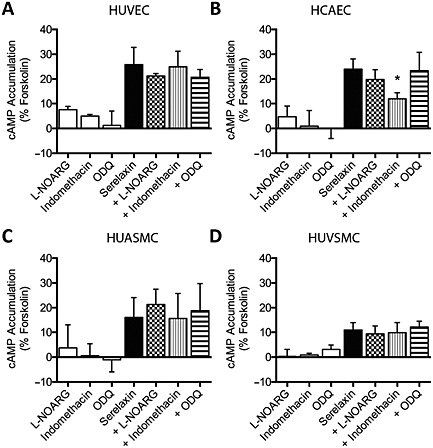
Serelaxin‐mediated cAMP accumulation in monocultures of human primary vascular cells (all n = 5). Serelaxin (30 nM, 30 min) increased cAMP accumulation in (A) HUVEC, (B) HCAEC, (C) HUASMC and (D) HUVSMC that was not significantly altered by pre‐incubation with l‐NOARG (30 μM, 30 min) or ODQ (1 μM, 30 min). Pre‐treatment with indomethacin (30 μM, 30 min) significantly inhibited serelaxin‐mediated (30 nM, 30 min) cAMP accumulation in (B) HCAEC but not in (A) HUVEC, (C) HUASMC or (D) HUVSMC. *P < 0.05; significantly different from serelaxin alone; one‐way anova with Dunnett's post hoc test.
In co‐cultures, pre‐treatment of HCAEC with l‐NOARG had no effect on cAMP accumulation in HUASMC (Figure 6C), HUVSMC (Figure 6D) or HCAEC (Figure 6B) suggesting that endothelial NO had no role in modulating cAMP accumulation (Table S1). Similarly, pre‐treatment of HUASMC or HUVSMC with ODQ (1 μM, 30 min) had no significant effect on serelaxin‐mediated (30 nM, 30 min) cAMP accumulation in HCAEC (Figure 6F), HUASMC (Figure 6G) and HUVSMC (Figure 6H), suggesting that GC activation in SMCs had no role in serelaxin‐mediated and HCAEC‐dependent cAMP accumulation (Table S2). By contrast, indomethacin pre‐treatment of HCAEC (Figure 6B) almost abolished the enhanced cAMP response observed in HUASMC (Figure 6C) and HUVSMC (Figure 6D) suggesting that serelaxin‐mediated prostanoid production in HCAEC was regulating cAMP production in both arterial and venous smooth muscle cells (Table S2).
Figure 6.
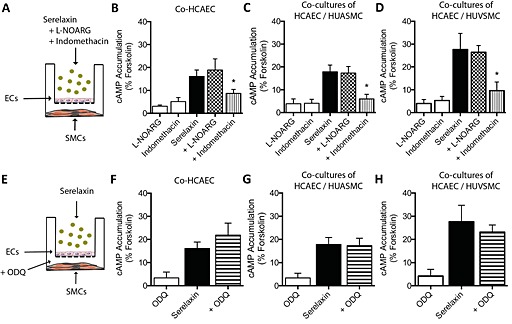
Serelaxin‐mediated cAMP accumulation in human primary vascular smooth muscle cells co‐cultured with HCAEC (A, E; all n = 5). Stimulation of HCAEC with serelaxin (30 nM, 30 min) increased cAMP accumulation not only in (B) HCAEC but also in co‐cultures of (C) HUASMC or (D) HUVSMC. Pre‐incubation of HCAEC with l‐NOARG (30 μM, 30 min) before addition of serelaxin (30 nM, 30 min) had no significant effect on cAMP accumulation in (B) HCAEC, (C) HUASMC or (D) HUVSMC. However, pre‐incubation of HCAEC with indomethacin (30 μM, 30 min) significantly inhibited serelaxin‐mediated (30 nM, 30 min) cAMP accumulation in (B) HCAEC and abolished cAMP accumulation in (C) HUASMC or (D) HUVSMC. Pre‐treatment of HUASMC or HUVSMC with ODQ (1 μM, 30 min) had no significant effect on serelaxin‐mediated (30 nM, 30 min) cAMP accumulation in (F) HCAEC, (G) HUASMC or (H) HUVSMC. *P < 0.05; significantly different from serelaxin alone; one‐way anova with Dunnett's post hoc test.
Discussion and conclusions
Serelaxin caused rapid and long‐lasting vasodilatory changes in patients with AHF (Ponikowski et al., 2013); however, the cellular and molecular mechanisms involved in humans remain poorly understood. In our previous study utilizing human primary vascular cells, we were able to show that serelaxin targeted cells of the human vasculature to cause short‐tern and long‐term signalling responses in human ECs, smooth muscle cells and fibroblasts (Sarwar et al., 2014). In this study, we demonstrate that the effects of serelaxin on vascular cells are enhanced by cellular crosstalk in an experimental paradigm that allows exchange of mediators between cells.
Vasodilation is a specific effect of relaxin that has been observed in many organs and tissues including the uterus (Bani et al., 1999, 1995b), mammary glands (Bani et al., 1995a), mesocaecum (Bigazzi et al., 1986), kidney (Danielson et al., 1999; Novak et al., 2001; Danielson and Conrad, 2003), liver (Bani et al., 2001), lung (Bani et al., 1997; Alexiou et al., 2013), brain (Chan and Cipolla, 2011; Chan et al., 2013) and heart (Bani Sacchi et al., 1995; Masini et al., 1997). These effects of relaxin can be chiefly ascribed to the stimulation of NO synthesis by cells of the vasculature. In vitro studies have shown that relaxin increases NO and/or intracellular cGMP levels in rat and human coronary artery endothelial cells, HUVEC, human umbilical artery and vein smooth muscle cells and bovine artery smooth muscle cells (Bani et al., 1998; Failli et al., 2002; Quattrone et al., 2004; Sarwar et al., 2014). This is in accord with our findings in HUVEC, HCAEC, HUASMC and HUVSMC where serelaxin‐mediated cGMP accumulation was blocked by the NOS inhibitor l‐NOARG and the GC inhibitor ODQ suggesting that serelaxin activated the NO/GC/cGMP pathway in human ECs and SMCs. To date, most cellular studies of signal transduction of serelaxin in vascular cells have been conducted in monocultures that provide no information on functional coupling between cells.
The vasodilating responses of relaxin have also been observed in a range of different intact blood vessels including rodent aorta, small renal and mesenteric arteries (Dschietzig et al., 2003; McGuane et al., 2011b), human s.c. (McGuane et al., 2011b) and human systemic resistance arteries (Fisher, 2009) suggesting that blood vessels are a prime target of relaxin. The different layers of blood vessels play distinct roles in blood vessel function and structure (Lüscher, 1990). Thus, the endothelium is in intimate contact with the bloodstream and regulates vascular tone by secretion of vasoactive substances such as NO, prostaglandins and EDHF (Lüscher and Tanner, 1992). However, the effects of relaxin on the secretion of these vasoactive substances and their effects on SMCs have not been reported. Administration of serelaxin to HUVEC or HCAEC produced an enhanced cGMP response in co‐cultured SMCs – typically 2 to 2.5 times than that observed in monocultures. cGMP responses in both ECs and SMCs were blocked by addition of l‐NOARG to ECs. Similarly, addition of the GC inhibitor, ODQ, to the SMCs blocked the response of serelaxin‐stimulated ECs, suggesting that serelaxin acted on the ECs to release NO that diffused to SMCs and activated guanylate cyclase to cause cGMP accumulation (Figure 7). This is in accord with previous findings as relaxin‐mediated vasodilation was blocked by NOS and GC inhibitors in uterine artery rings from mid‐pregnant rats (Longo et al., 2003) and human systemic resistance arteries (Fisher, 2009), suggesting a role of NO/cGMP in relaxin‐mediated vasodilation in rodents and humans. Interestingly, relaxin has been reported to be more potent than other vasodilators. In isolated and perfused rat and guinea pig heart, relaxin increased coronary flow to an extent that was significantly higher than that obtained with typical vasodilators such as ACh or sodium nitroprusside (Bani Sacchi et al., 1995), suggesting that perhaps relaxin may have additional vasodilatory mechanisms.
Figure 7.
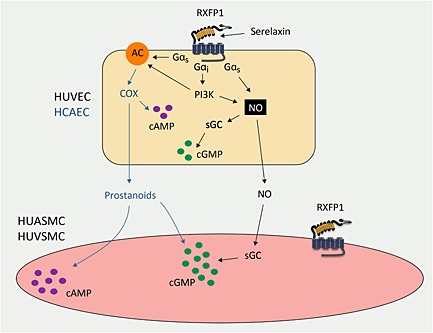
Signal transduction mechanisms activated by serelaxin in co‐cultures of human primary vascular cells. Activation of RXFP1 by serelaxin in HUVEC and HCAEC stimulates NO production and activates sGC and AC to produce cGMP and cAMP respectively. Endothelial NO also diffuses from the endothelial cells across the ThinCert membranes and activates sGC in both the arterial and venous smooth muscle cells. Additionally in HCAEC (blue lines) but not HUVEC, serelaxin stimulates prostanoid production that produces cAMP accumulation in both arterial and smooth muscle cells.
In some ECs such as HCAEC, serelaxin was shown, in addition to promoting NO‐dependent cGMP activation in SMCs, to promote the release of prostanoids to enhance both cGMP and cAMP accumulation (Figure 7). Thus, in HUVEC, indomethacin had no effect on serelaxin‐mediated cGMP and cAMP signalling (Figure 2), whereas significant inhibition of both pathways was observed in HCAEC (Figure 2). In HCAEC/SMC co‐cultures, indomethacin treatment of HCAEC significantly inhibited cGMP (Figure 3) and cAMP accumulation (Figure 6) in SMCs. Previous studies showed that indomethacin abolished (in patients taking ACE inhibitors) or reduced relaxin‐mediated vasodilation in human systemic resistance arteries (Fisher, 2009). Our study is the first to demonstrate this interaction between serelaxin and prostanoids in vitro in a system where signalling responses can be studied separately in endothelial and smooth muscle cells, which has important implications for understanding the mechanisms of actions of serelaxin in humans. Serelaxin‐mediated local prostanoid production may have paracrine and autocrine actions in particular regions and it is likely that in some tissues, serelaxin regulates vascular tone via both prostanoids and NO production. Thus, in rat mesenteric arteries, serelaxin enhanced bradykinin‐mediated vasodilation in a NO‐dependent manner (Jelinic et al., 2013), whereas serelaxin administration to rats increased the prostacyclin component of chronic bradykinin‐mediated vasorelaxation in small mesenteric arteries (Leo et al., 2013).
Cell surface expression of RXFP1 was shown to be essential for cAMP and cGMP responses (Sarwar et al., 2014) not only in ECs but also in co‐cultured SMCs because serelaxin treatment of HUAEC (non‐RXFP1 expressing cells) had no effect on cAMP and cGMP accumulation in co‐cultured SMCs. This further strengthens the notion that serelaxin is predominantly an endothelium‐dependent vasodilator that is governed by endothelial RXFP1 expression. This is in agreement with previous findings in human small resistance arteries where relaxin had no effect in endothelium‐denuded vessels (Fisher, 2009). Thus, serelaxin resembles other vasodilators such as ACh, bradykinin, ATP and substance P that cause endothelium‐dependent vasodilation (Furchgott and Zawadzki, 1980). Another finding in the time course experiments was that serelaxin failed to cause a response in SMCs when added to the inserts containing EC. This suggests that although it is likely that serelaxin penetrates the EC/ThinCert barrier, it fails to reach a concentration at the SMCs that can activate a signalling event. We also found that treatment of SMCs by ODQ reduced cGMP responses in these cells following addition of serelaxin to ECs but did not affect cGMP responses in ECs suggesting that ODQ like serelaxin does not pass the ThinCert barrier to produce concentrations high enough to be effective. Lastly, responses observed in smooth muscle cells followed the pattern of responses observed in the ECs. We have previously established that concentration–response relationships in HUASMC are sigmoidal (Sarwar et al., 2014), yet the concentration–response relationship in co‐cultures mirrors that found in the ECs (Figure 1), which for HUVEC and HCAEC were bell‐shaped, further strengthening the notion that serelaxin responses in the SMCs were governed by the ECs.
There were some limitations to our study. ECs and SMCs are physically separated by a small gap in our co‐culture model; however, in normal physiology, ECs and SMCs are in direct contact with each other. There are gap junctions not only between adjacent ECs and SMCs but also between ECs and SMCs that allow the passage of secreted substances. However, these gap junctions play an important role in vasorelaxation involving hyperpolarization of SMCs that is independent of NO and prostacyclin (Figueroa and Duling, 2009). So although there are clear advantages in working with a system that allows exchange of mediators together with examination of signalling pathways in endothelial and smooth muscle cells, there are other factors in an in vivo environment that are not accounted for in the co‐culture model including the presence of blood (proteins and cells), blood flow, shear stress and sympathetic innervation (Rodenwaldt et al., 2007). These important factors that are crucial for tissue function could be incorporated in future studies to determine their roles in serelaxin signalling.
Author contributions
M. S., C. S. S., R. A. B., D. R. S. and R. J. S. participated in research design. M. S. conducted experiments. R. A. B. contributed reagents or tools. M. S. and R. J. S. performed data analysis. M. S., C. S. S., R. A. B., D. R. S. and R. J. S. wrote or contributed to writing of the manuscript.
Conflict of interest
None.
Acknowledgements
We thank Corthera, Inc. (a subsidiary of Novartis AG, Switzerland) for the supply of serelaxin. This study was supported by Australian Research Council linkage grant (LP110100288) to R. J. S., C. S. S. and R. A. B. and Industry Partner Corthera Inc., a Novartis Company and National Health and Medical Research Council (NHMRC) of Australia senior research fellowships to C. S. S. (APP1041766) and R. A. D. B. (APP1042650).
Sarwar, M. , Samuel, C. S. , Bathgate, R. A. , Stewart, D. R. , and Summers, R. J. (2016) Enhanced serelaxin signalling in co‐cultures of human primary endothelial and smooth muscle cells. British Journal of Pharmacology, 173: 485–497. doi: 10.1111/bph.13371.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexiou K, Wilbring M, Matschke K, Dschietzig T (2013). Relaxin protects rat lungs from ischemia‐reperfusion injury via inducible NO synthase: role of ERK‐1/2, PI3K, and forkhead transcription factor FKHRL1. PLoS One 8: e75592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bani Sacchi T, Bigazzi M, Bani D, Mannaioni PF, Masini E (1995). Relaxin‐induced increased coronary flow through stimulation of nitric oxide production. Br J Pharmacol 116: 1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bani D, Baccari MC, Nistri S, Calamai F, Bigazzi M, Sacchi TB (1999). Relaxin up‐regulates the nitric oxide biosynthetic pathway in the mouse uterus: involvement in the inhibition of myometrial contractility. Endocrinology 140: 4434–4441. [DOI] [PubMed] [Google Scholar]
- Bani D, Ballati L, Masini E, Bigazzi M, Sacchi TB (1997). Relaxin counteracts asthma‐like reaction induced by inhaled antigen in sensitized guinea pigs. Endocrinology 138: 1909–1915. [DOI] [PubMed] [Google Scholar]
- Bani D, Failli P, Bello MG, Thiemermann C, Bani Sacchi T, Bigazzi M, et al. (1998). Relaxin activates the l‐arginine‐nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension 31: 1240–1247. [DOI] [PubMed] [Google Scholar]
- Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB (1995a). Relaxin activates the l‐arginine‐nitric oxide pathway in human breast cancer cells. Cancer Res 55: 5272–5275. [PubMed] [Google Scholar]
- Bani D, Nistri S, Quattrone S, Bigazzi M, Bani Sacchi T (2001). The vasorelaxant hormone relaxin induces changes in liver sinusoid microcirculation: a morphologic study in the rat. J Endocrinol 171: 541–549. [DOI] [PubMed] [Google Scholar]
- Bani G, Maurizi M, Bigazzi M, Bani Sacchi T (1995b). Effects of relaxin on the endometrial stroma. Studies in mice. Biol Reprod 53: 253–262. [DOI] [PubMed] [Google Scholar]
- Bigazzi M, Del Mese A, Petrucci F, Casali R, Novelli GP (1986). The local administration of relaxin induces changes in the microcirculation of the rat mesocaecum. Acta Endocrinol 112: 296–299. [DOI] [PubMed] [Google Scholar]
- Boccalini G, Sassoli C, Formigli L, Bani D, Nistri S (2015). Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: involvement of the Notch‐1 pathway. FASEB J 29: 239–249. [DOI] [PubMed] [Google Scholar]
- Bolton TB, Lang RJ, Takewaki T (1984). Mechanisms of action of noradrenaline and carbachol on smooth muscle of guinea‐pig anterior mesenteric artery. J Physiol 351: 549–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S‐L, Cipolla MJ (2011). Relaxin causes selective outward remodeling of brain parenchymal arterioles via activation of peroxisome proliferator‐activated receptor‐γ. FASEB J 25: 3229–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S‐L, Sweet JG, Cipolla MJ (2013). Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. FASEB J 27: 3917–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP, Debrah DO, Novak J, Danielson LA, Shroff SG (2004). Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology 145: 3289–3296. [DOI] [PubMed] [Google Scholar]
- Conrad KP, Shroff SG (2011). Effects of relaxin on arterial dilation, remodeling, and mechanical properties. Curr Hypertens Rep 13: 409–420. [DOI] [PubMed] [Google Scholar]
- Danielson LA, Conrad KP (2003). Time course and dose response of relaxin‐mediated renal vasodilation, hyperfiltration, and changes in plasma osmolality in conscious rats. J Appl Physiol 95: 1509–1514. [DOI] [PubMed] [Google Scholar]
- Danielson LA, Sherwood OD, Conrad KP (1999). Relaxin is a potent renal vasodilator in conscious rats. J Clin Invest 103: 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrah DO, Conrad KP, Jeyabalan A, Danielson LA, Shroff SG (2005). Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 46: 745–750. [DOI] [PubMed] [Google Scholar]
- Debrah DO, Novak J, Matthews JE, Ramirez RJ, Shroff SG, Conrad KP (2006). Relaxin is essential for systemic vasodilation and increased global arterial compliance during early pregnancy in conscious rats. Endocrinology 147: 5126–5131. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Bartsch C, Richter C, Laule M, Baumann G, Stangl K (2003). Relaxin, a pregnancy hormone, is a functional endothelin‐1 antagonist: attenuation of endothelin‐1‐mediated vasoconstriction by stimulation of endothelin type‐B receptor expression via ERK‐1/2 and nuclear factor‐kappaB. Circ Res 92: 32–40. [DOI] [PubMed] [Google Scholar]
- Failli P, Nistri S, Quattrone S, Mazzetti L, Bigazzi M, Sacchi TB, et al. (2002). Relaxin up‐regulates inducible nitric oxide synthase expression and nitric oxide generation in rat coronary endothelial cells. FASEB J 16: 252–254. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Duling BR (2009). Gap junctions in the control of vascular function. Antioxid Redox Signal 11: 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher CJ (2009). Relaxin: a new cardiovascular hormone in humans? Comparative potency and mechanisms of action. MD Thesis, University of Glasgow.
- Furchgott RF, Vanhoutte PM (1989). Endothelium‐derived relaxing and contracting factors. FASEB J 3: 2007–2018. [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376. [DOI] [PubMed] [Google Scholar]
- Ganz P, Davies PF, Leopold JA, Gimbrone MA, Alexander RW (1986). Short‐ and long‐term interactions of endothelium and vascular smooth muscle in coculture: effects on cyclic GMP production. Proc Natl Acad Sci U S A 83: 3552–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg SM (2007). Vasodilators in acute heart failure. Heart Fail Rev 12: 143–147. [DOI] [PubMed] [Google Scholar]
- Jelinic M, Leo C‐H, Post Uiterweer ED, Sandow SL, Gooi JH, Wlodek ME, et al. (2013). Localization of relaxin receptors in arteries and veins, and region‐specific increases in compliance and bradykinin‐mediated relaxation after in vivo serelaxin treatment. FASEB J 28: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyabalan A, Novak J, Danielson LA, Kerchner LJ, Opett SL, Conrad KP (2003). Essential role for vascular gelatinase activity in relaxin‐induced renal vasodilation, hyperfiltration, and reduced myogenic reactivity of small arteries. Circ Res 93: 1249–1257. [DOI] [PubMed] [Google Scholar]
- Leo C‐H, Jelinic M, Parkington HC, Tare M, Parry LJ (2013). Acute intravenous injection of serelaxin (recombinant human relaxin‐2) causes rapid and sustained bradykinin‐mediated vasorelaxation. J Am Heart Assoc 3: e000493–e000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo M, Jain V, Vedernikov YP, Garfield RE, Saade GR (2003). Effects of recombinant human relaxin on pregnant rat uterine artery and myometrium in vitro . Am J Obstet Gynecol 188: 9–9. [DOI] [PubMed] [Google Scholar]
- Lüscher TF (1990). Endothelium‐derived vasoactive factors and regulation of vascular tone in human blood vessels. Lung 168 (Suppl): 27–34. [DOI] [PubMed] [Google Scholar]
- Lüscher TF, Tanner FC (1992). Endothelial regulation of vascular tone and growth. Am J Hypertens 6: 283–293. [DOI] [PubMed] [Google Scholar]
- Majed BH, Khalil RA (2012). Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev 64: 540–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Berka V, Tsai AL, Murad F (2005). Soluble guanylyl cyclase: the nitric oxide receptor In: {0} (eds)Part E, Aec LP. Nitric Oxide. Academic Press, pp. 478–492. [DOI] [PubMed] [Google Scholar]
- Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, Sacchi TB (1997). Relaxin counteracts myocardial damage induced by ischemia‐reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology 138: 4713–4720. [DOI] [PubMed] [Google Scholar]
- Masini E, Zagli G, Ndisang JF, Solazzo M, Mannaioni PF, Bani D (2002). Protective effect of relaxin in cardiac anaphylaxis: involvement of the nitric oxide pathway. Br J Pharmacol 137: 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP (2011a). Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension 57: 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Debrah JE, Sautina L, Jarajapu YPR, Novak J, Rubin JP, et al. (2011b). Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 152: 2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak J, Danielson LA, Kerchner LJ, Sherwood OD, Ramirez RJ, Moalli PA, et al. (2001). Relaxin is essential for renal vasodilation during pregnancy in conscious rats. J Clin Invest 107: 1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG, Moncada S (1987). Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature 327: 524–526. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponikowski P, Mitrovic V, Ruda M, Fernandez A, Voors AA, Vishnevsky A, et al. (2013). A randomized, double‐blind, placebo‐controlled, multicentre study to assess haemodynamic effects of serelaxin in patients with acute heart failure. Eur Heart J 35: 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrone S, Chiappini L, Scapagnini G, Bigazzi B, Bani D (2004). Relaxin potentiates the expression of inducible nitric oxide synthase by endothelial cells from human umbilical vein in in vitro culture. Mol Hum Reprod 10: 325–330. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S (1987). The anti‐aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol 92: 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenwaldt B, Pohl U, de Wit C (2007). Endogenous and exogenous NO attenuates conduction of vasoconstrictions along arterioles in the microcirculation. Am J Physiol Heart Circ Physiol 292: H2341–H2348. [DOI] [PubMed] [Google Scholar]
- Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ (2014). Serelaxin‐mediated signal transduction in human vascular cells: bell‐shaped concentration–response curves reflect differential coupling to G proteins. Br J Pharmacol 172: 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal MS, Sautina L, Li S, Diao Y, Agoulnik AI, Kielczewski J, et al. (2012). Relaxin increases human endothelial progenitor cell NO and migration and vasculogenesis in mice. Blood 119: 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, et al. (2013). Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo‐controlled trial. Lancet 381: 29–39. [DOI] [PubMed] [Google Scholar]
- Voors A, Dahlke M, Meyer S, Stepinska J, Gottlieb S, Jones A, et al. (2014). Renal hemodynamic effects of serelaxin in patients with chronic heart failure: a randomized, placebo‐controlled study. Circ Heart Fail 7: 994–1002. [DOI] [PubMed] [Google Scholar]
- Voors A, Davison B, Felker M, Ponikowski P, Unemori E, Cotter G, et al. (2011). Early drop in systolic blood pressure and worsening renal function in acute heart failure: renal results of Pre‐RELAX‐AHF. Eur J Heart Fail 13: 961–967. [DOI] [PubMed] [Google Scholar]