

Abstract
Mechanosensory hair cells (HCs) residing in the inner ear are critical for hearing and balance. Precise coordination of proliferation, sensory specification, and differentiation during development is essential to ensure the correct patterning of HCs in the cochlear and vestibular epithelium. Recent studies have revealed that FGF20 signaling is vital for proper HC differentiation. However, the mechanisms by which FGF20 signaling promotes HC differentiation remain unknown. Here, we show that mitogen-activated protein 3 kinase 4 (MEKK4) expression is highly regulated during inner ear development and is critical to normal cytoarchitecture and function. Mice homozygous for a kinase-inactive MEKK4 mutation exhibit significant hearing loss. Lack of MEKK4 activity in vivo also leads to a significant reduction in the number of cochlear and vestibular HCs, suggesting that MEKK4 activity is essential for overall development of HCs within the inner ear. Furthermore, we show that loss of FGF20 signaling in vivo inhibits MEKK4 activity, whereas gain of Fgf20 function stimulates MEKK4 expression, suggesting that Fgf20 modulates MEKK4 activity to regulate cellular differentiation. Finally, we demonstrate, for the first time, that MEKK4 acts as a critical node to integrate FGF20-FGFR1 signaling responses to specifically influence HC development and that FGFR1 signaling through activation of MEKK4 is necessary for outer hair cell differentiation. Collectively, this study provides compelling evidence of an essential role for MEKK4 in inner ear morphogenesis and identifies the requirement of MEKK4 expression in regulating the specific response of FGFR1 during HC development and FGF20/FGFR1 signaling activated MEKK4 for normal sensory cell differentiation.
SIGNIFICANCE STATEMENT Sensory hair cells (HCs) are the mechanoreceptors within the inner ear responsible for our sense of hearing. HCs are formed before birth, and mammals lack the ability to restore the sensory deficits associated with their loss. In this study, we show, for the first time, that MEKK4 signaling is essential for the development of normal cytoarchitecture and hearing function as MEKK4 signaling-deficient mice exhibit a significant reduction of HCs and a hearing loss. We also identify MEKK4 as a critical hub kinase for FGF20-FGFR1 signaling to induce HC differentiation in the mammalian cochlea. These results reveal a new paradigm in the regulation of HC differentiation and provide significant new insights into the mechanism of Fgf signaling governing HC formation.
Keywords: cellular differentiation, cochlea, FGF20, MEKK4, mouse
Introduction
Normal development and function of the auditory-vestibular apparatus require precisely coordinated cellular processes, including induction, cell-type differentiation, pattern formation, and morphogenesis. This in turn requires spatial and temporal coordination of signaling molecules that elicit specific transcriptional responses and regulate inner ear form and function. The auditory portion of the inner ear, the cochlea, transduces sound to neural impulses via the organ of Corti, which is comprised of one row of inner hair cells (IHCs) and three rows of outer hair cells (OHCs). These two types of sensory HCs interdigitate with, and are separated by, distinct supporting cells and form a unique cellular pattern spanning the entire length of the cochlear duct. Several signaling pathways have been shown to regulate inner ear patterning (Groves and Fekete, 2012). In particular, Atoh1, which encodes for a basic helix-loop-helix transcriptional factor, is necessary and sufficient for generation of HCs within the inner ear. Also, previous studies have demonstrated that members of the fibroblast growth factor (FGF) family play a major role in inner ear development (Pirvola et al., 2000, 2002; Pauley et al., 2003; Wright and Mansour, 2003; Shim et al., 2005; Hayashi et al., 2007, 2008; Jacques et al., 2007; Schimmang, 2007; Huh et al., 2012; Yang et al., 2013; Zhang et al., 2014; Sai and Ladher, 2015; Urness et al., 2015; Wang et al., 2015). In particular, FGF20 and Fgf receptor (FGFR)1-mediated signaling positively regulates Atoh1-mediated HC differentiation as deletion of Fgf20/Fgfr1 leads to the significant reduction in the total number of cells that develop into HCs (Pirvola et al., 2002; Hayashi et al., 2008; Huh et al., 2012; Ono et al., 2014). However, exact mechanisms whereby Fgf signaling stimulates Atoh1 expression and subsequent HC differentiation remain unresolved. In contrast, FGFR3-mediated signaling is required for support cell development (Mueller et al., 2002; Puligilla et al., 2007). Together, these data indicate that Fgf signaling initially is required for inner ear induction and subsequently is required for HC and supporting cell differentiation.
It is generally accepted that FGF ligands signal via specific cell surface receptors that elicit specific cellular responses by stimulating intracellular signal transduction pathways. In particular, the growth factor-stimulated signaling pathways, such as the MAP kinase (MAPK) cascades, have been shown to regulate a wide variety of functions, including cell proliferation, adhesion, migration, differentiation, and survival by phosphorylating downstream kinases or transcriptional factors (Chen et al., 2001; Pearson et al., 2001). A diverse set of stimuli can lead to MAPK activation (Kyriakis and Avruch, 2001); and based on genetic and biochemical studies, the MAPK pathway is concomitantly activated by FGFs in most cell types (LaBonne and Whitman, 1997; Curran and Grainger, 2000; Abell et al., 2009).
The MAPK superfamily is comprised of a network of interacting kinases, which can be divided into subfamilies, including extracellular-signal-regulated kinases (ERKs), stress-activated protein kinases (JNKs), and p38s (Garrington and Johnson, 1999). The activation of each MAPK is mediated by phosphorylation by MAPK kinases (MAP2Ks), which in turn phosphorylate MAPK kinase kinase (MAP3Ks or MEKKs). MEKK4, commonly referred to as MAP3K4, has been shown to regulate skeletal and neural patterning during embryonic mouse development (Abell et al., 2005). In addition, MEKK4 phosphorylates and activates MAP2Ks, MKK3/MKK6, and MKK4/MKK7, leading to the activation of p38 and JNK pathways, respectively (Abell et al., 2007, 2009). However, upstream signaling molecules and molecular mechanisms that regulate MEKK4 activation are poorly defined. Given that MEKK4 activity regulates developmental epithelial cell patterning (Craig et al., 2008), we investigated the potential role of MEKK4 in inner ear development. We show that MEKK4 is highly expressed in the embryonic inner ear and ablation of its activity leads to aberrant differentiation of HCs and supporting cells and dysregulation of cellular patterning in the auditory and vestibular epithelium. We further demonstrate that MEKK4 expression is activated by FGF20-FGFR1-signaling in vivo and that FGF20-mediated signaling is activated via MEKK4 to influence sensory HC differentiation in the developing cochlea.
Materials and Methods
Mouse breeding and genotyping.
MEKK4K1361R/WT heterozygotic mice were obtained from MMMRC. The heterozygotic mice were intercrossed to obtain homozygous MEKK4K1361R mutant mice. All mice were genotyped by PCR as previously described (Abell et al., 2005). As noted previously, two-thirds of embryos homozygous for the MEKK4 kinase inactive mutation die perinatally due to open neural tube, exencephaly, and body wall defects (Abell et al., 2005); however, a small percentage of mice homozygous for MEKK4K1361R survived to adulthood (Table 1) and were used for auditory brainstem response (ABR) studies. CD-1 mice were obtained from Harlan Laboratories. For all mice, the day of mating plug observed was considered as E0.5 and the day of birth was considered P0. All mice were housed at the Medical University of South Carolina and maintained according to protocols approved by the Medical University of South Carolina Institutional Animal Care and Use Committee.
Table 1.
Total number of genotypes of offspring obtained from heterozygous MEKK4K1361R/+ mutant intercrosses
| Stage | Total no. of pups | No. of MEKK4WT | No. of MEKK4K1361R/+ | No. of MEKK4K1361R/K1361R |
|---|---|---|---|---|
| E14.5 | 74 | 9 | 39 | 26 |
| E18.5 | 193 | 41 | 93 | 59 |
| Postnatal | 266 | 61 | 143 | 62 |
Whole-mount in situ hybridization.
Digoxigenin-labeled probes were prepared from the plasmids provided as a gift to us by Dr. Amy Abell. Cochleae were collected at indicated ages from timed-pregnant mice and fixed in 4% PFA in PBS overnight at 4°C and stored in 100% methanol at −20°C. Tissue was washed three times in DEPC-PBS containing 0.5% Tween (PBST) and treated with detergent mix (50 mm Tris-HCl, pH 8.0, 1 mm EDTA, 150 mm NaCl, 1% IGEPAL, 1% SDS, and 0.5% sodium deoxycholate) for three times, 20 min each followed by three 10 min washes in PBST. Tissue was fixed in 4% PFA at room temperature (RT) followed by washing in DEPC-PBST for 10 min on a rocker. Tissue was incubated in 50% PBST and 50% freshly prepared hybridization mix (50% formamide, 1.3 × SSC, pH 5.0, 5 mm EDTA, pH 8.0, 50 μg/ml yeast t-RNA, 0.2% Tween 20, 0.5% CHAPS, 100 μg/ml heparin) with rocking for 10 min at RT followed by soaking in 100% hybridization mix for another 10 min. Tissue was then incubated in prewarmed hybridization mix for 2 h rocking at 65°C. Digoxigenin-labeled probe was mixed with fresh hybridization buffer, which was added to the tissue and incubated overnight at 65°C. The tissue was then washed with prewarmed hybridization buffer at 65°C rocking for 10 min followed by another two washes for 30 min each with 2 × SSC, pH 5.0, at 65°C and three washes for 10 min each with 1 × MABT (100 mm maleic acid, 150 mm NaCl, pH 7.5, 0.1% Tween) at RT on a rocker. Tissue was preblocked for 1 h in 2% BBR/MABT at RT followed by blocking in mix containing 2% BBR, 20% sheep serum in MABT at RT for 1 h. Tissue was then stained with anti-digoxigenin-alkaline phosphatase antibody (Roche) (1:2000) overnight at 4°C. After antibody incubation, the tissue was washed six times for 30 min each at RT with MABT followed by another three washes with freshly prepared NTMT buffer (100 mm Tris pH 9.5, 50 mm MgCl2, 100 mm NaCl, 1% PBST) containing 5 mm levamisole. The tissue was then developed in NTMT solution containing premixed NBT-BCIP solution (Roche) in the dark at RT until the purple reaction product had developed. The tissue was then rinsed and mounted on slides using fluoromount for analysis and imaging. Imaging was performed using Nikon Eclipse 360i or Zeiss AxioVision equipped with ERC5s camera.
RNA isolation and real-time quantitative PCR.
RNA was isolated using the TRIzol method from inner ears or cochlea micro-dissected down to the sensory epithelium at indicated ages. Briefly, 2–4 cochleae from genotype-matched embryos were pooled and RNA was isolated, which was then quantified using NanoDrop (Thermo Scientific), and ∼0.25–1 μg of RNA was transcribed to cDNA using Transcriptor first-stand cDNA synthesis kit (Roche Life Sciences). Three technical replicates of each of the at least three biological replicates were analyzed.
Quantification and statistical analysis.
HC counts within cochleae were made on processed images immunolabeled with Myo6 and actin using ImageJ software taken at distances, 25%, 50%, and 75% from basal regions of the cochlear duct. For determining the width of sensory HCs, we used AxioVision software by Carl Zeiss Imaging to generate images of the basal, mid-basal, mid-apical regions of whole-mount cochleae immunolabeled with Myo6 and actin and measured the width of 20 cells within each region. For utricular HC counts, three nonoverlapping boxes (50 × 100 μm) were digitally placed over the processed low-magnification z-stack images of utricles, and the total number of HCs were counted using ImageJ software. For total surface area measurements, we used Carl Zeiss AxioVision software to digitally measure the total area of the HCs within each utricle, and significance was determined using the Student's t test. Statistical analysis was performed using GraphPad Prism (3.03) or MS-Excel software. The differences in means between two groups were analyzed using two-tailed paired Student's t test to determine the significance of the difference between control and mutant samples. The difference in means more than two groups was done using one-way ANOVA followed by Tukey's comparison test. Values of p < 0.05 were considered to be statistically significant.
Cochlear outgrowth assay and explant culture.
Embryos were collected from timed pregnant females, and cochleae were dissected from the embryos at indicated ages and cultured as previously described (Haque et al., 2015). Cochlear explants were treated with anti-FGF20 or recombinant FGF20 protein (R&D Systems) or SU5402 (3-[4-methyl-2-(2-oxo-1,2-dihydro-indol-3-ylidenemethyl)-1H-pyrrol-3-yl]-proprionic acid) (Santa Cruz Biotechnology) at indicated concentrations. For the cochlear outgrowth assay, cochleae were bisected at ∼40% (mid-apex) of the cochlear length, and only the basal portion of the cochlea was cultured as previously described (Yamamoto et al., 2009).
Immunofluorescent labeling and imaging.
Staged inner ears or cochlear explants following the desired length of incubation were fixed in 4% PFA for 10 min at RT. After washing twice in PBS, samples were processed for immunolabeling. For sectioning, samples were cryo-protected in serial dilutions of 30% sucrose and were embedded in OCT compound (Tissue-Tek) and sectioned at 12 μm. For immunolabeling analysis, whole-mount or cross-sectioned cochleae were blocked with 10% normal horse serum or goat serum (Vector Labs) containing 0.5% Tween 20 in PBS (PBST) for 1 h at RT, then primary antibodies were applied and incubated at 4°C overnight. Primary antibodies used are as follows: Sox2 and Prestin (Santa Cruz Biotechnology); Myo6 and 7a (Proteus Biosciences); p75ntr (Millipore Bioscience Research Reagents); Islet1 (Abcam); p-ERK, p-AKT (Cell Signaling Technology), and p-JNK (Promega); p27kip1 (NeoMarkers); Parvalbumin (Sigma) and anti-MEKK4 (a gift from Dr. Vaillancourt). Tissue were washed three times with PBSTw and incubated with fluorescently conjugated secondary antibodies (Invitrogen) for 1 h at RT. Sections and/or cochlear explants were coverslipped, and imaging was performed using confocal microscope Zeiss LSM 880 or Zeiss Axioimager. All control and mutant samples were imaged using identical exposure times and capture settings.
ABR measurements.
Mice were anesthetized with an intraperitoneal injection of xylazine (10 mg/kg) and ketamine (100 mg/kg), and then placed in a sound-isolated and electrically shielded booth (Acoustic Systems). Body temperature was monitored and maintained near 37°C with a heating pad. Acoustic stimuli were delivered monaurally to a Beyer earphone attached to a customized plastic speculum inserted into the ear canal. Subdermal electrodes were inserted at the vertex of the skull, under the left ear and under the right ear (ground). ABRs were measured at 8, 16, and 32 kHz. Tucker Davis Technology System III hardware and SigGen/Biosig software were used to present the stimuli (15 ms duration tone bursts with 1 ms rise-fall time) and record the response. Up to 1024 responses were averaged for each stimulus level. Thresholds were determined for each frequency by reducing the intensity in 10 dB increments and then in 5 dB steps near threshold until no organized responses were detected. Thresholds were estimated between the lowest stimulus level where a response was observed and the highest level without response. All ABR measurements were conducted by the same experimenter. The ABR scores were assigned by an expert who was blinded to the treatment conditions.
Immunoblotting.
Briefly, cochleae were isolated from MEKK4WT and MEKK4K1361R mutant embryos and micro-dissected down to the sensory epithelium at indicated ages. Protein was isolated from 2 to 4 cochlea using RIPA lysis buffer (Sigma) containing protease and phosphatase inhibitors, pooled from similar genotypes. For efficient lysis, cochleae were crushed and homogenized using the pestle, and the tubes containing samples were placed on ice for 15–30 min, after which centrifugation was done at 12000 × g, and supernatant containing the protein was carefully collected. Protein was quantified using the Bradford method, and the same concentration of protein from WT and mutant samples was loaded onto any kDa (Bio-Rad) gel, and electrophoresis was done at 110 V for 1.5 h. Protein was then transferred from polyacrylamide gel to nitrocellulose membrane using iBlot (Invitrogen). Next, the membrane was blocked with 5% BSA in 1 × TBST for 1 h followed by overnight incubation with primary antibody. The blot was washed 3–5 times,15 min each with 1× TBST, and then incubated with secondary antibody solution (1:15,000 anti-mouse HRP) (Santa Cruz Biotechnology) prepared in 1 × TBST solution at RT. After secondary antibody incubation, blot was washed 3–5 times with 1 × TBST, and developed using Super signal West Dura (chemiluminescent reagent). Images were captured and analyzed using GE ImageQuant LAS 4000.
Results
MEKK4 is expressed in the developing inner ear
To determine whether MEKK4 is involved in the development of the mouse inner ear, we examined its expression by isolating RNA from inner ears of wild-type (WT) mice at E13, E15, and P0 and performed quantitative real-time PCR (qPCR) analysis. The results indicated that, although MEKK4 expression is initiated as early as embryonic day (E) 13, its expression steadily decreased between E13 and P0 (Fig. 1A). Because the temporal expression pattern of MEKK4 expression coincided with the development of the organ of Corti, we next analyzed its spatial expression pattern by performing in situ hybridization at various time-points during development. MEKK4 mRNA was broadly distributed throughout the floor of the cochlear duct (Fig. 1B,C) at E13.5. No detectable signal for MEKK4 mRNA was observed before E13. As differentiation progresses along the developing cochlear duct, by E16, its expression becomes restricted to developing HCs and supporting cells (Fig. 1D,E) extending from the base to the apex of the cochlea. In addition, expression also is seen in cells within Kolliker's organ (Fig. 1E). In contrast, cells within the lesser epithelial ridge domain are completely devoid of MEKK4 expression. By P0, expression becomes restricted to only one row of cells within the IHC region (Fig. 1F,G). However, no MEKK4 signal was seen in cells within the OHC domain (Fig. 1F,G, brackets). In addition, our immunolabeling analysis using anti-MEKK4 (Fig. 1H–J) demonstrates that, by P0, expression becomes restricted exclusively to inner phalangeal cells, a supporting cell type that interdigitates with IHCs located within the medial domain of the cochlear sensory epithelium. These results indicate that MEKK4 signaling is initially highly expressed in undifferentiated precursor cells and later in developing HCs and supporting cells. As differentiation progresses, its expression was significantly diminished. This spatial and temporal distribution of MEKK4 in the developing inner ear is consistent with previous data demonstrating its high expression levels in undifferentiated embryonic mouse trophoblast cells that is lost with differentiation (Abell et al., 2009). Based on these results, it can be suggested that MEKK4 signaling may play a role in cell fate determination.
Figure 1.
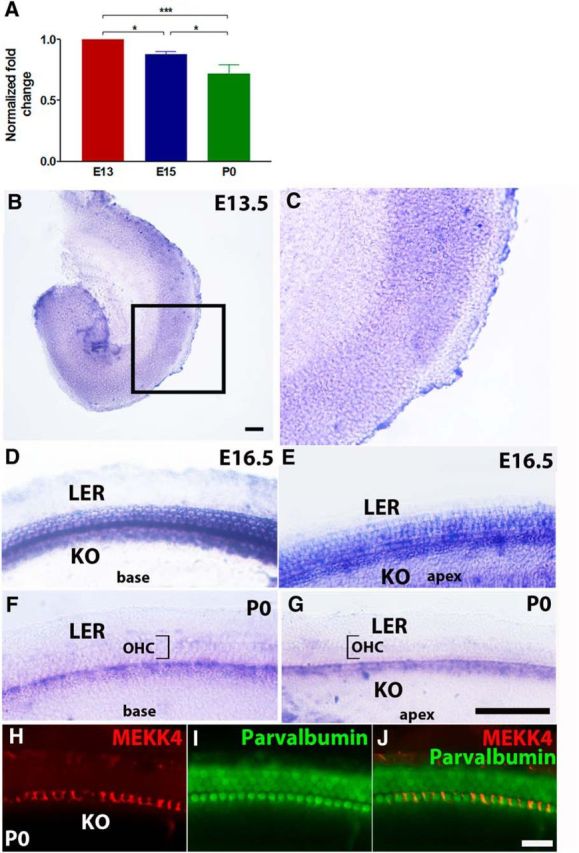
Spatiotemporal expression pattern of MEKK4 in the mouse inner ear. A, qPCR-based analysis reveals developmental changes in the levels of MEKK4 expression within WT mouse inner ears. Although MEKK4 is abundantly expressed in the developing inner ear as early as E13, its expression steadily decreased from E13 to P0. B–G, Whole-mount in situ hybridization for MEKK4 at E13.5 (B, C), E16.5 (D, E), and P0 (F, G) shows that MEKK4 is initially expressed broadly within organ of Corti; and by E16.5, expression is seen in developing HCs and supporting cells. By P0, expression becomes restricted to inner phalangeal cells within the medial domain of the organ of Corti. H–J, Immunolabeling of P0 cochlea using antibodies against MEKK4 (red) and parvalbumin (green) demonstrates the expression of MEKK4 in the soma of inner phalangeal cells. The results represent an average of six samples at each developmental age group. Data are mean ± SEM. KO, Kolliker's organ; LER, lesser epithelial ridge. Scale bars: (in B) B, C, 50 μm; (in G) D–G, 20 μm; (in J) H–J, 20 μm.
MEKK4 activity promotes HC and supporting cell differentiation
The MEKKs exhibit cell type specificity in signal transduction and play a central role in regulating cell fate during development (Schlesinger et al., 1998; Craig et al., 2008). To investigate whether MEKK4 is required for inner ear development, we analyzed MEKK4 kinase-inactive mutant mice in which the MEKK4 gene was altered by the knock-in mutation of the active lysine (K) to an arginine (R), thereby abolishing its phosphorylation activity (Abell et al., 2005). It has been shown that both conventional MEKK4 mutants and kinase-inactive mutant mice die during early embryonic development due to severe birth defects (Abell et al., 2005; Chi et al., 2005). To determine the function of MEKK4 in inner ear development, we examined the MEKK4WT and MEKK4K1361R mutant mice at different time-points. In contrast to the WT mice that had normal inner ears, the majority (>90%) of the mutant mice had clear inner ear abnormalities. Examination of E18.5 MEKK4K1361R mutant cochlea revealed the presence of two rows of OHCs (Fig. 2A–D) extending from the mid-base (10% from the base) to the apex. The third row of OHCs was seen only intermittently throughout the cochlear duct; and as a result, the precise cellular alignment was lost leading to patterning defects. Quantification of HC counts within cochlea at distances of 25%, 50%, and 75% from the basal region of the cochlea taken from 100 μm regions indicated that there was a significant decrease in the number of cells that differentiated into OHCs throughout the cochlear duct of mutants compared with controls (Fig. 2I). In most cases, these defects in patterning also were seen in mice heterozygous for the mutant MEKK4 allele (MEKK4K1361R/WT) because of the dominant negative properties of the kinase inactive MEKK4 protein (data not shown). However, we did not observe any significant reduction in the total number of OHCs in heterozygous mutants compared with WT littermates (data not shown). In contrast to OHCs, no alteration in the number or pattern of the IHC arrangement was observed in MEKK4K1361R cochleae compared with that in WT controls (Fig. 2A–D). It seems likely that the reduction in OHC number in homozygous mutants was due to a developmental delay, in which case it may lead to the rescue of HC numbers as differentiation progresses. To examine this possibility, the expression of HC markers is analyzed in the P7 cochlea. Immunolabeling for the OHC marker Prestin and phalloidin demonstrated that HCs were indeed not delayed in differentiation but rather lost in the absence of MEKK4 signaling (Fig. 2E–H). Indeed, analysis of HC number at postnatal day 7 and 15 (data not shown) shows comparable phenotype with that of E18.5 stage.
Figure 2.
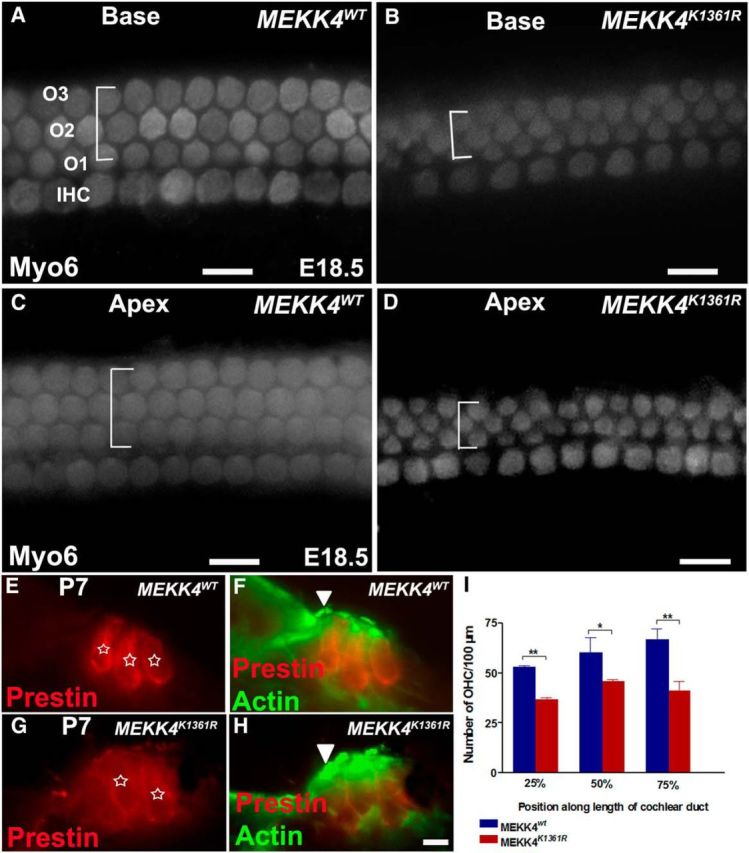
Disruption of MEKK4 pathway causes significant reduction in cells that differentiate into OHCs. A–D, Whole-mount surface views of the base (A, B) and apex (C, D) of a WT (A, C) and MEKK4K1361R mutant cochlea (B, D) at E18.5 immunolabeled with Myo6. In the WT cochlea, normal patterning of one row of IHCs and three rows of OHCs is present. However, in the mutants, the IHCs are unaffected; there are only two rows of OHCs with intermittent third row present in both basal and apical regions of the cochlear duct. The width of the lateral compartment of the sensory epithelium, as indicated by brackets, is diminished in the mutant cochlea compared with WT controls. E–H, Cross-sections of WT (E, F) and MEKK4K1361R cochleae (G, H) immunolabeled with Prestin (red) and actin (green) show that HCs (asterisks), still present at P7, are disorganized with less distinct separation between IHCs (F, H, arrowhead) and OHCs and closely packed OHCs in mutant cochlea as visualized by anti-Prestin (red). I, Quantification of HC counts shows significant decrease in the number of OHCs in homozygous MEKK4K1361R mutant cochlea, relative to MEKK4WT controls at distances 25%, 50%, and 75% from basal regions of the cochlear duct (n = 3). p < 0.001. Error bars indicate SEM. O1–O3, three rows of OHCs. Scale bars: (in D) A–D, 20 μm; (in H) E–H, 10 μm. * **
Because HC differentiation was disrupted in MEKK4 mutants, we analyzed the supporting cell phenotype by immunolabeling cochleae at P1 with antibodies against Sox2, p75ntr, Prox1 (data not shown), and β-tubulin I + II (Fig. 3). In WT cochlea, Sox2 is expressed in supporting cell nuclei, which include three rows of Deiters' cells, two rows of pillar cells, and one row of inner phalangeal cells. In contrast, only two rows of Deiters' cell nuclei were observed in MEKK4K1361R mutants (Fig. 3A,D). Immunolabeling of pillar cells and their nuclei by p75ntr and Sox2 labeling, respectively, showed that inner and outer pillar cells are aberrantly attached to each other, leading to the deformity of inner and outer pillar cell rows (Fig. 3A–F). Also, the elongated morphology of inner pillar cell nuclei in WT littermates (Fig. 3A, arrows) was completely lost in mutants. Despite these morphological defects, the pillar cell gap between IHCs and OHCs seemed normal in the mutants, as shown by p75ntr labeling. In addition, immunolabeling for β-tubulin I + II, which labels microtubules at the lumenal surface of pillar and Deiters' cells in WT cochlea (Fig. 3G–I), revealed disorganization of the normal stereotypical morphologies of these cell types in the absence of MEKK4 signaling (Fig. 3J–L). To quantify the observed differences, expression levels of the HC differentiation marker, Atoh1 and supporting cell markers, Sox2, Hey1, and Hey2 (Hume et al., 2007; Doetzlhofer et al., 2009; Tateya et al., 2011) were examined at P1 by qPCR-based analysis. A significant decrease in transcript levels of Atoh1, Sox2, Hey1, and Hey2 expression was observed in MEKK4K1361R mutants compared with MEKK4WT controls (Fig. 3M). Collectively, these observations suggest that broad distribution of MEKK4 activity in the developing inner ear is essential for normal differentiation of HCs and supporting cells.
Figure 3.

Supporting cell defects in MEKK4K1361R mutants. A–F, Whole-mount surface views of the mid-basal region of WT and mutant cochlea at P1 immunolabeled with Sox2 (A, D) or p75ntr (red) and actin (green) (B, C, E, F). A, In the WT, at least six rows of Sox2-positive supporting cells, including nuclei of inner and outer pillars, inner phalangeal cells, and three rows of Deiters' cells, are observed, which are arranged in organized rows. However, in mutants (D–F), disorganization of supporting cells is observed, and three rows of Deiters' cell nuclei are not discernible. Labeling of pillar cells is seen in the mutant organ of Corti with p75ntr comparable with WT control littermates (B, E, arrows) (B, C, E, F); however, the normal elongated shape of the nuclei of inner pillar cells (A, arrows) is not clearly seen, suggesting disruption of their morphological development. G–L, Labeling with β-tubulin I + β-tubulin II (green) reveals normal morphology of microtubule bundles within inner and outer pillar cell heads (arrows) and throughout the Deiters' cell rows in controls (G–I). J–L, In mutants, although microtubule bundles can be seen within inner pillar cells, microtubule bundle organization within outer pillar cells and Deiters' cells are indistinguishable. M, Relative expression levels of Atoh1, Sox2, Hey1, and Hey2 are significantly lower in mutant cochleae compared with controls (n = 3). p < 0.001. Error bars indicate SEM. D1–D3, Deiters' cell rows; OP, outer pillar cell row; IP, inner pillar cell row. Scale bars: A, D (in D), 20 μm; B, C, E, F (in F), 20 μm; G–L (in L), 20 μm.
To complement the cochlear HC analyses, we examined HCs within the utricle during development. As shown in Figure 4, HCs within the utricular maculae are less densely packed, and the total number of HCs is significantly decreased in MEKK4 kinase-defective inner ears in comparison with WT controls (Fig. 4A–F; n = 3, p < 0.0001). To determine whether this decrease in HC density was a result of a significant reduction in the number of HCs or decrease in the overall size of the epithelium, we measured the total surface area of the utricular epithelial region containing HCs. The data revealed a significant decrease in the size of the utricle of MEKK4 mutants compared with controls (Fig. 4F; n = 3, p < 0.003). These results suggest that the reduction in HC density can be attributed to a decrease in the total number of HCs per unit area. Together, these results suggest that MEKK4 activity regulates the total number of precursor cells that develop into auditory and utricular HCs.
Figure 4.

Disruption of MEKK4 pathway leads to reduction of vestibular HCs. A, B, Low-magnification surface views of utricular epithelium at P1 immunolabeled with Myo7a show fewer HCs in MEKK4K1361R mice (B) compared with MEKK4WT littermates (A). C, D, High-magnification views of WT (C) and mutant (D) utricles labeled with anti-Myo7a (red) and actin (green) show less dense packing of HCs. E, F, MEKK4K1361R utricles exhibit a significant decrease in HC number (E) and the overall size of the HC domain (F) compared with controls. Scale bars: A, 50 μm; B, 50 μm; (in D) C, D, 10 μm. **p < 0.01, ****p < 0.001.
Previous studies have demonstrated an antiapoptotic role for MEKK4 in the developing CNS (Chi et al., 2005), which prompted us to examine whether apoptosis may contribute to the significant reduction of HCs in MEKK4 kinase-deficient mutants. Using the TUNEL assay, we found no evidence of increased cell death in MEKK4K1361R mutants between E13 to P0 stages compared with that in WT littermate controls (data not shown). Together, these results suggest that MEKK4 signaling plays an essential role in cochlear and utricular sensory HC formation.
MEKK4 activity is dispensable for regulating prosensory cell number
The results presented above indicate a disruption in the development of sensory epithelial cells in MEKK4 kinase-defective mutants. It is possible that the significant reduction of HCs in MEKK4 mutants could be due to alterations in cell survival and/or proliferation. Previous studies have demonstrated a role for MEKK4 in cell survival and proliferation; and because the TUNEL assays did not show any signs of cell death in MEKK4 mutants, we next examined whether cell proliferation is affected in the mutant cochlea. No alterations in BrdU-positive cells within sensory epithelium were seen in MEKK4 kinase-defective cochlea at E12 (data not shown).
Because HCs are derived from the prosensory progenitor cells, we next investigated whether MEKK4 signaling plays a key role in the specification of prosensory cells. The expression of Sox2 in prosensory cells (Hume et al., 2007; Dabdoub et al., 2008), which is critical for the establishment of prosensory domain and subsequent HC formation (Kiernan et al., 2005), was examined in MEKK4WT control and MEKK4K1361R mutant inner ears at E11.5 (Fig. 5A–D) and E12.5 (data not shown). Our data reveal no change in the pattern of Sox2 expression within the cochlea, suggesting that MEKK4 does not influence the prosensory progenitor cell population. Furthermore, expression of prosensory markers, Sox2, S100 (Fig. 5E–J) (Buckiová and Syka, 2009), Islet1 (data not shown), and p27Kip1 (Chen and Segil, 1999; Lee et al., 2006) (data not shown) was examined at E14 in control and MEKK4K1361R cochleae. Our analysis on cochlear cross-sections (Fig. 5E–J) indicated comparable expression of prosensory markers between WT controls and MEKK4K1361R mutants. Both the width of the prospective sensory epithelium and relative positioning of these markers seem to be unaltered between control mice and their mutant littermates, suggesting that prosensory formation is relatively normal in MEKK4 mutants and that subsequent specification of these cells into HCs and supporting cells is disrupted within the cochlea.
Figure 5.

MEKK4 function is dispensable for prosensory induction or maintenance and is required for initial Atoh1 expression A–J, Inner ear cross-sections at E11.5 (A–D) and E14 (E–J) were immunolabeled for prosensory markers, Sox2 (red) and actin (green) (A–D) or S100 (green; E–J) demonstrates the intact presence of Sox2 expression within progenitor cells of MEKK4 mutant cochlea, and no significant differences in the size of the prosensory domain were observed between WT and mutant cochleae (indicated by brackets). K–N, Immunolabeling of E14 whole-mount cochlea using anti-Atoh1 (green) and actin (red) shows downregulation of Atoh1 in prospective sensory epithelial cells. O, qPCR-based analysis shows that Atoh1 transcript levels are dramatically reduced in E13.5 MEKK4K1361R cochleae compared with WT littermates; n = 3. ***p < 0.001. Error bars indicate SEM. Scale bars: (in D) A–D, 20 μm; (in J) E–J, 20 μm; (in N) K–N, 10 μm.
Because Atoh1 is one of the first genes expressed in HC and supporting cell precursors and is necessary and sufficient for their differentiation (Bermingham et al., 1999; Zheng and Gao, 2000; Yang et al., 2010; Driver et al., 2013; Jahan et al., 2015), immunolabeling was performed to analyze the expression of Atoh1 in E14 cochlea. As shown (Fig. 5K–N), Atoh1 expression is downregulated in MEKK4K1361R mutants compared with controls. In addition, qPCR-based analysis was performed to measure the levels of Atoh1 expression in the developing cochlear sensory epithelial cells. Although Atoh1 expression is found in MEKK4K1361R cochlea as early as E13.5, its levels were significantly lower than in WT controls (Fig. 5O), suggesting that MEKK4 activity influences initial levels of Atoh1 expression before HC differentiation in the developing cochlea.
MEKK4 signaling promotes cochlear outgrowth and patterning
Previous studies have shown that MEKK4 kinase-inactive mice display severe skeletal and neural patterning defects (Abell et al., 2005). To determine whether MEKK4 plays a role in patterning of the cochlea, we first analyzed the length and overall morphology of MEKK4-deficient and control cochleae at P0. Results indicated that cochleae from mutant mice were significantly shorter than those from WT (Fig. 6A,B) or heterozygous littermates (data not shown). As listed in Table 2, the average length of MEKK4K1361R cochlear duct was ∼20% shorter than their WT littermates. In addition, the width of the sensory epithelium, as measured along the mediolateral axis, was considerably narrower in mutant cochleae compared with WT controls (Table 2). Similarly, a modest but noticeable reduction in the average diameter of IHCs and OHCs was also observed in MEKK4 mutants compared with that in controls. A previous study has suggested that cochlear outgrowth, which is dependent on extension of the cochlear duct, is reliant on normal size and morphology of individual cells, specifically inner pillar cells within the developing sensory epithelium (Yamamoto et al., 2009). Based on these data, it could be suggested that disruption of normal elongated morphology of inner pillar cells in the absence of MEKK4 function may lead to the inhibition of cochlear duct extension.
Figure 6.
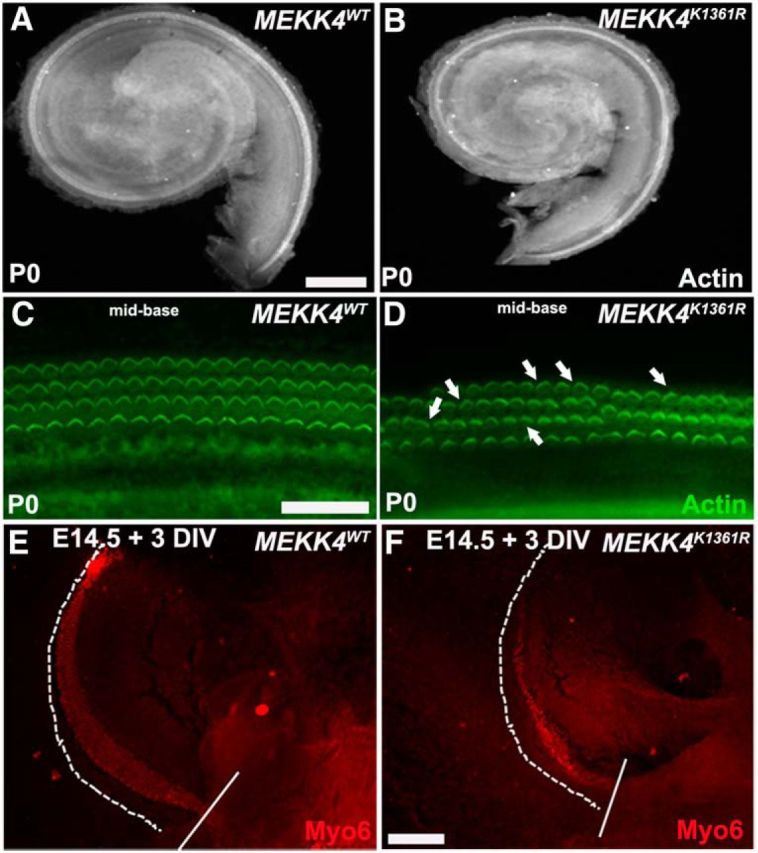
MEKK4 signaling is required for normal morphogenesis of the cochlea. A, B, Cochlear duct from MEKK4WT (A) and MEKK4K1361R mutants (B) immunolabeled with actin. The length and width of the mutant cochlea are considerably less than that in controls. C, D, High-magnification whole-mount surface views of control (C) and mutant (D) cochleae stained with actin (green) show disorganization of HCs with intermittent presence of third row of OHCs. In addition, although stereociliary bundles are intact in mutant cochlear HCs, a slight disorganization of their orientation was seen in mutants (D, arrows). E, F, The control (E) and mutant (F) cochleae were bisected in the mid-basal region (incision site indicated by solid white line), and the basal portion of cochlea from each genotype is explanted at E14.5 for 3 DIV and immunolabeled with Myo6 (red). The sensory epithelium is extended, as indicated by dotted line, in both controls and mutants. In addition, although one row of IHCs and three rows of OHCs are apparent in controls, it is less discernible in mutants and also the width of the extended sensory epithelium seems considerably smaller compared with controls. Scale bars: (in A) A, B, 50 μm; (in C) C, D, 20 μm; (in F) E, F, 20 μm.
Table 2.
Quantification of average length, width of the sensory epithelium, and the lumenal surface area of sensory HCs indicate that cochlear duct is shorter and that both OHCs and IHCs are smaller in MEKK4K1361R mutants compared with that in controls
| Sensory epithelium (SE)/sensory cell | MEKK4WT | MEKK4K1361R |
|---|---|---|
| Average length of SE (μm) | 4048.79 ± 67 | 3222.40 ± 59 |
| Average width of SE (μm) | 20.06 ± 0.02 | 17.16 ± 0.7 |
| Average width of inner hair cells (μm) | 4.78 ± 0.2 | 3.98 ± 0.1 |
| Average width of outer hair cells (μm) | 3.06 ± 0.1 | 2.43 ± 0.1 |
Moreover, it has recently been suggested that convergence extension (CE), which is mediated by planar cell polarity (PCP) process, plays a key role in cochlear patterning via influencing the normal morphology of sensory cells and elongation (Chen et al., 2002; Montcouquiol et al., 2003; McKenzie et al., 2004; Wang et al., 2005, 2006; Qian et al., 2007; Yamamoto et al., 2009). These observations led us to speculate that the MEKK4-deficient epithelium may display PCP and/or CE defects. To assess whether MEKK4 plays any role in regulating the polarity of hair bundles, we compared the organization of hair bundles in MEKK4-deficient epithelium to that in WT littermates. As seen in Figure 6 C and D, a slight disorganization of OHC bundles is observed in MEKK4 mutants. However, it is not clear whether this disruption is a direct effect of loss of MEKK4 activity or a secondary consequence of patterning defects, which in turn can be attributed to the significant reduction of OHCs in MEKK4 kinase-inactive mutants.
To examine whether MEKK4 kinase deficiency causes any CE defects within cochlear epithelium, we performed a cochlear outgrowth assay developed by Wang et al. (2005) at E14.5 on WT and MEKK4K1361R littermates. Although the mutant cochleae had fewer HCs generated from the apical portion of the incision site compared with controls, the average extension of the sensory epithelium was not significantly altered between WT and bisected mutant samples (Fig. 6E,F). In addition, HCs appeared smaller in mutants compared with controls, which was similar to that observed in the P1 mutant cochlea (Table 2). These observations strongly suggest that disruption in the polarity of hair bundles is unlikely to be a direct consequence of MEKK4 deficiency, but rather, an event induced by disruption in cell fate and patterning that may contribute to polarity defects. Together, these results imply that MEKK4 is not involved in cochlear extension but rather influences cellular morphology and that its disruption could contribute to the patterning defects observed within MEKK4-deficient sensory epithelium.
Neuronal cell patterning is disrupted in MEKK4K1361R mutants
It is generally accepted that, although processes from spiral ganglion neurons reach the sensory epithelium independent of HC differentiation, their ultimate pattern of innervation is dependent on normal cellular patterning within the organ of Corti (Yang et al., 2011). Therefore, because sensory HC and nonsensory supporting cell differentiation is disrupted in MEKK4K1361R cochlea, it seemed possible that neuronal cell patterning would be altered. To examine this, the innervation pattern was analyzed by immunolabeling using neurofilament marker, 2H3. Normally, two types of spiral ganglion neurons innervate sensory HCs: whereas fibers from Type I neurons innervate IHCs, fibers from Type II neurons cross the tunnel of Corti to innervate one or more OHCs and form outer spiral bundles before turning toward the base. Immunolabeling for 2H3, which recognizes both Type I and Type II fibers, demonstrated that Type I axon bundles are well organized in WT cochlea (Fig. 7A,B) but are highly disorganized in the MEKK4K1361R mutant epithelium (Fig. 7C,D). In addition, fewer Type II fibers were seen crossing the pillar cells and extending toward OHCs in the mutants, most of which displayed aberrant turning, and some just projected into the lateral space. However, because MEKK4 expression was not observed in spiral ganglion neurons during development (data not shown), the aberrant innervation patterns in the mutant cochleae are unlikely to be due to a direct requirement for MEKK4 signaling but rather are likely secondary to defective cellular patterning within the organ of Corti. Similarly, Fgfr3, although not expressed in spiral ganglion neurons, loss in mice, which causes HC differentiation defects, also shows a similar phenotype wherein the fibers are disorganized and fail to form bundles (Puligilla et al., 2007), consistent with the hypothesis that ultimate pattern of innervation is dependent on normal cellular differentiation. Loss of Prox1 also leads to failure of neuronal fiber patterning (Fritzsch et al., 2010); however, normal distribution pattern of Prox1 was seen in MEKK4K1431R mutant cochleae (data not shown). Previous studies have demonstrated a key role for MEKK4 signaling in neuronal proliferation and migration within developing and adult forebrain (Sarkisian et al., 2006, 2008; Ferland et al., 2009) and that its loss in mice causes neuronal migration defects and subsequent periventricular nodular heterotopia. Although its expression is not evident in embryonic spiral ganglion neurons, it remains to be seen whether MEKK4 is expressed in adult stages.
Figure 7.

Disruption of spiral ganglion neuron patterning in MEKK4K1361R mutants. Immunolabeling for Myo6 (red) and neurofilament 2H3 (green) in the mid-basal region of a WT (A, B) and mutant (C, D) cochlea at P0. In the WT cochlea, most of the afferent Type I fibers fasciculate to form bundles within otic mesenchyme (shown by clear arrows), before terminating on IHCs, whereas Type II afferent fibers, which constitute only 5% of afferent neurons, extend laterally to innervate OHCs and form outer spiral bundles before they turn toward the base (indicated by solid arrow) to form synapses with more than one OHC. C, In the mutants, disorganization of fibers from Type I neurons is quite evident and does not form bundles as indicated by clear arrows. Although most of these fibers terminate at IHC region, a smaller percentage of these fibers crossed the pillar cell region to reach OHCs compared with that in controls. Moreover, the fibers in the OHC region do not seem to turn toward the base (solid arrowheads); and instead, some fibers project laterally into the lesser epithelial ridge (asterisks). Scale bar: (in D) A–D, 20 μm.
Cellular differentiation defects in MEKK4 mutants occur in a JNK-independent manner
Our data indicate that MEKK4 signaling is required for the establishment of correct pattern of cellular differentiation. However, it is not clear how MEKK4 regulates the development of normal cellular phenotypes. It has been previously demonstrated that the JNK pathway is a downstream target of MEKK4 signaling and that MEKK4-mediated signals are transmitted via the JNK pathway to regulate embryonic mouse development (Abell and Johnson, 2005; Abell et al., 2009). Thus, to elucidate the mechanism of MEKK4 regulation in the developing inner ear, we examined whether JNK acts as a target of MEKK4 signaling to influence sensory HC differentiation. To determine whether this pathway is activated in developing sensory epithelial cells and whether the activity of phospho-JNK is affected in MEKK4 kinase-inactive mutants, we harvested protein from cochlear sensory epithelia of control MEKK4WT and homozygous MEKK4K1361R mutant littermates at E16.5 and performed immunoblot-based analysis. The data thus far indicate that, although JNK activity is present in the developing organ of Corti, its levels seem unaffected in MEKK4 signaling-defective mice and are comparable with that in WT controls (Fig. 8A). These results strongly suggest that the cellular differentiation defects observed in MEKK4 mutants are not due to insufficient JNK activity.
Figure 8.
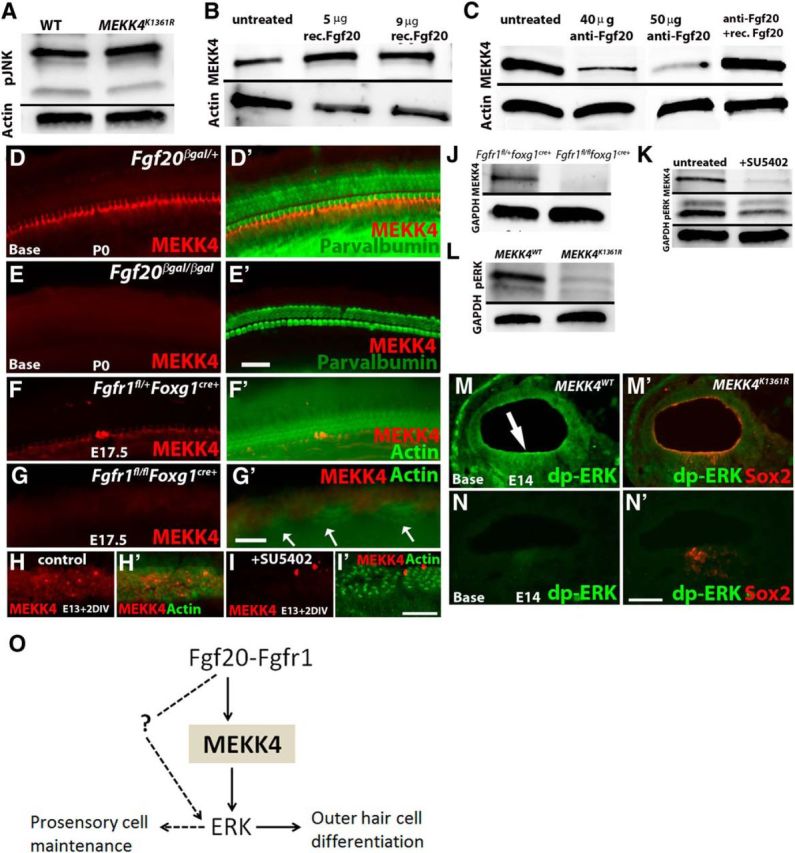
MEKK4 function in the inner ear is independent of JNK activity and that its expression is activated by FGF20-FGFR1 signaling. A, Protein harvested from cochleae at E16.5 and blotted with anti-phospho-JNK show no alteration in its activity between control and MEKK4K1361R mutant samples. Data shown are representative results from three independent experiments. B, C, Immunoblot analysis of proteins harvested from WT and mutant cochlear explants treated with recombinant Fgf20 (B) or function-blocking antibody (C) at E13 show stimulation (B) or inhibition (C) of MEKK4 expression, respectively. The inhibition of MEKK4 expression in cochlear explants cultured in the presence of anti-Fgf20 was rescued by the addition of recombinant Fgf20 protein for 2DIV (C). D–E′, Immunolabeling of Fgf20βgal/+ heterozygote control (D, D′) and Fgf20βgal/βgal homozygous mutant (E, E′) inner ears at P0 using anti-MEKK4 (red) and anti-parvalbumin (green) demonstrates normal expression of MEKK4 in the cochlear duct; however, its expression is downregulated in Fgf20βgal/βgal cochleae. F–G′, Immunolabeling of Fgfr1fl/+foxg1cre+ (F, F′) and Fgfr1fl/flfoxg1cre+ (G, G′) cochleae at E17.5 using anti-MEKK4 (red) and actin (green) shows normal distribution of MEKK4 expression in controls, whereas mutant cochlea display patchy HCs (G′, arrows) and a complete absence of MEKK4 expression. H–I′, Control DMSO-treated (H, H′) and SU5402-treated (I, I′) cochlear cultures established at E13 and after 2 DIV immunolabeled using anti-MEKK4 (red) and actin (green) show normal expression of MEKK4 throughout the sensory epithelial cells in control cochleae, although its expression is completely lost in treated cochleae. J, K, Immunoblot-based assays show complete absence of MEKK4 expression within Fgfr1fl/flfoxg1cre+ mutant cochlea at E13 (J). In addition, E13 cochlear cultures treated with SU5402 for 12 h show rapid downregulation of MEKK4 and pERK levels (K). L–N′, Phospho-ERK levels (green) were also significantly downregulated in MEKK4K1361R compared with controls in the developing cochlear duct as shown by immunoblot (L) and immunolabeling (M–N′) (arrow in M indicates sensory patch) assays at E13 and E14 stages, respectively. O, Summary of the molecular signaling modules that control sensory cell differentiation during inner ear morphogenesis. FGF20 signals are transduced through FGFRs, most likely FGFR1, to MEKK4 and ERK to regulate differentiation of OHCs, whereas FGF signaling activation via yet unknown MAPK molecule, possibly another MEKK, controls cochlear progenitor cell population. Solid arrows indicate direct interaction between molecular components. Dotted line and arrows indicate hypothetical direct regulation. Scale bars: (in E′) D–E′, 20 μm; (in G′) F–G′, 20 μm; (in I′) H–I′, 20 μm; (in N′) M–N′, 10 μm.
FGF20 signaling via FGFR1 regulates expression of MEKK4 in the developing sensory epithelium
Previous studies have shown that Fgf20 is broadly expressed in the developing cochlear duct as early as E13 and by P0 its expression becomes restricted to inner phalangeal cells (Huh et al., 2012). In addition, lack of Fgf20-mediated receptor tyrosine kinase signaling in vivo and ex vivo leads to defects in the differentiation of OHCs and Deiters' cells (Hayashi et al., 2008; Huh et al., 2012). Based on these results, we hypothesized that the FGF20-mediated signaling pathway may act upstream to regulate MEKK4 expression in the developing sensory epithelium. To test this, we analyzed whether gain of Fgf20 function could stabilize MEKK4 expression. To analyze this, we performed overexpression analysis by treating embryonic cochlear explants at E13 with recombinant Fgf20 protein and analyzing for MEKK4 expression after 1 DIV by immunoblotting and immunohistochemistry (data not shown). As shown in Figure 8B, MEKK4 expression is upregulated in Fgf20-treated cochleae, compared with normal expression levels in untreated controls. To test the inverse, whether loss of Fgf20 function could disrupt MEKK4 expression, embryonic cochlear explants were established at E13 and exposed with function-blocking antibody against Fgf20, which has been shown to disrupt Fgf20 function (Hayashi et al., 2008). After 1 DIV, cultures were subjected to immunoblot assays. While untreated control cochlea show normal levels of MEKK4 expression, anti-FGF20 treated cochlea show dose-dependent inhibition of MEKK4 expression. Moreover, to test whether MEKK4 expression could be rescued in Fgf20-inhibited cochleae, recombinant Fgf20 protein was added to the explants after 1 DIV and incubated for 1 additional DIV. Immunoblotting using anti-MEKK4 demonstrates the rescue of MEKK4 expression to levels comparable with untreated cochleae (Fig. 8C), suggesting that FGF20 stabilizes MEKK4 expression in the developing cochlea.
To confirm the above results in vivo, we analyzed heterozygous Fgf20βgal/+ (controls) and homozygous mutant Fgf20βgal/βgal inner ears by immunolabeling for MEKK4. Our data indicate that MEKK4 was strongly expressed in controls, whereas Fgf20βgal/βgal mutant cochlea was completely devoid of expression (Fig. 8D,E′). These data indicate that FGF20 regulates MEKK4 expression in vivo and influences differentiation of sensory epithelial cells within the developing inner ear.
Previous studies have demonstrated that Fgf20 binds to and activates Fgf receptor (Fgfr) 1 during the early stages of cochlear development, including prosensory specification and differentiation of HCs (Hayashi et al., 2008; Huh et al., 2012; Ono et al., 2014). Conditional deletion of Fgfr1 in the developing otocyst leads to severe reduction of HCs and support cells (Pirvola et al., 2002). Similarly, treatment of cochlear cultures with Fgf receptor-specific inhibitor, SU5402 (Mohammadi et al., 1997) at early stages of development also causes a significant reduction of HC and supporting cell number (Hayashi et al., 2008), demonstrating the necessity of Fgfr1 activity in generation of correct pattern of HCs and supporting cells. However, the underlying mechanisms by which FGFR1 signaling regulate sensory cell differentiation in the mammalian cochlea remain unknown. To examine whether MEKK4 is required for FGFR1 to promote HC differentiation, we generated conditional Fgfr1 mutant mice using Foxg1cre driver and analyzed for the expression of MEKK4. Our analysis indicates that the expression levels of MEKK4 in Fgfr1fl/flfoxg1cre+ mutant cochlea are significantly downregulated, whereas control Fgfr1fl/+foxg1cre+ mice displayed a normal pattern of MEKK4 expression (Fig. 8F–G′,J). To confirm that the observed downregulation of MEKK4 in Fgfr1 mutant mice in vivo is specific to the loss of Fgfr1 activity and not a secondary consequence due to loss of protein occurring at a more global level, we used pharmacological inhibition of Fgfr1 ex vivo using SU5402 in cochlear cultures as early as E13 (Fig. 8H–I′), E14 or E15 (data not shown) stages. Consistent with a previous study (Hayashi et al., 2008), treatment with SU5402 for 2 DIV led to a significant reduction of HCs, specifically OHCs at all three stages (data not shown). In addition, we observed a significant downregulation of MEKK4 expression levels in treated cochlea, whereas the expression is intact in DMSO-treated controls. Moreover, our immunoblot assays indicate a complete absence of MEKK4 expression in Fgfr1fl/flfoxg1cre+ mutant cochleae as early as E13 (Fig. 8J) and a rapid downregulation in SU5402-treated cultures (Fig. 8K), demonstrating that MEKK4 expression is dependent on active FGFR1 signaling and that FGFR1 signaling through activation of MEKK4 is necessary for driving OHC differentiation.
To determine the mechanisms that dictate the signaling specificity of the MEKK4 cascade by FGF20-FGFR1 signaling and regulate its downstream activity, we investigated ERK activity in MEKK4 kinase-defective inner ears. Diphosphorylated ERK has been shown to be expressed as early as E10.5 in the ventral half of the developing mouse otic vesicle (Ono et al., 2014). Our data using immunoblot and immunolabeling-based assays showed that phospho-ERK1/2 activity is significantly diminished in MEKK4K1361R inner ears at E13 (Fig. 8L) and E14 (Fig. 8M–N′) stages, respectively. In contrast, we detected the expression of phosphorylated ERK in the developing cochlear duct of controls. Moreover, the levels of pERK are also reduced in SU5402-treated cochleae (Fig. 8K). On the other hand, phospho-AKT levels were unaltered in mutants (data not shown). Thus, inhibition of MEKK4 activity in mutant inner ears specifically affects ERK phosphorylation in the developing cochlea, and MEKK4-ERK signaling possibly functions to influence cellular differentiation.
Loss of MEKK4 kinase activity in vivo leads to congenital hearing loss
The aberrant cellular differentiation and patterning observed in MEKK4-deficient cochleae strongly suggested that MEKK4 may be required for normal auditory function. To determine this, ABR measurements were conducted at postnatal day 25 in WT, heterozygous MEKK4K1361R/WT, and homozygous MEKK4K1361R mutant littermate mice. Auditory thresholds differed significantly between the three groups at 8 (F(2,6) = 13.66, p = 0.0058), 16 (F(2,6) = 21.12, p < 0.0019), and 32 kHz (F(2,6) = 9.103, p = 0.0152). Further, Tukey's Multiple Comparison test confirmed a significant difference in auditory thresholds between WT littermates and MEKK4K1361R mutant mice at all measured three frequencies, but there were no differences between WT and heterozygous mice. The mutant mice showed an average 20 dB elevation of auditory thresholds at both 8 (p = 0.018) and 16 kHz (p = 0.004), and an average 40 dB at 32 kHz (p = 0.04) compared with WT littermates. The auditory thresholds of WT littermates remained within normal hearing ranges (Fig. 9). These data indicate a role for MEKK4 in the development of normal ABR responses in mice and that disruption in sensory and/or neuronal patterning could contribute to the overall level of auditory dysfunction in the absence of MEKK4 signaling. Future studies would be required to distinguish whether the hearing phenotype originated at the level of the auditory nerve or within the cochlear sensory epithelium.
Figure 9.

Loss of MEKK4 signaling causes elevations in auditory threshold responses ABR thresholds obtained from WT (blue), heterozygous MEKK4K1361/+ (green), and MEKK4K1361R (red) mutant mice. Thresholds were significantly elevated in MEKK4K1361R mutant mice compared with WT mice at all three tested frequencies. Data are mean ± SEM; n = 3. *p < 0.05. **p < 0.01.
Discussion
Previous reports have demonstrated a role for cochlear MAPK signaling in protection from noise-induced or aminoglycoside-induced ototoxic damage (Zine and van de Water, 2004; Dinh et al., 2015). The MAPK pathway also has been shown to be involved in chick otic placode induction (Yang et al., 2013). However, the role of specific MAPK signaling components, including MAP3Ks in inner ear development, remains unknown. The data presented here demonstrate previously unreported roles for MEKK4 signaling in the development and function of the mouse inner ear. We found that inhibition of MEKK4 kinase activity leads to disruption of normal cochlear morphogenesis. In particular, MEKK4-kinase defective mutants display a shortened cochlea and smaller sensory HCs compared with WT controls. It is well established that disruption in PCP pathway leads to neural tube defects (Wallingford et al., 2000; Wallingford, 2006; Nishimura et al., 2012); and given that MEKK4K1361R mutants display neural tube closure defects, these data point to a potential disruption of the PCP pathway in the MEKK4K1361R cochlea. Both CE and growth were shown to play a significant role in cochlear patterning (Chen and Segil, 1999; Cantos et al., 2000; McKenzie et al., 2004; Yamamoto et al., 2009). Moreover, a recent study by Huh et al. (2015) suggested that the overall length of the cochlear duct is dependent on normal sensory cell differentiation. Therefore, it is possible that the defects in HC differentiation and their organization observed in MEKK4 mutants may result in shorter cochlear duct. On the other hand, although all HCs die in Atoh1 mutants, the elongation of the cochlear duct is normal (Fritzsch et al., 2005). The cochlear extension also seems intact in the absence of progenitor cells in Sox2 mutants (Kiernan et al., 2005). Because the normal morphology of inner pillar cells is crucial for cochlear extension (Yamamoto et al., 2009), it is also possible that loss of stereotypical morphology of pillar cells in MEKK4 mutants may lead to disruption in cochlear extension.
We show here, for the first time, that MEKK4 kinase activity is required for normal sensory cell development in the mouse inner ear. MEKK4-kinase defective mutants display a significant reduction of sensory HCs within the developing utricle and organ of Corti. In addition, we found aberrant differentiation of pillar cells and Deiters' cells in the absence of MEKK4 signaling. Based on the expression pattern, together with cellular differentiation defects in MEKK4 kinase-deficient epithelium, it could be suggested that initial broad expression pattern of MEKK4 is required for normal HC and supporting cell fate specification and that the subsequent downregulation of MEKK4 in cells within the OHC domain may be essential to their normal differentiation. Also, MEKK4 is not required for induction or maintenance of prosensory cells during sensory patch formation. Together, these data suggest us that MEKK4 signaling regulates HC but not prosensory cell development.
Furthermore, levels of Atoh1 transcripts within precursor cells in MEKK4 mutants were found to be significantly lower compared with WT controls, suggesting that although MEKK4 is dispensable for prosensory induction and/or maintenance, it may act as a critical component of the molecular machinery that positively regulates Atoh1 levels within HC precursors during differentiation. The reduction in the Atoh1 transcript levels by nearly 70% may explain the decreased OHC number in the homozygous MEKK4 mutants, consistent with Pan et al. (2012) data showing a direct correlation of Atoh1 expression levels and HC development. Also, the presence of residual levels of Atoh1 expression in MEKK4 mutants suggests that MEKK4 signaling-independent mechanisms may compensate for the loss of MEKK4 activity. It also is likely that MEKK4 kinase activity may act as a permissive factor to regulate Atoh1 expression levels and subsequent differentiation of HCs. In addition, although these possibilities cannot be completely discounted, it remains to be seen whether MEKK4 influences Atoh1 post-translationally via phosphorylation to regulate its expression levels during development.
Recent findings have shown that signaling mediated by the Fgf20 and/or Fgfr1 pathways influences Atoh1-mediated HC development (Pirvola et al., 2002; Hayashi et al., 2008; Huh et al., 2012; Ono et al., 2014). These studies have also suggested that Fgf20 acts as a ligand to mediate Fgfr1 signaling during cellular fate specification and differentiation in the developing organ of Corti. However, it is not clear how signals from the Fgf20-Fgfr1 pathway are integrated to influence Atoh1 expression and subsequent HC differentiation. In particular, Fgf20, similar to MEKK4, is highly expressed throughout the sensory epithelium as early as E13, but by P0 its expression is restricted to inner phalangeal cells within the medial domain of the sensory epithelium. Genetic deletion of FGF20 leads to aberrant differentiation of sensory cells within the lateral compartment, which includes OHCs and Deiters' cells (Huh et al., 2012). Remarkably, the phenotype of FGF20 knock-out mice is strikingly similar to MEKK4 mutant mice in most aspects of differentiation defects within the inner ear. However, the alterations in HC patterning in MEKK4 mutants in vivo are less severe when FGF20 is deleted. Nonetheless, based on these observations, it is plausible that the two factors share a common signaling pathway during embryonic inner ear development. Consistent with this hypothesis, our loss- and gain-of-Fgf20 function analyses using in vivo and ex vivo approaches demonstrate that Fgf20 acts to regulate MEKK4 expression in the developing sensory epithelial cells. This conclusion is further supported by the demonstration of a similar expression pattern of MEKK4 and FGF20 in the developing cochlea. Together, these data indicate that Fgf20 controls the spatial and temporal pattern of MEKK4 expression in the developing cochlea and that the Fgf20-MEKK4 axis acts to influence specification and subsequent differentiation of OHCs and Deiters' cells.
Similarly, FGFR1, which acts as a likely receptor for FGF20-mediated signaling (Hayashi et al., 2008, 2010; Huh et al., 2012), has also been shown to be important for the development of cochlear HCs and supporting cells (Pirvola et al., 2002; Huh et al., 2012; Ono et al., 2014). In addition, recent studies demonstrate that FGF20-FGFR1 signaling also regulates the total number of prosensory progenitor cells (Ono et al., 2014; Huh et al., 2015). These studies, together, imply a dual role for Fgf signaling: initially, it is required for the maintenance of cochlear progenitor cells; and subsequently, it promotes sensory HC differentiation. However, the molecular mechanism by which Fgf signaling controls these processes during sensory specification is unclear. Previous studies have suggested that Fgf signaling may act via MAPK or mesenchymal FGFR2 or its downstream transcriptional targets Pea3 and Erm to influence cochlear progenitor cell population or sensory cell differentiation, respectively (Hayashi et al., 2008; Ono et al., 2014; Huh et al., 2015). Our data demonstrate that MEKK4 is a key kinase that acts as a “hub” in integrating FGF20-FGFR1-mediated signaling responses in inducing OHC differentiation and that MEKK4 expression dictates specific response of Fgfr1 signaling during HC development. These data also suggest that Fgf signaling is activated via yet unidentified distinct molecule, probably another MEKK, to regulate the total number of cochlear progenitor cells.
Interestingly, recent studies have revealed that MEKK4 activity stimulates the JNK pathway (Abell et al., 2007, 2009) in trophoblast stem cells during placental development, thus maintaining trophoblast stem cells in an undifferentiated state. In the inner ear, JNK activity is reported to be involved in spiral ganglion neurite outgrowth; however, its role in the sensory cell development, if any, is not known (Bodmer et al., 2002; Atkinson et al., 2011). Our data here demonstrate the presence of active JNK in the developing organ of Corti. However, loss of MEKK4 function does not affect the levels of JNK activity within the organ of Corti, suggesting that the cellular phenotypes observed in MEKK4-deficient inner ears are not due to insufficient active JNK. On the other hand, we found that phosphorylated ERK levels are repressed in inner ears of MEKK4 signaling-defective mutants, supporting a novel JNK-independent role for MEKK4 during development. The data thus suggest that MEKK4-ERK signaling mediates inner ear development and that FGFR1 acts upstream to regulate MEKK4-ERK signaling. In agreement with this hypothesis, studies have demonstrated that phospho-ERK is activated by FGF and participates in the regulation of proliferation, differentiation, survival, and apoptosis within CNS, among other systems (Sato et al., 2004; Gotoh, 2009). The effects of inhibition of MEKK4-ERK signaling on OHC development can be explained by Atoh1 downregulation observed in MEKK4 mutants. Given that downregulation of Atoh1 levels is seen in Fgf20-inhibited and MEKK4K1361R cochleae, it is likely that FGF signaling via MEKK4-ERK might disrupt the Atoh1 feedback loop through post-translational modifications. Also, phospho-ERK activity is downregulated in Fgfr1 mutants within the developing otocyst, suggesting that FGFR1 signaling is mediated via ERK to regulate prosensory development (Ono et al., 2014). Together, these data indicate that the FGF20, possibly by binding to FGFR1, signals via MEKK4 to activate ERK activity to influence HC differentiation. This, in turn, suggests that FGF20-FGFR1 signaling is dependent on MEKK4 expression levels to regulate HC development and differentiation and that FGF20-FGFR1 is activated via MEKK4-ERK to regulate inner ear HC development. We summarize these signaling interactions in Figure 8O.
Footnotes
This work was supported by Grant C06 RR014516 (NIH) and conducted in the renovated space of the Walton Research Building at Medical University of South Carolina. We thank Drs. David Ornitz and Sung-Ho Huh (Washington University, St. Louis, Missouri) for providing us with Fgf20 mutant samples; Drs. Doris Wu (National Institute on Deafness and Other Communication Disorders, National Institutes of Health), Bradley Schulte (Medical University of South Carolina, Charleston, South Carolina), and Alain Dabdoub (Sunnybrook Research Institute, Toronto, Ontario, Canada) for critical reading of the manuscript; Dr. Sandeep Kumar for technical assistance; and members of the Medical University of South Carolina Hearing Research Program for comments throughout this study.
The authors declare no competing financial interests.
References
- Abell AN, Johnson GL. MEKK4 is an effector of the embryonic TRAF4 for JNK activation. J Biol Chem. 2005;280:35793–35796. doi: 10.1074/jbc.C500260200. [DOI] [PubMed] [Google Scholar]
- Abell AN, Rivera-Perez JA, Cuevas BD, Uhlik MT, Sather S, Johnson NL, Minton SK, Lauder JM, Winter-Vann AM, Nakamura K, Magnuson T, Vaillancourt RR, Heasley LE, Johnson GL. Ablation of MEKK4 kinase activity causes neurulation and skeletal patterning defects in the mouse embryo. Mol Cell Biol. 2005;25:8948–8959. doi: 10.1128/MCB.25.20.8948-8959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abell AN, Granger DA, Johnson GL. MEKK4 stimulation of p38 and JNK activity is negatively regulated by GSK3beta. J Biol Chem. 2007;282:30476–30484. doi: 10.1074/jbc.M705783200. [DOI] [PubMed] [Google Scholar]
- Abell AN, Granger DA, Johnson NL, Vincent-Jordan N, Dibble CF, Johnson GL. Trophoblast stem cell maintenance by fibroblast growth factor 4 requires MEKK4 activation of Jun N-terminal kinase. Mol Cell Biol. 2009;29:2748–2761. doi: 10.1128/MCB.01391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson PJ, Cho CH, Hansen MR, Green SH. Activity of all JNK isoforms contributes to neurite growth in spiral ganglion neurons. Hear Res. 2011;278:77–85. doi: 10.1016/j.heares.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- Bodmer D, Gloddek B, Ryan AF, Huverstuhl J, Brors D. Inhibition of the c-Jun N-terminal kinase signaling pathway influences neurite outgrowth of spiral ganglion neurons in vitro. Laryngoscope. 2002;112:2057–2061. doi: 10.1097/00005537-200211000-00028. [DOI] [PubMed] [Google Scholar]
- Buckiová D, Syka J. Calbindin and S100 protein expression in the developing inner ear in mice. J Comp Neurol. 2009;513:469–482. doi: 10.1002/cne.21967. [DOI] [PubMed] [Google Scholar]
- Cantos R, Cole LK, Acampora D, Simeone A, Wu DK. Patterning of the mammalian cochlea. Proc Natl Acad Sci U S A. 2000;97:11707–11713. doi: 10.1073/pnas.97.22.11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Segil N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development. 1999;126:1581–1590. doi: 10.1242/dev.126.8.1581. [DOI] [PubMed] [Google Scholar]
- Chen P, Johnson JE, Zoghbi HY, Segil N. The role of Math1 in inner ear development: uncoupling the establishment of the sensory primordium from hair cell fate determination. Development. 2002;129:2495–2505. doi: 10.1242/dev.129.10.2495. [DOI] [PubMed] [Google Scholar]
- Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- Chi H, Sarkisian MR, Rakic P, Flavell RA. Loss of mitogen-activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proc Natl Acad Sci U S A. 2005;102:3846–3851. doi: 10.1073/pnas.0500026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig EA, Stevens MV, Vaillancourt RR, Camenisch TD. MAP3Ks as central regulators of cell fate during development. Dev Dyn. 2008;237:3102–3114. doi: 10.1002/dvdy.21750. [DOI] [PubMed] [Google Scholar]
- Curran KL, Grainger RM. Expression of activated MAP kinase in Xenopus laevis embryos: evaluating the roles of FGF and other signaling pathways in early induction and patterning. Dev Biol. 2000;228:41–56. doi: 10.1006/dbio.2000.9917. [DOI] [PubMed] [Google Scholar]
- Dabdoub A, Puligilla C, Jones JM, Fritzsch B, Cheah KS, Pevny LH, Kelley MW. Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc Natl Acad Sci U S A. 2008;105:18396–18401. doi: 10.1073/pnas.0808175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh CT, Goncalves S, Bas E, Van De Water TR, Zine A. Molecular regulation of auditory hair cell death and approaches to protect sensory receptor cells and/or stimulate repair following acoustic trauma. Front Cell Neurosci. 2015;9:96. doi: 10.3389/fncel.2015.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetzlhofer A, Basch ML, Ohyama T, Gessler M, Groves AK, Segil N. Hey2 regulation by FGF provides a Notch-independent mechanism for maintaining pillar cell fate in the organ of Corti. Dev Cell. 2009;16:58–69. doi: 10.1016/j.devcel.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driver EC, Sillers L, Coate TM, Rose MF, Kelley MW. The Atoh1-lineage gives rise to hair cells and supporting cells within the mammalian cochlea. Dev Biol. 2013;376:86–98. doi: 10.1016/j.ydbio.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland RJ, Batiz LF, Neal J, Lian G, Bundock E, Lu J, Hsiao YC, Diamond R, Mei D, Banham AH, Brown PJ, Vanderburg CR, Joseph J, Hecht JL, Folkerth R, Guerrini R, Walsh CA, Rodriguez EM, Sheen VL. Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum Mol Genet. 2009;18:497–516. doi: 10.1093/hmg/ddn377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Pauley S, Matei V, Katz DM, Xiang M, Tessarollo L. Mutant mice reveal the molecular and cellular basis for specific sensory connections to inner ear epithelia and primary nuclei of the brain. Hear Res. 2005;206:52–63. doi: 10.1016/j.heares.2004.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Dillard M, Lavado A, Harvey NL, Jahan I. Canal cristae growth and fiber extension to the outer hair cells of the mouse ear require Prox1 activity. PLoS One. 2010;5:e9377. doi: 10.1371/journal.pone.0009377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/S0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Gotoh N. Control of stemness by fibroblast growth factor signaling in stem cells and cancer stem cells. Curr Stem Cell Res Ther. 2009;4:9–15. doi: 10.2174/157488809787169048. [DOI] [PubMed] [Google Scholar]
- Groves AK, Fekete DM. Shaping sound in space: the regulation of inner ear patterning. Development. 2012;139:245–257. doi: 10.1242/dev.067074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque KD, Pandey AK, Kelley MW, Puligilla C. Culture of embryonic mouse cochlear explants and gene transfer by electroporation. J Vis Exp. 2015;95:52260. doi: 10.3791/52260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Cunningham D, Bermingham-McDonogh O. Loss of Fgfr3 leads to excess hair cell development in the mouse organ of Corti. Dev Dyn. 2007;236:525–533. doi: 10.1002/dvdy.21026. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Ray CA, Bermingham-McDonogh O. Fgf20 is required for sensory epithelial specification in the developing cochlea. J Neurosci. 2008;28:5991–5999. doi: 10.1523/JNEUROSCI.1690-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Ray CA, Younkins C, Bermingham-McDonogh O. Expression patterns of FGF receptors in the developing mammalian cochlea. Dev Dyn. 2010;239:1019–1026. doi: 10.1002/dvdy.22236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh SH, Jones J, Warchol ME, Ornitz DM. Differentiation of the lateral compartment of the cochlea requires a temporally restricted FGF20 signal. PLoS Biol. 2012;10:e1001231. doi: 10.1371/journal.pbio.1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh SH, Warchol ME, Ornitz DM. Cochlear progenitor number is controlled through mesenchymal FGF receptor signaling. Elife. 2015:4. doi: 10.7554/eLife.05921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume CR, Bratt DL, Oesterle EC. Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr Patterns. 2007;7:798–807. doi: 10.1016/j.modgep.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques BE, Montcouquiol ME, Layman EM, Lewandoski M, Kelley MW. Fgf8 induces pillar cell fate and regulates cellular patterning in the mammalian cochlea. Development. 2007;134:3021–3029. doi: 10.1242/dev.02874. [DOI] [PubMed] [Google Scholar]
- Jahan I, Pan N, Fritzsch B. Opportunities and limits of the one gene approach: the ability of Atoh1 to differentiate and maintain hair cells depends on the molecular context. Front Cell Neurosci. 2015;9:26. doi: 10.3389/fncel.2015.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- LaBonne C, Whitman M. Localization of MAP kinase activity in early Xenopus embryos: implications for endogenous FGF signaling. Dev Biol. 1997;183:9–20. doi: 10.1006/dbio.1996.8497. [DOI] [PubMed] [Google Scholar]
- Lee YS, Liu F, Segil N. A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development. 2006;133:2817–2826. doi: 10.1242/dev.02453. [DOI] [PubMed] [Google Scholar]
- McKenzie E, Krupin A, Kelley MW. Cellular growth and rearrangement during the development of the mammalian organ of Corti. Dev Dyn. 2004;229:802–812. doi: 10.1002/dvdy.10500. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature. 2003;423:173–177. doi: 10.1038/nature01618. [DOI] [PubMed] [Google Scholar]
- Mueller KL, Jacques BE, Kelley MW. Fibroblast growth factor signaling regulates pillar cell development in the organ of Corti. J Neurosci. 2002;22:9368–9377. doi: 10.1523/JNEUROSCI.22-21-09368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Honda H, Takeichi M. Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell. 2012;149:1084–1097. doi: 10.1016/j.cell.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Ono K, Kita T, Sato S, O'Neill P, Mak SS, Paschaki M, Ito M, Gotoh N, Kawakami K, Sasai Y, Ladher RK. FGFR1-Frs2/3 signalling maintains sensory progenitors during inner ear hair cell formation. PLoS Genet. 2014;10:e1004118. doi: 10.1371/journal.pgen.1004118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan N, Jahan I, Kersigo J, Duncan JS, Kopecky B, Fritzsch B. A novel Atoh1 “self-terminating” mouse model reveals the necessity of proper Atoh1 level and duration for hair cell differentiation and viability. PLoS One. 2012;7:e30358. doi: 10.1371/journal.pone.0030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauley S, Wright TJ, Pirvola U, Ornitz D, Beisel K, Fritzsch B. Expression and function of FGF10 in mammalian inner ear development. Dev Dyn. 2003;227:203–215. doi: 10.1002/dvdy.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Spencer-Dene B, Xing-Qun L, Kettunen P, Thesleff I, Fritzsch B, Dickson C, Ylikoski J. FGF/FGFR-2(IIIb) signaling is essential for inner ear morphogenesis. J Neurosci. 2000;20:6125–6134. doi: 10.1523/JNEUROSCI.20-16-06125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hébert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/S0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Puligilla C, Feng F, Ishikawa K, Bertuzzi S, Dabdoub A, Griffith AJ, Fritzsch B, Kelley MW. Disruption of fibroblast growth factor receptor 3 signaling results in defects in cellular differentiation, neuronal patterning, and hearing impairment. Dev Dyn. 2007;236:1905–1917. doi: 10.1002/dvdy.21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian D, Jones C, Rzadzinska A, Mark S, Zhang X, Steel KP, Dai X, Chen P. Wnt5a functions in planar cell polarity regulation in mice. Dev Biol. 2007;306:121–133. doi: 10.1016/j.ydbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sai X, Ladher RK. Early steps in inner ear development: induction and morphogenesis of the otic placode. Front Pharmacol. 2015;6:19. doi: 10.3389/fphar.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, Flavell RA, Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801. doi: 10.1016/j.neuron.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Rakic P. Trouble making the first move: interpreting arrested neuronal migration in the cerebral cortex. Trends Neurosci. 2008;31:54–61. doi: 10.1016/j.tins.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Sato T, Joyner AL, Nakamura H. How does Fgf signaling from the isthmic organizer induce midbrain and cerebellum development? Dev Growth Differ. 2004;46:487–494. doi: 10.1111/j.1440-169x.2004.00769.x. [DOI] [PubMed] [Google Scholar]
- Schimmang T. Expression and functions of FGF ligands during early otic development. Int J Dev Biol. 2007;51:473–481. doi: 10.1387/ijdb.072334ts. [DOI] [PubMed] [Google Scholar]
- Schlesinger TK, Fanger GR, Yujiri T, Johnson GL. The TAO of MEKK. Front Biosci. 1998;3:D1181–D1186. doi: 10.2741/a354. [DOI] [PubMed] [Google Scholar]
- Shim K, Minowada G, Coling DE, Martin GR. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev Cell. 2005;8:553–564. doi: 10.1016/j.devcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Tateya T, Imayoshi I, Tateya I, Ito J, Kageyama R. Cooperative functions of Hes/Hey genes in auditory hair cell and supporting cell development. Dev Biol. 2011;352:329–340. doi: 10.1016/j.ydbio.2011.01.038. [DOI] [PubMed] [Google Scholar]
- Urness LD, Wang X, Shibata S, Ohyama T, Mansour SL. Fgf10 is required for specification of non-sensory regions of the cochlear epithelium. Dev Biol. 2015;400:59–71. doi: 10.1016/j.ydbio.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB. Planar cell polarity, ciliogenesis and neural tube defects. Hum Mol Genet. 2006;15:R227–R234. doi: 10.1093/hmg/ddl216. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Rowning BA, Vogeli KM, Rothbächer U, Fraser SE, Harland RM. Dishevelled controls cell polarity during Xenopus gastrulation. Nature. 2000;405:81–85. doi: 10.1038/35011077. [DOI] [PubMed] [Google Scholar]
- Wang J, Mark S, Zhang X, Qian D, Yoo SJ, Radde-Gallwitz K, Zhang Y, Lin X, Collazo A, Wynshaw-Boris A, Chen P. Regulation of polarized extension and planar cell polarity in the cochlea by the vertebrate PCP pathway. Nat Genet. 2005;37:980–985. doi: 10.1038/ng1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hamblet NS, Mark S, Dickinson ME, Brinkman BC, Segil N, Fraser SE, Chen P, Wallingford JB, Wynshaw-Boris A. Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development. 2006;133:1767–1778. doi: 10.1242/dev.02347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wu Y, Zhao F, Dong W, Zhao J, Zhu Z, Liu D. Fgf-signaling-dependent Sox9a and Atoh1a regulate otic neural development in zebrafish. J Neurosci. 2015;35:234–244. doi: 10.1523/JNEUROSCI.3353-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright TJ, Mansour SL. Fgf3 and Fgf10 are required for mouse otic placode induction. Development. 2003;130:3379–3390. doi: 10.1242/dev.00555. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Okano T, Ma X, Adelstein RS, Kelley MW. Myosin II regulates extension, growth and patterning in the mammalian cochlear duct. Development. 2009;136:1977–1986. doi: 10.1242/dev.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xie X, Deng M, Chen X, Gan L. Generation and characterization of Atoh1-Cre knock-in mouse line. Genesis. 2010;48:407–413. doi: 10.1002/dvg.20633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, O'Neill P, Martin K, Maass JC, Vassilev V, Ladher R, Groves AK. Analysis of FGF-dependent and FGF-independent pathways in otic placode induction. PLoS One. 2013;8:e55011. doi: 10.1371/journal.pone.0055011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Kersigo J, Jahan I, Pan N, Fritzsch B. The molecular basis of making spiral ganglion neurons and connecting them to hair cells of the organ of Corti. Hear Res. 2011;278:21–33. doi: 10.1016/j.heares.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wright KD, Mahoney Rogers AA, Barrett MM, Shim K. Compensatory regulation of the size of the inner ear in response to excess induction of otic progenitors by fibroblast growth factor signaling. Dev Dyn. 2014;243:1317–1327. doi: 10.1002/dvdy.24148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JL, Gao WQ. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci. 2000;3:580–586. doi: 10.1038/75753. [DOI] [PubMed] [Google Scholar]
- Zine A, van de Water TR. The MAPK/JNK signalling pathway offers potential therapeutic targets for the prevention of acquired deafness. Curr Drug Targets CNS Neurol Disord. 2004;3:325–332. doi: 10.2174/1568007043337166. [DOI] [PubMed] [Google Scholar]