

Abstract
Objective
Fever is associated with improved survival, although its beneficial mechanisms are poorly understood. Previous studies indicate that the thermal element of fever augments lymphocyte migration across high endothelial venules (HEVs) of lymphoid organs by increasing the intravascular display of a gatekeeper trafficking molecule, intercellular adhesion molecule-1 (ICAM-1). Here, we evaluated the spatio-temporal relationship between the thermal induction of intravascular ICAM-1 and lymphocyte trafficking.
Methods
Intravascular ICAM-1 density was quantified by immunofluorescence staining in mice exposed to fever-range whole-body hyperthermia (39.5±.5°C). ICAM-1-dependent lymphocyte trafficking was measured in short-term homing assays.
Results
A linear relationship was observed between the duration of heat treatment and intravascular ICAM-1 density in HEVs with maximal responses requiring sustained (i.e., five hours) thermal stress. Circulating lymphocytes were found to sense incremental changes in ICAM-1 on HEVs, such that trafficking is proportional to the intravascular density of ICAM-1. We further identified a hydroxamate-sensitive shedding mechanism that restores ICAM-1 expression to homeostatic levels following the cessation of thermal stress.
Conclusions
The time-dependent response to thermal stress indicates that ICAM-1 density governs the efficiency of lymphocyte interactions with HEVs in vivo. These studies highlight the dynamic role of the microcirculation in promoting immune surveillance during febrile inflammatory responses.
Keywords: fever-range thermal stress, high endothelial venules, lymphocyte trafficking, intercellular adhesion molecule-1
The adaptive immune response depends on the continuous migration of blood-borne lymphocytes into secondary lymphoid organs where naïve and central memory lymphocytes encounter antigen-presenting cells. The major site of entry of bloodborne lymphocytes into lymph nodes and Peyer patches is across postcapillary high endothelial venules (HEVs) [9, 66]. Cuboidal HEVs are distinguished from squamous endothelium lining the majority of blood vessels throughout the body by their ability to support efficient trafficking under homeostatic conditions [25]. Lymphocyte migration across HEVs depends on a highly ordered sequence of adhesion events that includes: (1) primary tethering and rolling along vessel walls, (2) chemokine-dependent activation, (3) secondary firm arrest, and (4) transendothelial migration [9,47,66].
Recent studies have shown that lymphocyte trafficking across HEVs in secondary lymphoid organs is augmented by the thermal element of fever [15–18, 23]. Fever has been linked with improved survival during acute infection in vertebrate species, although its physiological benefit is poorly understood [3, 27]. The duration and temperature range of the hyperthermic component of physiological fever has been simulated in vivo by the administration of whole-body hyperthermia (WBH) to raise core temperatures to 39.590.58C [8, 16]. Fever-range WBH treatment of both mice and advanced cancer patients causes a transient decrease in the number of circulating lymphocytes [23, 29, 43]. In mice, this corresponds with an ~twofold increase in lymphocyte trafficking selectively across the specialized microvasculature of HEVs [16, 17, 23]. This change represents a profound effect on an already efficient trafficking mechanism, whereby >105 cells are estimated to transit across HEVs hourly in a single lymph node under normothermic conditions [65]. Heightened trafficking across HEVs is predicted to improve immune surveillance by enhancing the probability that rare antigen-restricted lymphocytes will encounter cognate antigens in organs strategically positioned to be the first line of defense against pathogens that enter via the skin or the respiratory, urogenital, or gastrointestinal tracts.
Systemic thermal stress improves lymphocyte trafficking by acting at discrete steps in the adhesion cascade required for extravasation across HEVs. Intravital microscopy revealed that thermal stress causes an ~twofold increase in the ability of HEVs to support the firm arrest of lymphocytes on vessel walls, which leads to transendothelial migration into the underlying parenchyma [15,16,23]. Enhanced firm arrest in response to fever-range thermal stress is coordinated by increases in the intravascular density of the chemokine CCL21 and intercellular adhesion molecule-1 (ICAM-1) on HEVs [16,54,64]. CCL21 contributes to lymphocyte trafficking through the engagement of CCR7 receptors on apposing lymphocytes, resulting in increased affinity and avidity of leukocyte function-associated antigen-1 (LFA-1) for its endothelial counter-receptors, ICAM-1 and ICAM-2 [9,16,33,66]. Interleukin-6 (IL-6) is the sole cytokine found to regulate HEV expression of ICAM-1 during thermal stress, whereas CCL21 is induced through an IL-6–independent mechanism [16,64]. Thus, ICAM-1 upregulation was shown to be crucial for the thermal response based on evidence that disruption of IL-6 signaling prevents thermal stimulation of lymphocyte trafficking across HEVs [16].
The effects of systemic thermal stress on ICAM-1 density were previously examined at a fixed time point (i.e., six hours), at which a marked increase in ICAM-1 density was detected, similar to levels induced by potent inflammatory stimuli including tumor necrosis factor (TNF) or IL-6 [16,54,64]. These observations raised fundamental questions regarding the spatio-temporal relationship between thermally induced changes in the intravascular density of ICAM-1 and the efficiency of lymphocyte trafficking, as well as the mechanisms leading to the restoration of homeostatic trafficking after the cessation of thermal stress. Here, we present evidence of a linear relationship between the duration of heat treatment and the density of ICAM-1 expressed on the lumenal surface of HEVs. Further, circulating lymphocytes are capable of sensing incremental increases in ICAM-1 presented by HEVs, such that their trafficking response is proportional to the intravascular density of ICAM-1. Finally, the restoration of ICAM-1 to homeostatic levels following the cessation of thermally-induced inflammation was shown to require the activity of a zinc-dependent metalloproteinase. These findings support the notion that dynamic changes in intravascular ICAM-1 density in HEVs act as a rheostat to regulate the immune surveillance of secondary lymphoid organs during inflammation and infection.
MATERIALS AND METHODS
Mice
Age-matched (8–12 weeks) BALB/c and C57BL/6 female mice were from the National Cancer Institute (Frederick, Maryland, USA) and Taconic (Hudson, New York, USA) and were maintained under pathogen-free barrier conditions. All animal protocols were approved by the Roswell Park Cancer Institute Institutional Animal Care and Use Committee (Buffalo, New York, USA). Since it was essential to process all mice and tissue specimens identically within a given time-course experiment (i.e., during heating, adoptive transfer of splenocytes, injection of ICAM-specific antibody [Ab], harvesting tissues, and staining/photographing tissue sections), each experiment included one mouse per time point (totaling four to seven mice per experiment), and each independent experiment was repeated two to five times.
Treatment with Fever-Range WBH or TAPI-1 Protease Inhibitor
Mice were treated with fever-range WBH (core temperature of 39.5±0.5°C) for the indicated time periods in an environmental chamber (model BE5000; Memmert, Schwabach, Germany), as described [8,16,23,45]. Normothermic (NT) control mice (core temperature, 36.8±0.2°C) were maintained at room temperature in a darkened cabinet for the experimental period. Heated and control mice were injected intraperitoneally with 1 mL of sterile saline prior to the initiation of experiments to avoid dehydration. Core temperatures of sentinel mice in all experimental groups were measured by using a subcutaneously implanted microchip thermotransponder and a programmable data-acquisition system (Bio Medic Data Systems, Seaford, Delaware, USA). Heat treatment was maintained for the designated time period, once the mice reached the target temperature of 39.5±0.5°C [45]. The TAPI-1 protease inhibitor (Calbiochem, San Diego, California, USA) was injected via the tail vein at a dose reported to block the sheddase-dependent release of TNF in vivo by ~50-fold (0.5 mg in 0.25 mL saline) [38].
Analysis of Intravascular Display of Adhesion Molecules and HEV Density
Quantitative analysis of intravascular staining was performed as described [16]. Primary monoclonal antibodies (mAbs) specific for ICAM-1 (3E2, 25 μg/mouse) and ICAM-2 (3C4, 50 μg/mouse) (BD Biosciences, San Jose, California, USA) or isotype control Ab were injected via the tail vein of experimental mice 20 minutes after the cessation of heat treatment. Organs (pooled peripheral lymph nodes [paired inguinal, brachial, axillary, sciatic, superficial, and deep cervical nodes], spleen, pancreas, all mesenteric lymph nodes, and Peyer patches) were harvested 20 minutes later and embedded in an optimum cutting temperature compound (OCT; Sakura Finetek, Torrance, California, USA). Cryosections (9 mm) were fixed in methanol:acetone (3:1), rehydrated in phosphate-buffered saline (PBS), and incubated with goat serum (1:10 dilution) to block Fc receptors. Some tissue cryosections were subsequently stained with primary mAb specific for PNAd (MECA79, 20 μg/mL), CD31 (MEC13.3, 5 μg/mL), or isotype controls (BD Biosciences). After washing, primary Abs were detected by fluorochrome-conjugated (fluorescein or rhodamine) secondary Ab (Jackson ImmunoResearch, West Grove, Pennsylvania, USA). In selected experiments, frozen tissues were stained with primary anti-ICAM-1 Ab (3E2; 5 μg/mL) after sectioning.
Digital images were captured with an Olympus BX50 upright fluorescence microscope equipped with a SPOT RT camera (Spectra Services, Rochester, New York, USA), using identical exposure times and image settings within each experiment. Cuboidal HEV structures (in lymph nodes, Peyer patches) or CD31+ vessels (in pancreas or spleen) were encircled by using an Intuos3 Tablet (Wacom, Vancouver, Washington, USA) and fluorescence analyzed by using ImageJ software [1] was expressed as fluorescence intensity/pixels (i.e., reflecting a fixed unit area) [16]. Histograms represent the data from all pixels analyzed (ranging from 1 to 1.7 ×106) for the total number of HEVs per cryosection of pooled 14 peripheral lymph nodes (ranging from 100 to 175); similar numbers of HEVs and pixels were analyzed for each condition within individual experiments. Presentation of the data as a distribution measures population shifts and can detect subtle differences between conditions. HEV density in pooled peripheral lymph node cryosections was determined by enumerating the total HEVs per field in ≥ 25 fields (unit area of each field =0.34 mm2).
Intravital Analysis of Vessel Diameter and Blood Cell Velocity
Intravital microscopy of inguinal lymph nodes was performed as described [16,65]. C57BL/6 mice were anesthetized by an intraperitoneal injection of 1 μg/mL of xylazine and 10 μg/mL of ketamine (10 mL/kg body weight). The postcapillary vascular structures were observed under fluorescent microscopy, and each venule was assigned an order (from I to V). High-order (III–V) venules within the paracortical region represent HEVs, whereas low-order (I and II) venules are flat-walled vessels that drain into the superficial epigastric vein [65]. The diameters of order III venules were measured at order II/III branching points. To measure blood cell velocity, approximately 2.5 ×107 calcein-labeled splenocytes were injected into a catheter inserted into the right femoral artery and fluorescent microscopy was used to measure the velocity of noninteracting (i.e., fast moving) cells in order III venules, as described [16,57,65]. All observations were captured by using an EB CCD camera (Hamamatsu Photonics, Bridgewater, New Jersy, &USA) and recorded by using a digital videocassette recorder (DSR-11; Sony, New York, New York, USA) for analysis.
Determination of Vascular Permeability
Quantitative analysis of vascular integrity/permeability was performed essentially as described [7,22]. For this analysis, 2.5 mg of TRITC-conjugated bovine serum albumin (BSA; Invitrogen, Carlsbad, California, USA) was injected via the tail vein of experimental mice 20 minutes after cessation of WBH treatment. Organs were harvested three minutes later and fixed for four hours in Carnoy’s solution (ethanol:chloroform:acetic acid, 6:3:1), followed by graded ethanol (100%, 90%, and 70%) and PBS prior to embedding and freezing in OCT. HEVs of frozen cryosections (9 mm) were identified by MECA-79 Ab staining (25 μg/mL) [16]. Leakage of TRITC-BSA across HEVs was analyzed as described [7], in paired regions of 11 ×12 mm (equivalent to 30 ×33 pixels) positioned 50 mm apart, one within the lumen of HEVs and the second within the surrounding lymph node stroma. Each pixel within these defined regions (total, ~80 HEVs) was assigned a fluorescence intensity value on a scale from 0 to 255 (generated by ImageJ). The permeability index is the ratio of lumenal mean fluorescence intensity (MFI) to stromal MFI.
In vivo Homing Assay
Lymphocyte homing to lymphoid and nonlymphoid organs was assessed in short-term (i.e., one hour) homing assays [16,23]. Mouse splenocytes were labeled with 4 μM TRITC (Molecular Probes, Carlsbad, California, USA) for 20 minutes at 378C and the labeling was stopped by centrifugation through a fetal calf serum cushion (Invitrogen). Within individual experiments, equivalent numbers of labeled cells (ranging from 1 to 2 ×107) were adoptively transferred by intravenous injection into mice 20 minutes after the cessation of WBH and organs were harvested one hour after cell transfer. The number of fluorescent-labeled cells was quantified (i.e., blinded) in ≥10 fields (unit area of each field =0.34 mm2) of nonsequential 9-mm cryosections, using an Olympus BX50 (Spectra Services, Rochester, New York, USA) upright fluorescence microscope. In selected experiments, mice were treated 30 minutes before cell transfer with neutralizing mAb specific for ICAM-1 (3E2, 50 μg/mouse; BD Biosciences), ICAM-2 (3C4, 50 μg/mouse; BD Biosciences), or isotype control Ab. Alternatively, TRITC-labeled splenocytes were pretreated with mAb specific for LFA-1 (M17/4, 50 μg/mL; eBioscience, San Diego, California, USA) or isotype control Ab 30 minutes before transfer.
Analysis of Plasma-Soluble (s)ICAM-1
Concentrations of soluble ICAM-1 (sICAM-1) in heparinized plasma were quantified by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, Minneasota, USA).
Statistical Analysis
Statistical analysis of short-term homing, vessel diameter, blood cell velocity, vascular permeability, and soluble ICAM-1 was performed by using the unpaired, two-tailed Student’s t-test, where P ≤0.05 was significant [16]; data are expressed as the mean±standard error of the mean (SEM).
For the analysis of ICAM-1 and ICAM-2 density, Kolmogorov Smirnov tests [19] were used to evaluate the null hypothesis that paired datasets were from the same underlying distribution. A permutation scheme was performed (1000 runs) to determine the P-values for significance for the Kolmogorov Smirnov statistic, D. To generate a threshold D-value that represents intermouse variability, we compared ICAM-1 immunofluorescence data from five nonheated control mice in a single experiment. We found that MFI values were highly consistent in these replicate mice analyzed under identical Ab staining conditions and camera settings (MFIs: 45.6, 42.9, 42.1, 42.9, and 42.8; mean = 43.3, SEM =0.06). The largest observed Kolmogorov Smirnov statistic (D =0.09) represents mouse-to-mouse variation and was applied to all pair-wise comparisons of immunofluorescence results, such that D 50.09 indicates that the two datasets are not different from each other, whereas D >0.09 indicates that the datasets are different. The R-programming language was used to perform all tests and all permutation simulations [59].
RESULTS
ICAM-1 Induction Depends on Prolonged and Sustained Exposure to Fever-Range Thermal Stress
Kinetics studies were performed to identify spatiotemporal changes in the intravascular density of ICAM-1 in peripheral lymph node HEVs of BALB/c mice after treatment with fever-range WBH (Figure 1). ICAM-1 expression was detected by in situ immunofluorescence analysis following hyperthermia treatment for various time periods (two, four, and six hours of continuous elevation of core temperature to 39.5±0.5°C). ICAM-1 was detected on the lumenal surface of HEVs (i.e., the site of lymphocyte contact during extravasation) by injecting ICAM-1–specific mAb into the vascular compartment and then counter-staining frozen tissue sections with fluorescence-labeled detection antibody.
Figure 1.
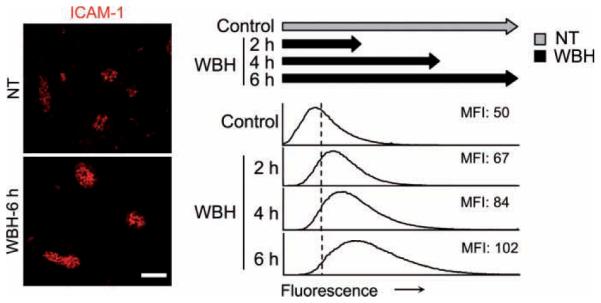
Thermal induction of intercellular adhesion molecule-1 (ICAM-1) depends on the duration of heat treatment. Intravascular staining of ICAM-1 in peripheral lymph nodes was performed by intravenous injection of primary monoclonal antibody (mAb) in normothermic control (NT) mice and mice treated with whole-body hyperthermia (WBH) for various periods. Peripheral lymph node cryosections were counter-stained with fluorochrome-conjugated secondary Ab. Immunofluorescence staining was not observed using isotype-matched control Ab (not shown). Photomicrographs show typical images from NT mice and mice with WBH treatment for six hours. Scale bar, 50 mm. Histograms indicate the immunofluorescence intensity of ICAM-1 staining quantified in cuboidal high endothelial venules by image analysis. The x-axis indicates fluorescence intensity, and the y-axis indicates the number of pixels with each intensity; the mean fluorescence intensity (MFI) is indicated in each condition. Data represent ≥3 independent experiments.
Quantitative image analysis revealed a linear relationship between ICAM-1 intravascular density and the duration of inflammatory cues provided by thermal stress. Maximal ICAM-1 induction was observed following continuous heat treatment for six hours, reflected by the ~twofold increase in the mean fluorescence intensity (MFI) of ICAM-1 detected in HEVs of heated mice, compared with the steady-state levels of the normothermic control (NT) (Figure 1). Incremental increases observed in intravascular ICAM-1 density after treatment with WBH for each of the experimental intervals (two, four, and six hours) were all different from normothermal control as well as from every other time point (D >0.09, by Kolmogorov Smirnov distribution analysis). Similar kinetics of ICAM-1 induction were observed in Peyer patch and mesenteric lymph node HEVs (data not shown). In contrast, thermal stress did not alter intravascular ICAM-1 density in CD31+ vessels of extralymphoid organs that lack HEV structures, such as pancreas or spleen (i.e., MFIs for NT and mice treated six hours with WBH, respectively, were 34.7 and 36.1 for pancreas and 17.9 and 19.3 for spleen; D 50.09). Intravascular display of the functionally and structurally related adhesion molecule, ICAM-2 was measured in HEVs after WBH treatment as an internal control, since ICAM-2 is not inducible under inflammatory conditions [24]. No change was detected in ICAM-2 staining (MFI: 67 [NT] and 69 [WBH]; D ≤0.09), demonstrating that the observed increases for ICAM-1 cannot be attributed to trivial mechanisms, such as improved access of Ab to adhesion molecules on the surface of HEVs.
A possible explanation for the limited ICAM-1 induction detected after short-term (i.e., two-hour) heat treatment was that insufficient time was allowed for de novo synthesis and expression of ICAM-1 on the lumenal surface of HEVs. In this regard, studies in primary endothelial cells in vitro have shown that ICAM-1 mRNA levels are detectable as early as two hours following stimulation by inflammatory cytokines (TNF, IL-1β), while expression of ICAM-1 protein at high density on the endothelial surface requires six to eight hours [40]. To determine if shortterm exposure to fever-range thermal stress was sufficient to initiate ICAM-1 induction on HEVs, mice were treated with continuous WBH for variable periods, ranging from one to six hours. Mice were then allowed to revert to normothermal temperature and intravascular ICAM-1 density was detected after a total six-hour period (Figure 2). ICAM-1 staining after WBH treatment for one to two hours was indistinguishable from baseline levels of ICAM-1 (D ≤0.09). ICAM-1 expression after either three or four hours of heat exposure was moderately elevated, compared to control (D >0.09), but not different from each other (D ≤0.09). Maximal lumenal display of ICAM-1 (~twofold upregulation) was observed after WBH treatment for five or six hours; ICAM-1 densities at these time points were significantly different from all other conditions (D >0.09), but were indistinguishable from each other (D ≤0.09). Collectively, these results demonstrate that prolonged and sustained thermal stress is required for optimal expression of ICAM-1 in HEVs.
Figure 2.

Sustained heat treatment is required for maximal thermal induction of intercellular adhesion molecule-1 (ICAM-1) expression. Experimental mice were treated with whole-body hyperthermia (WBH) for various time periods and then allowed to revert to normothermal temperature (NT). At the end of the six-hour period, intravascular density of ICAM-1 in peripheral lymph nodes was quantified by image analysis, as shown in histograms. The x-axis indicates fluorescence intensity and the y-axis indicates the number of pixels with each intensity; the mean fluorescence intensity (MFI) is indicated in each condition. Data represent ≥ 3 independent experiments.
Fever-Range Thermal Stress Enhances Lymphocyte Trafficking Across HEVs in an ICAM-1-Dependent Manner
Since maximal induction of ICAM-1 expression in HEVs was detected after continuous WBH treatment for six hours (Figures 1 and 2), this time point was used in short-term (i.e., one-hour) homing studies to investigate the contribution of ICAM-1 density to lymphocyte trafficking. For these experiments, fluorescent (TRITC)-labeled splenocytes were adoptively transferred into mice 20 minutes after the cessation of WBH treatment. This allowed mice to revert to normal core body temperature (Figure 3A), thereby avoiding the reported effects of direct heat on the homing potential of transferred lymphocytes [23,18].
Figure 3.

Thermal enhancement of trafficking across high endothelial venules (HEVs) is dependent on leukocyte function associated antigen-1/intercellular adhesion molecule-1 (LFA-1/ICAM-1) interactions. (A) Core temperatures of sentinel mice were monitored every 30 minutes during 6 hour whole-body hyperthermia (WBH) and every minute after cessation of WBH. The dotted line represents baseline normothermic temperature (NT); measurements were started when the target temperature range was reached (39.5±0.5°C). Arrow indicates the time point at which monoclonal antibodies (mAbs) or adoptive transfer cells were injected. Data represent ≥3 independent experiments. (B, C) in vivo short-term (one-hour) homing of TRITC (red)-labeled splenocytes to peripheral lymph nodes (PLN), spleen (SPL), and pancreas (Panc) was assessed in NT control or WBH-treated (six-hour) BALB/c mice. (B) PLN cryosections were stained with MECA-79 mAb specific for PNAd to visualize HEVs (green). Photomicrographs show typical images of TRITC-labeled cells either associated with HEVs or infiltrated into the tissue parenchyma. The total number of TRITC-labeled cells and the percentage of cells within the lymph node parenchyma were quantified in tissue cryosections by fluorescence microscopy (n=10 fields; bar=50 μm). (C) Splenocytes were treated with LFA-1 function-blocking mAb or control mAb prior to transfer. Alternatively, 20 minutes prior to adoptive transfer, recipient mice were treated with function-blocking mAb specific for ICAM-1 or −2 or with isotype-matched control Ab. The number of TRITC-labeled cells in tissue cryosections was quantified by fluorescence microscopy (n =10 fields). P-values represent the difference between NT and WBH conditions in (B) and represent the difference between function-blocking reagents and controls in (C). *P <0.0001. Data represent ≥3 independent experiments.
An ~twofold increase in trafficking of TRITC-labeled lymphocytes was detected in response to thermal stress exclusively in organs with HEVs, including peripheral lymph nodes (Figure 3B and 3C), mesenteric lymph nodes, and Peyer patches (data not shown). Enumeration of fluorescent-labeled transferred cells within or outside HEVs (demarked by staining for the HEV-specific molecule, PNAd [58]) revealed that thermal stress did not alter the relative proportion ( ~70%) of cells that completed the adhesion cascade leading to extravasation into the lymph node stroma (Figure 3B; P =0.67). Labeled cells within the vascular space of HEVs (Figure 3B) appear to be actively interacting with vessel walls, rather than merely passing through the local vasculature. This conclusion is based on comparisons with organs such as the pancreas (Figure 3C), in which adoptively transferred cells are rarely detected, despite high vascular density (i.e., 131.9±16.5 CD31+ vessels per unit area in pancreas versus 5.3±1.6 PNAd+ HEVs per unit area in lymph nodes; P <0.0001). Consistent with findings that thermal stress did not augment ICAM-1 expression on non-HEV vessels in spleen or pancreas, no increase in lymphocyte trafficking was detected in these organs following WBH (Figure 3C; P >0.4). The constant level of trafficking observed in these organs provided an internal control for the transfer of equivalent number of lymphocytes in individual mice within a given experiment.
The role of ICAM-1 in thermally regulated trafficking across HEVs was previously examined by using function-blocking mAb directed against ICAM-1 [16]. Here, we extended these findings by blocking the ICAM-1 counter-receptor, LFA-1, on lymphocytes prior to adoptive transfer into control or heat-treated mice. Under normothermic conditions, lymphocyte homing across HEVs was substantially inhibited by LFA-1-blocking mAb, whereas mAb specific for either ICAM-1 or −2 had little effect (Figure 3C). This result is consistent with prior findings showing that ICAM-1 and −2 are functionally redundant ligands for LFA-1 during homeostatic trafficking in lymphoid organs [16,31]. Transferred cells detected within HEVs in the presence of LFA-1-blocking mAb likely reflect L-selectin/PNAd-dependent tethering and rolling interactions. Improved lymphocyte trafficking across HEVs in response to thermal stress was further shown to be dependent on cognate interactions between LFA-1 and ICAM-1 (Figure 3C). In this regard, thermal enhancement of lymphocyte trafficking to peripheral lymph nodes was fully prevented by the mAb blockade of LFA-1 on lymphocytes prior to adoptive transfer as well as by neutralizing mAb specific for ICAM-1, but not ICAM-2 (Figure 3C). Heat treatment did not influence LFA-1/ICAM-1–dependent lymphocyte homing to organs lacking HEV structures including spleen and pancreas (Figure 3C; P >0.4).
We formally excluded the possibility that lymphocyte homing detected following WBH was influenced by changes in hemodynamic parameters by measuring vessel diameter, blood cell velocity, HEV density, and vascular integrity/permeability at the time of the initiation of homing assays (i.e., 20 minutes following cessation of WBH). In these studies, vessel diameter and blood cell velocity were determined to be equivalent in normothermal controls and WBH-pretreated mice (Figure 4A; P =0.10 and 0.36, respectively) by intravital microscopic analysis of HEVs in inguinal lymph nodes. Moreover, there was no difference in the overall density of HEVs in heated versus nonheated lymph nodes (Figure 4B; P =0.77). To rule out the possibility that thermal stress enhanced trafficking by compromising the integrity of the vascular barrier, we measured the permeability of HEVs following the intravenous injection of TRITC-conjugated BSA [7,22]. Quantitative image analysis of the relative fluorescence within the vascular space of PNAd+ HEVs versus fluorescence within the surrounding lymph node stroma revealed that there was no difference in the permeability of HEVs from WBH-treated mice, compared to normothermal control mice (Figure 4C; P =0.34). Taken together, these results establish that changes in lymphocyte trafficking detected in response to thermal stress in adoptive transfer experiments can be attributed to the induction of ICAM-1 expression in HEVs.
Figure 4.

Hemodynamic parameters are normal in high endothelial venules (HEVs) after whole-body hyperthermia (WBH) treatment. (A) Vascular diameter and blood cell velocity were quantified by intravital microscopy in level III venules of inguinal lymph nodes. For vascular diameter (left), data were generated from 15 order III venules (i.e., HEVs) of normothermic (NT) control mice and 11 order III venules of WBH-treated (six hours) mice (n =3 per group). For blood cell velocities (right), the velocity of >60 noninteracting (i.e., fast moving) cells in order III venules was measured in NT control and WBH-treated (six hours) mice (n =3 per group). (B) HEV density was quantified in pooled peripheral lymph nodes from NT control and WBH-treated (six hours) mice. (C) TRITC-conjugated bovine serum albumin was introduced into the vascular compartment of mice 20 minutes after WBH treatment and allowed to circulate for three minutes. Peripheral lymph node cryosections were counterstained for PNAd (green) to demark HEVs. Permeability was determined by measuring the relative fluorescence within the vascular space of ~80 PNAd+ HEVs versus fluorescence within the surrounding lymph node stroma. Permeability index =lumenal MFI/stromal MFI. Bar =50 mm. Data in (B) and (C) represent ≥ 2 independent experiments. Differences between NT control and WBH-treated mice are not significant (P ≥ 0.1).
Maximal ICAM-1 Density is Required for Optimal Lymphocyte Homing Across HEVs
Evidence that ICAM-1 density in HEVs is spatiotemporally regulated by thermal stress provided a unique in vivo model system to investigate the relationship between ICAM-1 density and the efficiency of lymphocyte homing to secondary lymphoid organs. The contribution of ICAM-1 density to lymphocyte-endothelial adhesion has previously been restricted mainly to in vitro systems, in which ICAM-1 was immobilized on artificial surfaces (e.g., plastic, beads) or where ICAM-1 was induced in cytokine (TNF)-stimulated cultured endothelial cells [20,37,46,50]. A key question in the current study was whether moderate increases in ICAM-1 density reached the threshold required for optimal lymphocyte trafficking, or if homing is further improved when ICAM-1 is maximally induced on HEVs.
This issue was addressed by performing short-term homing assays in mice pretreated with WBH for two, four, or six hours to achieve variable levels of ICAM-1 on HEVs, following the heating schemas shown in Figures 2 and 5. Paralleling the lack of change in ICAM-1 density after two hours of WBH treatment (Figure 2), there was no difference in lymphocyte homing to peripheral lymph nodes at this time point (Figure 5; P =0.34). Intermediate levels of homing were observed following WBH pretreatment for four hours, at a point when ICAM-1 density is moderately increased, but suboptimal. Maximal homing was observed after six hours of continuous heat treatment when ICAM-1 levels were greatest (Figure 5). No change in lymphocyte trafficking was observed in spleen or pancreas at any of these time points (Figure 5). Thus, circulating lymphocytes sense incremental differences in ICAM-1 density presented on the lumenal surface of HEVs, such that their trafficking response is proportional to the intravascular density of ICAM-1.
Figure 5.
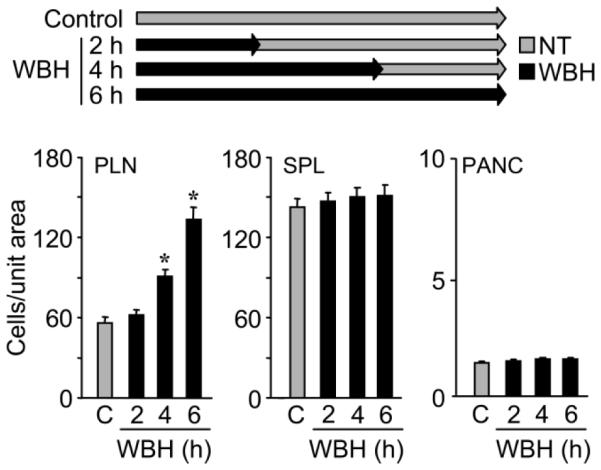
Prolonged heat treatment is required for optimal lymphocyte trafficking in peripheral lymph nodes. Experimental mice were treated with whole-body hyperthermia (WBH) for varying amounts of time and then allowed to revert to normothermal temperature, as indicated. At the end of the six-hour period, in vivo shortterm (one-hour) homing of fluorescent (TRITC)-labeled splenocytes to peripheral lymph nodes (PLN), spleen (SPL), and pancreas (Panc) was assessed in normothermic (NT) control (C) or WBH-treated mice. Organs were harvested one hour after adoptive transfer, and the number of TRITC-labeled cells was quantified in tissue cryosections (n =10 fields) by fluorescence microscopy. P-values represent the difference between NT control and WBH treatment (*P B0.0001). Data represent ≥ 2 independent experiments.
Thermal Induction of ICAM-1 is Restored to Normal Following Cessation of Heat Treatment by a Zinc-Dependent Metalloproteinase
A hallmark of the febrile response is that it is self-limiting, thereby restoring homeostasis [28]. We, therefore, determined the duration of increased ICAM-1 intravascular density in HEVs following the cessation of WBH. Quantitative image analysis revealed a linear decrease in ICAM-1 density, reaching homeostatic levels within six hours of cessation of WBH (Figure 6A). In this regard, statistical analysis showed that ICAM-1 densities at two, four, or six hours after the cessation of WBH were all different from each other and, further, were different from the highly induced levels detected following continuous WBH treatment for six hours (D >0.09). By six hours after the cessation of WBH, ICAM-1 levels were indistinguishable from normothermal control (D ≤0.09). Consistent with the downregulation of ICAM-1 expression, lymphocyte trafficking in peripheral lymph nodes returned to normothermic levels at six hours after the cessation of heat treatment (Figure 6B).
Figure 6.

Intercellular adhesion molecule-1 (ICAM-1) expression and lymphocyte trafficking is reversible after whole-body hyperthermia (WBH) treatment. (A) Intravascular staining of ICAM-1 in peripheral lymph nodes was performed in mice at the indicated time points after a six-hour heat treatment. The histograms indicate the immunofluorescence intensity of ICAM-1 staining quantified in cuboidal high endothelial venules (HEVs) by image analysis. The x-axis indicates fluorescence intensity and the y-axis indicates the number of pixels with each intensity; the mean fluorescence intensity (MFI) is indicated in each condition. Data represent ≥ 3 independent experiments. (B) Short-term (one-hour) homing of fluorescent (TRITC)-labeled splenocytes to peripheral lymph nodes (PLN), spleen (SPL), and pancreas (Panc) was assessed in mice with different treatments: normothermic (NT) control, immediately after six-hour WBH treatment, and six hours after cessation of heat treatment. Organs were harvested one hour after adoptive transfer and the number of TRITC-labeled cells was quantified in tissue cryosections (n =10 fields) by fluorescence microscopy. P-values represent the difference between NT control and WBH treatment (*P B0.0001). Data represent ≥ 2 independent experiments.
These results raised the question of the fate of ICAM-1 following the cessation of WBH. One possibility was that ICAM-1 was internalized following ligation by LFA-1 on adherent cells, which occurs in cultured human endothelial cells only after extensive ICAM-1 cross-linking. [30,39]. Alternatively, ICAM-1 could be actively shed from the lumenal surface of HEVs by proteases. The zinc-dependent proteolytic enzymes, ADAM-17/TACE (a disintegrin and metalloproteinase/TNF-alpha converting enzyme) and MT1-MMP (membrane type 1-matrix metalloproteinase) have been implicated in the cleavage of ICAM-1 from the surface of TNF-activated cultured endothelial cells in vitro [53,63], although the contribution of sheddases to the regulation of endothelial ICAM-1 in vivo has not been previously investigated. To determine if the restoration of ICAM-1 density to basal levels in HEVs involves a shedding mechanism, we used TAPI-1, a peptidomimetic zinc-ligating metalloproteinase inhibitor that has been shown to inhibit ICAM-1 release by endothelial cells in vitro [53,63]. In initial studies performed in normothermic mice, we determined that administration of TAPI-1 at a concentration reported to block ADAM-17/TACE activity in vivo [38] had no effect on basal levels of ICAM-1 density over a six-hour period, compared to DMSO vehicle controls (MFIs: 42 [NT], 43 [DMSO], and 39 [TAPI-1]; D ≤0.09). Findings that the rate of constitutive TAPI-1-sensitive shedding of ICAM-1 by HEVs is limited under homeostatic conditions suggest that the observed increase in ICAM-1 density that occurs during fever-range thermal stress cannot be attributed solely to the inhibition of constitutive ICAM-1 shedding from high endothelial cells.
Treatment with TAPI-1 immediately after cessation of WBH inhibited the loss of ICAM-1 over a six-hour period by 89% (Figure 7). Statistical analysis indicated that ICAM-1 staining in HEVs of mice treated with TAPI-1 post-WBH was different from mice treated with DMSO post-WBH (D >0.09). Identical results were obtained when intravascular staining was performed using higher amounts of anti-ICAM-1 primary Ab (50 μg instead of 25 μg/mouse), or when frozen tissues were stained with primary anti-ICAM-1 Ab after sectioning. These results excluded the possibility that shed ICAM-1 in the vascular compartment was interfering with intravascular staining of ICAM-1 in HEVs. Independent confirmation that TAPI-1 affects cellular release of ICAM-1 in vivo was provided by findings that administration of TAPI-1 immediately after cessation of thermal stress significantly decreased circulating levels of soluble (s)ICAM-1 over a six-hour period (i.e., plasma sICAM-1 concentrations were 375.6±24.2 ng/mL for TAPI-1 treatment vs. 490.7±44.8 ng/mL for non-TAPI-1–treated mice when measured six hours post-WBH; P <0.05, data are the mean9SEM; n =3 mice per group). Moreover, a trend toward an increase in plasma sICAM-1 was detected six hours after cessation of thermal stress, although the results did not reach statistical significance (i.e., 490.7944.8 ng/mL for six hours post-WBH vs. 448.2922.3 ng/mL for normothermal controls, P =0.41; n =3 mice). The failure to detect an increase in sICAM-1 during the period that ICAM-1 was shed from HEVs may reflect the limited number of high endothelial cells contributing to the circulating pool as well as clearance of sICAM-1 by the liver and kidneys [6,61]. Collectively, these data indicate that the rapid reduction in ICAM-1 display on HEVs after cessation of WBH is due to active shedding by ADAM-17/TACE or a related zinc-dependent metalloproteinase.
Figure 7.

Loss of intercellular adhesion molecule-1 (ICAM-1) after cessation of whole-body hyperthermia (WBH) treatment is due to proteolytic cleavage. Mice were subjected to WBH treatment for six hours to induce maximal ICAM-1 expression in high endothelial venules (HEVs). Mice were then treated with either the hydroxamic acid-based metalloproteinase inhibitor, TAPI-1, or with vehicle alone (dimethyl sulfoxide; DMSO). Photomicrographs show typical images of intravascular staining of ICAM-1 under the indicated conditions. Scale bar, 50 mm. Histograms indicate the immunofluorescence intensity of ICAM-1 staining in cuboidal HEVs, as determined by quantitative image analysis. The x-axis indicates fluorescence intensity and the y-axis indicates the number of pixels with each intensity; the mean fluorescence intensity (MFI) is indicated in each condition. Data represent ≥ 2 independent experiments.
DISCUSSION
The continuous recirculation of naïve lymphocytes across HEVs of secondary lymphoid organs plays a critical role in immune surveillance. Under homeostatic conditions, the cellular content of a single mouse lymph node (comprised of ~106 cells) is replaced ~three times each day [65]. This high-throughput process favors the chance that rare antigen-specific T cells, present at a frequency of ~1 in 105–106 cells [5,44], will encounter dendritic cells presenting cognate antigen from peripheral tissues. It has long been recognized that the dynamics of lymphocyte trafficking through regional lymph nodes is dramatically altered during the classical “shutdown” response that occurs 3–18 hours after local infection or inflammation [26,10]. Exit from lymph nodes during the shutdown phase is prevented by transient downregulation of the sphingosine-1-phosphate receptor on antigen-stimulated lymphocytes, thereby allowing sufficient opportunity for these cells to undergo proliferation within the supportive environment of the lymph node [52]. A concomitant increase in entry of naïve T cells across HEVs results from dilation of the main arterioles that supply the microcirculation in lymph nodes [55]. The total number of HEVs is also elevated in inflamed lymph nodes, providing additional surface area for naïve lymphocytes to initiate the multistep adhesion cascade leading to extravasation [55,68].
Although the adhesion mechanisms controlling homeostatic lymphocyte trafficking in HEVs are well understood, little is known about the molecular changes in HEV adhesion that lead to increased entry of naïve T cells during inflammation. Here, we used thermal stress as a model of systemic febrile inflammation to investigate the relationship between ICAM-1 density on HEVs and lymphocyte trafficking in lymphoid organs. The major findings of this report are: (1) The surface density of the gatekeeper homing molecule ICAM-1 in HEVs is temporally controlled by the thermal element of the febrile inflammatory response; (2) ICAM-1 density on HEVs is a critical determinant of the efficiency of lymphocyte trafficking; and (3) Restoration of ICAM-1-dependent trafficking to homeostatic levels following cessation of thermal stress requires the activity of a zinc-dependent metalloproteinase.
Progressive increases in ICAM-1 density on HEVs following heat treatment were linked to enhanced LFA-1/ICAM-1-dependent trafficking of lymphocytes across this microvascular barrier. Hemodynamic parameters were normal at the time point at which homing assays were performed, strongly suggesting that improved trafficking is attributed to increased availability of ICAM-1 to circulating lymphocytes as they pass through HEVs. These observations do not exclude the possibility that transient hemodynamic changes occur during the actual heating period that contribute to increased intravascular ICAM-1, particularly in view of reports that shear stress stimulates ICAM-1 synthesis and surface display on endothelial cells in vitro [41,62]. The temporal effects of heat on microvascular adhesion are consistent with the sustained time interval of physiological fever during inflammation and infection. Of particular note, we have shown that strong HEV responses are triggered by inflammatory cues provided by systemic thermal stress in the absence of antigenic stimulation. Targeted regulation of trafficking across HEVs in lymphoid organs throughout the body in the context of febrile temperatures, as shown in the present study, would be immunologically advantageous, since immune surveillance would be amplified locally in sentinel lymph nodes (i.e., the site of antigen deposition) as well as systemically.
The sensitivity of circulating lymphocytes to incremental changes in intravascular ICAM-1 is remarkable given that HEVs constitutively express ICAM-1 at high density (i.e., at ~13,800±1500 sites/mm2 [20]). Elevation of ICAM-1 density on the lumenal surface of HEVs by thermal stress is predicted to augment trafficking by enhancing the probability of encounters with a fixed number of LFA-1 molecules on circulating lymphocytes. Our observations regarding ICAM-1–dependent trafficking in HEVs in vivo are in line with in vitro studies showing that ICAM-1 density affects lymphocyte adhesion [20,50]. In this regard, significantly greater lymphocyte adhesion occurs on immobilized ICAM-1 at higher, compared to lower, density (6000 versus 500 sites/μm2 [20] or 1900 versus 190 sites/μm2 [50]). Ligand interactions under conditions of high ICAM-1 density have been shown in vitro to stabilize adhesion via outside-in signaling that converts LFA-1 to a high-affinity conformer [11,50]. Notably, ICAM-1, but not ICAM-2, forms dimers when expressed at high density on cultured endothelial cells and bond lifetimes of dimeric ICAM-1 for LFA-1 are ~10 times longer than the monomeric form [37,46,48]. High endothelial expression of ICAM-1 could contribute to the preferential role demonstrated in vitro for this LFA-1 ligand in activation of Rho GTPase, which is required for cytoskeletal reorganization, generation of endothelial docking structures, and transendothelial migration of lymphocytes [2,4,12,13,34,36,42,51,60].
The rapid reduction in ICAM-1 density detected in HEVs following cessation of thermal stress is reminiscent of the kinetics of the early phase of resolution of inflammation, which aims for a return to homeostasis. Normalization of ICAM-1 density in HEVs within six hours restores trafficking to constitutive levels, thus reducing the metabolic demands associated with new ICAM-1 synthesis and active lymphocyte migration. Evidence that the loss of ICAM-1 from HEVs is inhibited by the hydroxamic acid-based metalloproteinase inhibitor, TAPI-1, as reported for endothelial cells in vitro [53,63], strongly implicates ADAM-17/TACE and/ or MT1-MMP in ICAM-1 ectodomain cleavage during the return to vascular homeostasis following febrile episodes. Recent findings [35] that ICAM-1 is shed as functionally active dimers in vivo raise the possibility that sICAM-1 could locally dampen lymphocyte-endothelial adhesion in HEVs during the resolving phase of inflammation.
Our data further demonstrate a differential effect of TAPI-1 on ICAM-1 expression in HEVs of normothermic mice and heat-treated mice. In this regard, baseline steady-state levels of ICAM-1 on HEVs were not altered by TAPI-1 treatment in normothermic mice, whereas TAPI-1 had a profound effect when administered following WBH treatment. These findings suggest that the rate of ICAM-1 shedding in high endothelial cells was accelerated during the resolving phase following the cessation of fever-range thermal stress. ADAM-17 activity in neutrophils or epithelial cells has been shown in vitro to be induced in response to PMA activation or phagocytosis of bacterial pathogens within a similar time frame as our studies (≤ six hours) by multiple mechanisms, including control of mRNA levels, processing of the proenzyme to the catalytically active mature form, and translocation from intracellular stores (endoplasmic reticulum) to the cell surface [14,49,56,67,69]. It remains to be determined whether ICAM-1 cleavage in high endothelial cells following thermal stress is regulated by these intrinsic mechanisms or if extrinsic mechanisms are involved in which sheddases on adherent leukocytes act in trans to release ICAM-1 substrate from HEVs.
CONCLUSIONS
In summary, these findings highlight the dynamic role of the microcirculation in regulating immune surveillance under acute inflammatory conditions associated with fever and the early phase of resolution of inflammation. These results suggest that an important function of fever is to act as a rheostat to augment lymphocyte migration through secondary lymphoid organs by controlling ICAM-1-dependent adhesion in the lymphocyte–HEV axis throughout the body. The complex mechanisms regulating ICAM-1 density in vascular endothelium are likely to also be relevant to inflammation associated with autoimmune disorders and in cancer patients during local and systemic thermal therapy [21,32,47].
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- 1.Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with Image. J. Biophoton Int. 2004;11:36–42. [Google Scholar]
- 2.Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. J Immunol. 1999;162:2964–2973. [PubMed] [Google Scholar]
- 3.Appenheimer MM, Chen Q, Girard RA, Wang WC, Evans SS. Impact of fever-range thermal stress on lymphocyte-endothelial adhesion and lymphocyte trafficking. Immunol Invest. 2005;34:295–323. doi: 10.1081/imm-200064501. [DOI] [PubMed] [Google Scholar]
- 4.Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–664. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonomini M, Reale M, Santarelli P, Stuard S, Settefrati N, Albertazzi A. Serum levels of soluble adhesion molecules in chronic renal failure and dialysis patients. Nephron. 1998;79:399–407. doi: 10.1159/000045084. [DOI] [PubMed] [Google Scholar]
- 7.Bossi F, Fischetti F, Pellis V, Bulla R, Ferrero E, Mollnes TE, Regoli D, Tedesco F. Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. J Immunol. 2004;173:6921–6927. doi: 10.4049/jimmunol.173.11.6921. [DOI] [PubMed] [Google Scholar]
- 8.Burd R, Dziedzic TS, Xu Y, Caligiuri MA, Subjeck JR, Repasky EA. Tumor cell apoptosis, lymphocyte recruitment, and tumor vascular changes are induced by low temperature, long duration (fever-like) whole body hyperthermia. J Cell Physiol. 1998;177:137–147. doi: 10.1002/(SICI)1097-4652(199810)177:1<137::AID-JCP15>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 9.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–6. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 10.Cahill RN, Frost H, Trnka Z. The effects of antigen on the migration of recirculating lymphocytes through single lymph nodes. J Exp Med. 1976;143:870–888. doi: 10.1084/jem.143.4.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carman CV, Springer TA. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr Opin Cell Biol. 2003;15:547–556. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cernuda-Morollon E, Ridley AJ. Rho GTPases and leukocyte adhesion receptor expression and function in endothelial cells. Circ Res. 2006;98:757–767. doi: 10.1161/01.RES.0000210579.35304.d3. [DOI] [PubMed] [Google Scholar]
- 14.Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–1755. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 15.Chen Q, Clancy KA, Wang WC, Evans SS. Inflammatory Cues Controlling Lymphocyte-Endothelial Interactions During Fever-Range Thermal Stress. In: Aird W, editor. Endothelial Biomedicine. Cambridge University Press; 2007. pp. 471–479. [Google Scholar]
- 16.Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, Rose-John S, von Andrian UH, Baumann H, Evans SS. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol. 2006;7:1299–1308. doi: 10.1038/ni1406. [DOI] [PubMed] [Google Scholar]
- 17.Chen Q, Fisher DT, Kucinska SA, Wang WC, Evans SS. Dynamic control of lymphocyte trafficking by fever-range thermal stress. Cancer Immunol Immunother. 2006;55:299–311. doi: 10.1007/s00262-005-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Q, Wang WC, Bruce R, Li H, Schleider DM, Mulbury MJ, Bain MD, Wallace PK, Baumann H, Evan SS. Central role of IL-6 receptor signal-transducing chain gp130 in activation of L-selectin adhesion by fever-range thermal stress. Immunity. 2004;20:59–70. doi: 10.1016/s1074-7613(03)00358-3. [DOI] [PubMed] [Google Scholar]
- 19.Conver WJ. Practical Nonparametric Statistics. John Wiley & Sons; New York: 1971. pp. 309–314. [Google Scholar]
- 20.Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, Laudanna C. Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–769. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 21.Corry PM, Dewhirst MW. Thermal medicine, heat shock proteins, and cancer. Int J Hyperthermia. 2005;21:675–677. doi: 10.1080/02656730500272856. [DOI] [PubMed] [Google Scholar]
- 22.Dafni H, Landsman L, Schechter B, Kohen F, Neeman M. MRI and fluorescence microscopy of the acute vascular response to VEGF165: vasodilation, hyper-permeability, and lymphatic uptake, followed by rapid inactivation of the growth factor. NMR Biomed. 2002;15:120–131. doi: 10.1002/nbm.724. [DOI] [PubMed] [Google Scholar]
- 23.Evans SS, Wang WC, Bain MD, Burd R, Ostberg JR, Repasky EA. Fever-range hyperthermia dynamically regulates lymphocyte delivery to high endothelial venules. Blood. 2001;97:2727–2733. doi: 10.1182/blood.v97.9.2727. [DOI] [PubMed] [Google Scholar]
- 24.de Fougerolles AR, Stacker SA, Schwarting R, Springer TA. Characterization of ICAM-2 and evidence for a third counter-receptor for LFA-1. J Exp Med. 1991;174:253–267. doi: 10.1084/jem.174.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Girard JP, Springer TA. High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol Today. 1995;16:449–457. doi: 10.1016/0167-5699(95)80023-9. [DOI] [PubMed] [Google Scholar]
- 26.Hall JG, Morris B. The immediate effect of antigens on the cell output of a lymph node. Br J Exp Pathol. 1965;46:450–454. [PMC free article] [PubMed] [Google Scholar]
- 27.Hasday JD, Fairchild KD, Shanholtz C. The role of fever in the infected host. Microbes Infect. 2000;2:1891–1904. doi: 10.1016/s1286-4579(00)01337-x. [DOI] [PubMed] [Google Scholar]
- 28.Kluger MJ. Fever: role of pyrogens and cryogens. Physiol Rev. 1991;71:93–127. doi: 10.1152/physrev.1991.71.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraybill WG, Olenki T, Evans SS, Ostberg JR, O’Leary KA, Gibbs JF, Repasky EA. A phase I study of fever-range whole body hyperthermia (FR-WBH) in patients with advanced solid tumours: correlation with mouse models. Int J Hyperthermia. 2002;18:253–266. doi: 10.1080/02656730110116704. [DOI] [PubMed] [Google Scholar]
- 30.Kuijpers TW, Raleigh M, Kavanagh T, Janssen H, Calafat J, Roos D, Harlan JM. Cytokine-activated endothelial cells internalize E-selectin into a lysosomal compartment of vesiculotubular shape. A tubulin-driven process. J Immunol. 1994;152:5060–5069. [PubMed] [Google Scholar]
- 31.Lehmann JC, Jablonski-Westrich D, Haubold U, Gutierrez-Ramos JC, Springer T, Hamann A. Overlapping and selective roles of endothelial intercellular adhesion molecule-1 (ICAM-1) and ICAM-2 in lymphocyte trafficking. J Immunol. 2003;171:2588–2593. doi: 10.4049/jimmunol.171.5.2588. [DOI] [PubMed] [Google Scholar]
- 32.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 33.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyck R, Reiss Y, Gerwin N, Greenwood J, Adamson P, Engelhardt B. T-cell interaction with ICAM-1/ICAM-2 double-deficient brain endothelium in vitro: the cytoplasmic tail of endothelial ICAM-1 is necessary for transendothelial migration of T cells. Blood. 2003;102:3675–3683. doi: 10.1182/blood-2003-02-0358. [DOI] [PubMed] [Google Scholar]
- 35.Melis M, Pace E, Siena L, Spatafora M, Tipa A, Profita M, Bonanno A, Vignola AM, Bonsignore G, Mody CH, Gjomarkaj M. Biologically active intercellular adhesion molecule-1 is shed as dimers by a regulated mechanism in the inflamed pleural space. Am J Respir Crit Care Med. 2003;167:1131–1138. doi: 10.1164/rccm.200207-654OC. [DOI] [PubMed] [Google Scholar]
- 36.Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- 37.Miller J, Knorr R, Ferrone M, Houdei R, Carron CP, Dustin ML. Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function associated-1. J Exp Med. 1995;182:1231–1241. doi: 10.1084/jem.182.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohler KM, Sleath PR, Fitzner JN, Cerretti DP, Alderson M, Kerwar SS, Torrance DS, Otten-Evans C, Greenstreet T, Weerawarna K, et al. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature. 1994;370:218–220. doi: 10.1038/370218a0. [DOI] [PubMed] [Google Scholar]
- 39.Murciano JC, Muro S, Koniaris L, Christofidou-Solomidou M, Harshaw DW, Albelda SM, Granger DN, Cines DB, Muzykantov VR. ICAM-directed vascular immunotargeting of antithrombotic agents to the endothelial luminal surface. Blood. 2003;101:3977–3984. doi: 10.1182/blood-2002-09-2853. [DOI] [PubMed] [Google Scholar]
- 40.Myers CL, Wertheimer SJ, Schembri-King J, Parks T, Wallace RW. Induction of ICAM-1 by TNF-alpha, IL-1 beta, and LPS in human endothelial cells after downregulation of PKC. Am J Physiol. 1992;263:C767–C772. doi: 10.1152/ajpcell.1992.263.4.C767. [DOI] [PubMed] [Google Scholar]
- 41.Nagel T, Resnick N, Atkinson WJ, Dewey CF, Jr., Gimbrone MA., Jr. Shear stress selectively upregulates intercellular adhesion molecule-1 expression in cultured human vascular endothelial cells. J Clin Invest. 1994;94:885–891. doi: 10.1172/JCI117410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oh HM, Lee S, Na BR, Wee H, Kim SH, Choi SC, Lee KM, Jun CD. RKIKK motif in the intracellular domain is critical for spatial and dynamic organization of ICAM-1: functional implication for the leukocyte adhesion and transmigration. Mol Biol Cell. 2007;18:2322–2335. doi: 10.1091/mbc.E06-08-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostberg JR, Repasky EA. Comparison of the effects of two different whole body hyperthermia protocols on the distribution of murine leukocyte populations. Int J Hyperthermia. 2000;16:29–43. doi: 10.1080/026567300285402. [DOI] [PubMed] [Google Scholar]
- 44.Pabst R, Binns RM. Heterogeneity of lymphocyte homing physiology: several mechanisms operate in the control of migration to lymphoid and non-lymphoid organs in vivo. Immunol Rev. 1989;108:83–109. doi: 10.1111/j.1600-065x.1989.tb00014.x. [DOI] [PubMed] [Google Scholar]
- 45.Pritchard MT, Ostberg JR, Evans SS, Burd R, Kray-bill W, Bull JM, Repasky EA. Protocols for simulating the thermal component of fever: preclinical and clinical experience. Methods. 2004;32:54–62. doi: 10.1016/s1046-2023(03)00187-7. [DOI] [PubMed] [Google Scholar]
- 46.Reilly PL, Woska JR, Jr, Jeanfavre DD, McNally E, Rothlein R, Bormann BJ. The native structure of intercellular adhesion molecule-1 (ICAM-1) is a dimer. Correlation with binding to LFA-1. J Immunol. 1995;155:529–532. [PubMed] [Google Scholar]
- 47.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 48.Sarantos MR, Raychaudhuri S, Lum AF, Staunton DE, Simon SI. Leukocyte function-associated antigen 1-mediated adhesion stability is dynamically regulated through affinity and valency during bond formation with intercellular adhesion molecule-1. J Biol Chem. 2005;280:28290–28298. doi: 10.1074/jbc.M501662200. [DOI] [PubMed] [Google Scholar]
- 49.Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE) Biochem J. 2000;347(Pt 1):131–138. [PMC free article] [PubMed] [Google Scholar]
- 50.Shamri R, Grabovsky V, Gauguet JM, Feigelson S, Manevich E, Kolanus W, Robinson MK, Staunton DE, von Andrian UH, Alon R. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol. 2005;6:497–506. doi: 10.1038/ni1194. [DOI] [PubMed] [Google Scholar]
- 51.Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, Parkos CA, Luscinskas FW. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 53.Sithu SD, English WR, Olson P, Krubasik D, Baker AH, Murphy G, D’Souza SE. Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J Biol Chem. 2007;282:25010–25019. doi: 10.1074/jbc.M611273200. [DOI] [PubMed] [Google Scholar]
- 54.Skitzki JJ, Chen Q, Wang WC, Evans SS. Primary immune surveillance: some like it hot. J Mol Med. 2007;85:1361–1367. doi: 10.1007/s00109-007-0245-7. [DOI] [PubMed] [Google Scholar]
- 55.Soderberg KA, Payne GW, Sato A, Medzhitov R, Segal SS, Iwasaki A. Innate control of adaptive immunity via remodeling of lymph node feed arteriole. Proc Natl Acad Sci U S A. 2005;102:16315–16320. doi: 10.1073/pnas.0506190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soond SM, Everson B, Riches DW, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci. 2005;118:2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- 57.Stein JV, Cheng G, Stockton BM, Fors BP, Butcher EC, von Andrian UH. L-selectin-mediated leukocyte adhesion in vivo: microvillous distribution determines tethering efficiency, but not rolling velocity. J Exp Med. 1999;189:37–50. doi: 10.1084/jem.189.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Streeter PR, Berg EL, Rouse BT, Bargatze RF, Butcher EC. A tissue-specific endothelial cell molecule involved in lymphocyte homing. Nature. 1988;331:41–46. doi: 10.1038/331041a0. [DOI] [PubMed] [Google Scholar]
- 59.Team RDC . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2006. [Google Scholar]
- 60.Thompson PW, Randi AM, Ridley AJ. Intercellular adhesion molecule (ICAM)-1, but not ICAM-2, activates RhoA and stimulates c-fos and rhoA transcription in endothelial cells. J Immunol. 2002;169:1007–1013. doi: 10.4049/jimmunol.169.2.1007. [DOI] [PubMed] [Google Scholar]
- 61.Thomson AW, Satoh S, Nussler AK, Tamura K, Woo J, Gavaler J, van Thiel DH. Circulating intercellular adhesion molecule-1 (ICAM-1) in auto-immune liver disease and evidence for the production of ICAM-1 by cytokine-stimulated human hepatocytes. Clin Exp Immunol. 1994;95:83–90. doi: 10.1111/j.1365-2249.1994.tb06019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Topper JN, Gimbrone MA., Jr. Blood flow and vascular gene expression: fluid shear stress as a modulator of endothelial phenotype. Mol Med Today. 1999;5:40–46. doi: 10.1016/s1357-4310(98)01372-0. [DOI] [PubMed] [Google Scholar]
- 63.Tsakadze NL, Sithu SD, Sen U, English WR, Murphy G, D’Souza SE. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1) J Biol Chem. 2006;281:3157–3164. doi: 10.1074/jbc.M510797200. [DOI] [PubMed] [Google Scholar]
- 64.Vardam TD, Zhou L, Appenheimer MM, Chen Q, Wang WC, Baumann H, Evans SS. Regulation of a lymphocyte-endothelial-IL-6 trans-signaling axis by fever-range thermal stress: hot spot of immune surveillance. Cytokine. 2007;39:84–96. doi: 10.1016/j.cyto.2007.07.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.von Andrian UH. Intravital microscopy of the peripheral lymph node microcirculation in mice. Microcirculation. 1996;3:287–300. doi: 10.3109/10739689609148303. [DOI] [PubMed] [Google Scholar]
- 66.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 67.Walcheck B, Herrera AH, Hill C, Mattila PE, Whitney AR, Deleo FR. ADAM17 activity during human neutrophil activation and apoptosis. Eur J Immunol. 2006;36:968–976. doi: 10.1002/eji.200535257. [DOI] [PubMed] [Google Scholar]
- 68.Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT. Regulation of lymph node vascular growth by dendritic cells. J Exp Med. 2006;203:1903–1913. doi: 10.1084/jem.20052272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Q, Thomas SM, Lui VW, Xi S, Siegfried JM, Fan H, Smithgall TE, Mills GB, Grandis JR. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci U S A. 2006;103:6901–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]