

Abstract
Aging is associated with cognitive decline and increasing risk of neurodegeneration. Perturbation of mitochondrial function, dynamics, and trafficking are implicated in the pathogenesis of several age-associated neurodegenerative diseases. Despite this fundamental importance, the critical understanding of how organismal aging affects lifetime neuronal mitochondrial maintenance remains unknown, particularly in a physiologically relevant context. To address this issue, we performed a comprehensive in vivo analysis of age-associated changes in mitochondrial morphology, density, trafficking, and stress resistance in individual Caenorhabditis elegans neurons throughout adult life. Adult neurons display three distinct stages of increase, maintenance, and decrease in mitochondrial size and density during adulthood. Mitochondrial trafficking in the distal neuronal processes declines progressively with age starting from early adulthood. In contrast, long-lived daf-2 mutants exhibit delayed age-associated changes in mitochondrial morphology, constant mitochondrial density, and maintained trafficking rates during adulthood. Reduced mitochondrial load at late adulthood correlates with decreased mitochondrial resistance to oxidative stress. Revealing aging-associated changes in neuronal mitochondria in vivo is an essential precedent that will allow future elucidation of the mechanistic causes of mitochondrial aging. Thus, our study establishes the critical foundation for the future analysis of cellular pathways and genetic and pharmacological factors regulating mitochondrial maintenance in aging- and disease-relevant conditions.
SIGNIFICANCE STATEMENT Using Caenorhabditis elegans as a model, we address long-standing questions: How does aging affect neuronal mitochondrial morphology, density, trafficking, and oxidative stress resistance? Are these age-related changes amenable to genetic manipulations that slow down the aging process? Our study illustrates that mitochondrial trafficking declines progressively from the first day of adulthood, whereas mitochondrial size, density, and resistance to oxidative stress undergo three distinct stages: increase in early adulthood, maintenance at high levels during mid-adulthood, and decline during late adulthood. Thus, our study characterizes mitochondrial aging profile at the level of a single neuron in its native environment and establishes the critical foundation for the future genetic and pharmacological dissection of factors that influence long-term mitochondrial maintenance in neurons.
Keywords: age-related changes, Caenorhabditis elegans, in vivo microscopy, mitochondrial trafficking, neuronal mitochondria
Introduction
Aging is a key risk factor in the development of neurodegenerative diseases (de Lau et al., 2004; Yankner et al., 2008; Niccoli and Partridge, 2012). Neurons face unique challenges in maintaining energy supply in distal processes over the life span of an individual in the absence of self-renewal by cell division. Local energy demands in axons and dendrites are met by dynamic distribution of mitochondria along these processes through a complex pattern of bidirectional trafficking and stationary docking (Frederick and Shaw, 2007; MacAskill et al., 2010; Sheng, 2014). Perturbation of mitochondrial function and integrity, membrane dynamics, and trafficking are implicated in the pathogenesis of several major age-associated neurodegenerative diseases (Chen et al., 2009; Morfini et al., 2009; Schon and Przedborski, 2011; Sheng and Cai, 2012). Loss of mitochondrial function is also implicated in cognitive decline that accompanies healthy aging. Despite this fundamental importance, how organismal aging affects lifetime neuronal mitochondrial maintenance at the levels of morphology, density, trafficking, and response to oxidative stress remains largely unknown. In vivo investigation into the neuronal mitochondrial aging process is fundamental to our understanding of the biological basis of aging-associated decline of neuronal function and susceptibility to neurodegeneration.
In the past decades, cultured neurons were widely used for investigations into mitochondria morphology, dynamics, transport, and integrity. However, in vitro cell models have their obvious limitations in characterizing aging-associated changes. Studies in mouse models are challenging due to the relatively long life span and difficulties in visualizing mitochondrial maintenance and transport in vivo. We set out to address this fundamental issue by imaging live Caenorhabiditis elegans and characterizing the effect of organismal aging on neuronal mitochondrial maintenance in the native physiological context. Under laboratory conditions at 20°C, C. elegans nematodes live ∼2 weeks upon reaching adulthood (Huang et al., 2004). The simple and well-characterized nervous system, translucent body, amenability to genetic manipulation, and short life span of C. elegans provide significant advantages for examining mitochondria in individual neurons in vivo throughout animal life. C. elegans life span can be genetically extended by reduction-of-function mutations in daf-2 gene, the C. elegans homolog of insulin-like growth factor receptor that regulates the aging rate from yeast to mammals (Tatar et al., 2003; Kenyon 2001). daf-2(e1370) mutants live twice as long as the control wild-type animals (WTs; Kenyon et al., 1993; Kimura et al., 1997). This dramatic life span extension is dependent on FOXO (Forkhead box protein O)-type transcription factor DAF-16 (Kenyon et al., 1993), although the life span of daf-16(mu86) mutants alone is only slightly shortened compared to wild-type controls (Li et al., 2007). Life span extension was also reported in wild-type strains that overexpress sir-2.1 deacetylase (Viswanathan and Guarente, 2011) and in the eat-2(ad465) mutant strain, which is a genetic model of dietary restriction (Lakowski and Hekimi, 1998; Huang et al., 2004; Bansal et al., 2015). Many reports illustrate similarities of age-related changes between C. elegans and vertebrates at the organismal, cellular, and subcellular levels in neurons (Collins et al., 2008; Pan et al., 2011; Tank et al., 2011; Toth et al., 2012).
Using C. elegans as a model, we address two long-standing questions: How does aging affect neuronal mitochondrial morphology, density or load, trafficking, and oxidative stress resistance? Are these age-related changes amenable to genetic manipulations that slow down the aging process? Our study illustrates that age-related changes in neuronal mitochondria are complex: mitochondrial trafficking declines progressively from the first day of adulthood (DA), whereas mitochondrial size, density, and resistance to oxidative stress undergo three distinct stages during animal life: increase in early adulthood, maintenance at high levels during midadulthood, and decline during late adulthood. Our study provides in vivo evidence revealing unexpected peak of neuronal mitochondrial maintenance at animal midlife rather than in early adulthood and establishes the critical foundation for genetic and pharmacological dissection of factors that influence long-term mitochondrial maintenance in neuronal processes.
Materials and Methods
Strains.
We crossed genomically integrated mitochondria-GFP reporter jsIs609 [Pmec-7::MLS::GFP] (Fatouros et al., 2012) into the various genetic backgrounds to create PTN54 N2; jsIs609, PTN61 daf-16(mu86); jsIs609, PTN63 daf-2(e1370); jsIs609, PTN74 eat-2(ad465); jsIs609, and PTN76 geIs3 [sir-2.1(+) + rol-6(su1006)]; jsIs609. Mitochondrial redox experiments were done using genomically integrated mitoROGFP2 reporter in a wild-type background (strain OR3193 odIs111[Pglr-1::MLS::ROGFP2] from Ghose et al., 2013) and a daf-2(e1370) background (PTN77 daf-2(e1370); odIs111[Pglr-1::MLS::ROGFP2]). RNA interference (RNAi) experiments were performed in the PTN20 strain, which was created by crossing the jsIs609 reporter into the TU3401 sid-1(pk3321); uIs69 [pCFJ90(myo-2p::mCherry) + unc-119p::sid-1] strain (Calixto et al., 2010). C. elegans species consist of two sexes: hermaphrodites and males. Only hermaphrodites were used in the experiments.
Worm aging.
Nematodes were maintained under standard conditions in a 20°C incubator on 5-cm-diameter Petri dishes with nematode growth media (NGM) agar coated with monoxenic culture of live Escherichia coli OP50 bacterial lawn, as described previously (Brenner, 1974). E. coli OP50 is a uracil auxotroph whose growth is limited on NGM plates, allowing for easier observation and handling of the animals. All animals, except for those used in experiments in Figure 5, were synchronized at egg stage, and 3 d (∼72 h) later animals were imaged as 1 d adults (1DA). All animals in Figure 5 experiments were age synchronized at the last larval stage (L4) to circumvent the developmental delay of eat-2(ad465) animals. In all the experiments, aging animals were maintained by manual transfer to fresh NGM OP50 plates every 2 d to avoid contamination by progeny. No fluorodeoxyuridine (FUdR), an inhibitor of DNA synthesis, was used in this study.
Figure 5.

Mitochondrial trafficking progressively declines with age. A, B, Representative kymographs showing mitochondrial motility along the distal ALM processes in 1DA and 11DA WT (A) and daf-2 (B) animals during a 5 min time-lapse imaging. Vertical lines represent stationary mitochondria; lines slanted to the right (negative slope) represent retrograde movement; those to the left (positive slope) indicate anterograde transport. An organelle is considered stationary if it remained immotile (net displacement of ≤3 μm) during the 5 min time-lapse imaging. Note that kymographs (A) display an increased mitochondrial density in WT neurons at 11DA relative to 1DA, whereas the change in density is not readily observed in daf-2. C–E, Quantitative analysis of motile mitochondria frequency and run length (anterograde, red; retrograde, green) in the distal ALM processes of WT (C), daf-2(e1370) (D), and eat-2(ad465) (E) animals from 1DA to 11DA. Age-associated changes in motile mitochondrial frequency and run lengths are visualized as linear regression slopes fitted to the averages of each age group within the 1DA–11DA age range. Progressive decline in the rates of anterograde and retrograde mitochondrial motile frequency and run length was readily observed in WT, but not in long-lived daf-2 and eat-2 animals within the 1DA–11DA age period. Scale bars: 10 μm. The total numbers of neurons analyzed (indicated below the bar graphs) were pooled from three independent trials. Data represent means ± SEM. Slopes of decay, goodness-of-fit R2 values, and p values of zero slope probability were calculated based on linear regression analysis in GraphPad Prism 6.
Epifluorescent microscopy.
A Nikon Eclipse Ti wide-field microscope with a 40× oil objective and Photometrics Coolsnap ES2 12-bit 20 MHz digital monochrome camera were used for all imaging except the eat-2 experiment in Figure 5E, which was done using a Zeiss Axioplan II microscope with 63× (1.4 numerical aperture) PlanApo objective and an ORCA charge-coupled device camera (Hamamatsu). Animals were mounted on 5% agar pads and anesthetized for 4 min in 10 mm levamisol solution in an M9 buffer drop before imaging. All animals were imaged within 20 min of mounting, and only one ALM neuron (closest to the coverslip) was imaged for each animal. Each neuron was imaged as a Z-stack with a 0.3 μm step size at the distal portion of the ALM neurite, covering ∼220 μm of the distalmost neurite as measured from the distal tip (∼150 μm for eat-2 in Fig. 5E). Twenty-five to thirty animals were imaged per trial, per genotype, per time point. For each experimental trial, the control “wild-type” strain PTN54 was prepared, maintained, and monitored strictly in parallel with the mutant test strains. To quantify mitochondrial movements, we performed single-plane time-lapse live imaging at 1 s/frame of the distalmost ∼220 μm section of ALM axon in levamisole-immobilized animals, for 5 min total time.
Image analysis.
All image analysis was done using ImageJ software. For quantitative characterization of stationary mitochondria size and density, image z-stacks were flattened via the maximum fluorescence projection function and background subtracted (rolling ball 30), and the resulting images were thresholded at the default setting. Resulting particles were analyzed to collect the following parameters for each mito GFP-labeled particle: area, perimeter, major axis (length), minor axis (width), aspect ratio (major/minor axis), and X–Y coordinates. Particles >0.15 μm2 were entered into an Excel spreadsheet and sorted for each neuron based on its x-axis coordinate. Intermitochondrial distance (IMD) was calculated based on X–Y coordinates of neighboring mitochondria: IMD = √[(Xn+1 − Xn)2 + (Yn+1 − Yn)2]. The total mitochondrial load of each neurite was calculated as follows: sum (particle area)/total distance * 100.
To quantify mitochondrial trafficking, time-lapse stacks of 300 distal ALM images collected over a 5 min period (1 image per second) were converted to 2D kymographs using the “Kymograph” macro of ImageJ. Trafficking frequency was scored as the number of motile mito-GFP particles in a kymograph, later averaged by animal to get a population mean for that age and genotype. A particle was considered motile if it moved >3 μm during acquisition. Run length was measured as the distance between the starting and last stopping points (or end of the kymograph, whichever comes first) of a motile mito-GFP particle.
Chronological versus physiological aging.
Animals were age synchronized by egg lay and passaged every 2 d until reaching an 11DA age (as described above). Each animal was imaged in two settings: (1) with a 40× objective under a 488 nm fluorescent light for anterior lateral microtubule (ALM) mitochondria imaging and (2) with a 4× objective with differential interference contrast settings for whole-body imaging. First, all neuronal mitochondria were analyzed as described above. Then the animals were sorted based on their body image into one of three groups, “youthful,” “decrepit,” or “intermediate,” and their corresponding neuronal mitochondrial parameters were averaged to compare these groups.
Mitochondrial redox measurements.
The age-synchronized population was sampled at 1DA, 4DA, 8DA, 11DA, and 15DA time points to acquire images of ROGFP2-tagged mitochondria in four glr-1-positive head neuronal processes at 405 nm (150 ms) and 488 nm (50 ms) stimulation. To measure response to oxidative stress, another sample of same-age animals was incubated in 200 mm H2O2 M9 solution for 30 min and imaged immediately after.
Electron microscopy.
Synchronized animals were fixed and embedded for transmission electron microscopy (TEM) following standard chemical immersion protocol (Hall, 1995). Briefly, thin sections were collected onto formvar-coated slot grids, poststained with uranyl acetate and lead citrate, and examined using a Philips CM10 electron microscope. Images were collected on Kodak 4489 film or using an Olympus Morada digital camera.
RNAi experiments.
RNAi was administered by feeding the animals with IPTG (isopropyl-beta-D-thiogalactopyranoside)-induced dsRNA-expressing bacteria and restricted to the nervous system by neuron-specific expression of dsRNA channel SID-1 in the sid-1 mutant background (Calixto et al., 2010). RNAi cultures of HT115 bacteria with RNAi vectors were grown overnight at 37°C in LB medium with 100 μg/ml ampicillin. Two hundred microliters of the fresh cultures were spread on 5-cm-diameter NGM plates supplemented with 10 μl IPTG (1 m) and allowed to grow overnight in the dark at room temperature. To parse out the gene-targeted RNAi effect from nonspecific effects of different genetic backgrounds and RNAi experimental procedures, all animals were grown on empty vector (EV) RNAi plates from egg to 1DA and then transferred to gene-targeted RNAi plates from 1DA to 4DA (including EV as a negative control). All animals were transferred to fresh RNAi plates at 3DA to avoid food depletion and progeny crowding, and imaged at 4DA age.
Statistical analysis.
Statistical analyses were performed in GraphPad Prism 6 software using mitochondrial data averaged by animal. For characterization of the basic neuronal aging phenotypes (Fig. 1), we pooled selected control measurements from eight full-length experiments and performed statistical analysis on this large sample. Age groups were compared to each other using one-way ANOVA with Tukey's post hoc multiple comparison test of the mean of each column with the mean of every other column at familywise significance level α = 0.01. To compare aging phenotypes of daf-2 and daf-16 mutants to WTs, we used two-way ANOVA of mitochondrial data averaged by neuron. In each age category, the mean of each mutant phenotype was compared to the corresponding age-matched WT control using either Dunnett's or Sidak's (where appropriate) post hoc multiple comparison test with familywise significance level α = 0.01. To determine the slope of age-related decline in mitochondrial trafficking frequency and run length, we performed linear regression analysis in GraphPad Prism 6 and checked that the corresponding R2 value for goodness of fit was >0.9 and p value for nonzero slope was <0.05.
Figure 1.
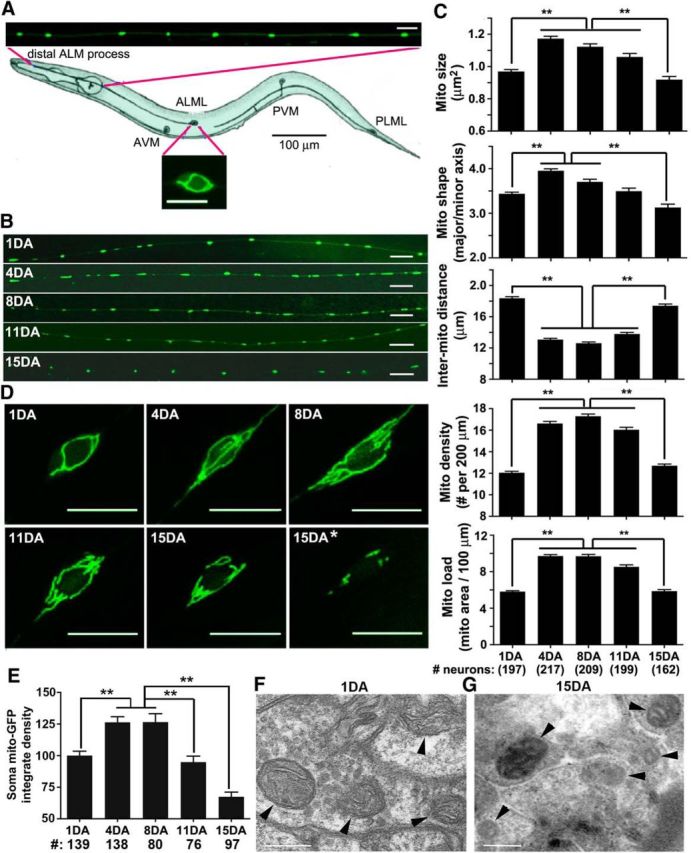
Neuronal mitochondria undergo three phasic changes in morphology and density during adulthood. A, Diagram showing the C. elegans ALM neurons and GFP-labeled mitochondria in the distal ALM process and cell body [adapted from Goodman (2006)]. B, C, Representative images (B) and quantitative analysis (C) of mitochondrial size, shape (major/minor axis), intermitochondrial distance, density, and total load in distal ALM processes at five time points from 1DA to 15DA. Note that mitochondrial size, load, and density undergo three phases: a rapid increase during young adulthood (1DA–4DA), steady maintenance at midlife (4DA–11DA), and a fast decline during late adulthood (11DA–15DA; Table 1). D, E, Representative images (D) and quantitative analysis (E) of integrated density of mito-GFP signal in maximum intensity z-projections of ALM cell bodies over animal adulthood. Data were normalized by 1DA group. Note that mitochondria are redistributed toward polar ends of the soma in late adulthood (11DA–15DA); 48% of 15DA animals (15DA*; D) display a dramatic reduction in mitochondrial content in the soma. F, G, TEM images showing ultrastructural changes of neuronal mitochondria in young (1DA; F) and aged (15DA; G) animals. Mitochondria were viewed in cross-section. Note that mitochondria in young adult (1DA) animals showed prominent internal cristae structures. At 15DA, mitoplasm was electron dense, and some of cristae were difficult to resolve. Arrowheads indicate mitochondria. The total numbers of animals analyzed are indicated below the bars. Data were pooled from eight independent trials and represent means ± SEM with one-way ANOVA test and Tukey's multiple comparisons post hoc test. **p = 0.005. Scale bars: B, D, 10 μm; F, G, 0.2 μm.
Results
Neuronal mitochondria undergo three phasic stages in morphology and density throughout adulthood
To visualize mitochondria in a single neuron in vivo, we used transgenic C. elegans strain jsIs609, which expresses mitochondria-targeted GFP in mechanosensory ALM, AVM, PLM, and PVM neurons (anterior lateral microtubule, anterior ventral microtubule, posterior lateral microtubule, and posterior ventral microtubule, respectively) (Fatouros et al., 2012). GFP-labeled mitochondria in ALM neuronal processes appear as sphere and rod-shaped organelles of varying size distributed at distinct intervals (Fig. 1A), similar to axonal mitochondria in mammalian neurons (Kang et al., 2008; Chen and Sheng, 2013). Mitochondria in the adult neuronal cell bodies form a filamentous tubular network that encircles the soma perimeter (Fig. 1A). To characterize age-related changes in neuronal mitochondria, we conducted a cross-sectional study and imaged mitochondria in vivo in the distal ALM processes (Fig. 1B) in animals sampled from age-synchronized populations at 1DA, 4DA, 8DA, 11DA, and 15DA. The observations of neuronal mitochondria using wide-field epifluorescent microscopy were confirmed by confocal microscopy (data not shown). To characterize mitochondria at distal neuronal process, we measured individual mitochondrion size, shape (expressed as an aspect ratio, major/minor axis), spacing (expressed as an IMD), density (total number of mitochondria per 200 μm length), and mitochondrial load (summed mitochondrial area per 100 μm process length).
We observed a nonlinear pattern of age-related changes in mitochondrial size, shape, density, and total load in distal ALM neuronal processes (Fig. 1C, Table 1). The first stage of adulthood (1DA–4DA) is characterized by a dramatic increase (67%) of mitochondrial load from 5.8 ± 0.1 μm2 (at 1DA) to 9.7 ± 0.2 μm2 (at 4DA) per 100 μm of neurite (mean ± SEM, p = 0.001), attributed to a combinatorial effect of increased mitochondrial size, number, and density. The increase in mitochondrial size is contributed mainly by elongation of mitochondria as reflected by an increased aspect ratio from 3.4 to 4.0 (Fig. 1C, Table 1). During the second stage of adulthood (4DA–8DA), mitochondrial load, size, and density remain relatively stable. The third stage of adulthood (11DA–15DA) is characterized by progressive decline of mitochondrial load due to combinatorial effects of decreased mitochondrial size, number, and density. By 15DA, mitochondrial load in the distal ALM processes is reduced to 5.9 ± 0.1 μm2 per 100 μm of neurite (p = 0.001), a 39% decrease from its peak at middle adulthood (8DA). We conclude that mitochondrial size and total load in neuronal processes undergo three phasic changes throughout adulthood: a rapid increase during young adulthood (1DA–4DA), steady maintenance at midlife (4DA–8DA), and progressive decline during late adulthood (11DA–15DA). Aging is a stochastic process characterized by the increasing heterogeneity of phenotypes with age even within a chronologically synchronized population (Herndon et al., 2002). Consistently, we observed an age-related increase in the variability of neuronal mitochondrial phenotypes, as reflected by an age-related increase in SDs (Table 1).
Table 1.
Age-related changes of mitochondrial morphology and density in WT distal ALM processes
| 1DA |
4DA |
8DA |
11DA |
15DA |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Mito area (μm2) | 0.97 | 0.17 | 1.17 | 0.23 | 1.12 | 0.26 | 1.06 | 0.31 | 0.92 | 0.25 |
| Major axis (μm) | 1.95 | 0.26 | 2.30 | 0.35 | 2.17 | 0.48 | 2.05 | 0.55 | 1.82 | 0.49 |
| Minor axis (μm) | 0.63 | 0.06 | 0.63 | 0.05 | 0.64 | 0.05 | 0.64 | 0.05 | 0.64 | 0.06 |
| Aspect ratio | 3.4 | 0.5 | 4.0 | 0.6 | 3.7 | 0.9 | 3.5 | 1.0 | 3.1 | 1.0 |
| IMD (μm) | 18.4 | 2.9 | 13.1 | 2.3 | 12.6 | 2.3 | 13.8 | 2.9 | 17.4 | 2.8 |
| # mitos/200 μm | 12.1 | 1.8 | 16.6 | 2.7 | 17.3 | 3.1 | 16.0 | 3.2 | 12.7 | 2.0 |
| Mito load (μm2/100 μm) | 5.82 | 1.2 | 9.71 | 2.3 | 9.70 | 2.7 | 8.53 | 3.0 | 5.87 | 2.1 |
| Neurite measured (μm) | 222 | 21 | 228 | 20 | 228 | 21 | 228 | 21 | 232 | 20 |
| Number of animals | 197 | 217 | 209 | 199 | 162 | |||||
Quantitative analysis of mitochondrial size, shape (expressed as an aspect ratio of major to minor axis), intermitochondrial distance, density (total number of mitochondria per 200 μm length), and mitochondrial load (summed mitochondrial area per 100 μm process length) in distal ALM processes at five time points from 1DA, 4DA, 8DA, 11DA, and 15DA-old animals. Data were pooled in eight independent trials from the total number of animals as indicated and are expressed as mean ± SD.
Age-related changes of the mitochondrial network in the ALM cell body mirror the phasic pattern in neuronal processes. Mitochondria in ALM soma at young adulthood (1DA) display a single continuous filament that loops around the cell body (Fig. 1D). As animals mature into midadulthood (4DA–8DA), the filamentous network becomes more expansive and complex. As animals transition into late adulthood (11DA–15DA), the mitochondrial network becomes smaller and fragmented, some of which often redistributes toward polar ends of the soma. Forty-eight percent of 15DA animals display a dramatic reduction in mitochondrial content in the soma (Fig. 1D, 15DA*). Compared to 1DA levels, the integrated density of mito-GFP signal in cell bodies increased to 126% by middle age (4DA–8DA) and then dropped to 67% at 15DA (Fig. 1E).
To further confirm our findings, we examined neuronal mitochondria with TEM in WT C. elegans. Because TEM images do no represent serial sections, full dimensions of a mitochondrion could not be determined, so we measured only cross-sectional diameters of mitochondria. In young adulthood (1DA), neuronal mitochondria contain clearly visible cristae structures (Fig. 1F) with an average cross-section diameter of 0.18 ± 0.01 μm (mean ± SEM). At 15DA, neuronal mitochondria exhibit dramatic ultrastructural changes, including loss of visible membrane and cristae structures and an increase in electron-dense staining of mitoplasm (Fig. 1G; mean mitochondrial diameter, 0.17 ± 0.005 μm). Although mitochondria in 1DA and 15DA animals are similar in size, they appear markedly different in their ultrastructure. Since mitochondrial cristae organization is relevant to functional integrity, these changes are likely reflective of aging-associated mitochondrial degeneration. Altered mitochondrial ultrastructure in late adulthood, combined with reduced mito-GFP size and density in ALM cell bodies and distal neurites, support the notion that organismal aging ultimately has a negative effect on neuronal mitochondria maintenance.
Increased mitochondrial size during early adulthood (1DA–4DA) may occur through mitochondrial fusion/fission dynamics (Chan, 2012). To test this hypothesis, we knocked down transcripts of C. elegans homologs of the conserved mitochondrial fusion proteins fzo-1 (Mfn1 and Mfn2) and eat-3 (Opa1) and fission protein drp-1 (Drp1) in a strain that restricts RNAi effect to neurons only (Calixto et al., 2010). We chose our gene-specific treatments in early adulthood stages from 1DA to 4DA because RNAi-treated animals age at a different rate, and some gene-specific treatments cause faster age-related deterioration in late stages. RNAi treatment against fzo-1 and eat-3 from 1DA to 4DA abolished the mitochondrial size increase seen in RNAi control animals; their neuronal mitochondria at 4DA were the same size as at 1DA (Fig. 2A). Because mitochondrial density was not affected (Fig. 2B), the effect of fzo-1 and opa-1 knockdown on total mitochondrial load was moderately lower than that in 4DA controls (Fig. 2C). Interestingly, drp-1-targeted RNAi treatment had no observable impact on this phasic increase in mitochondrial size (Fig. 2A). Our data suggest that knockdown of mitochondrial fusion proteins can suppress mitochondrial elongation during early adulthood (1DA–4DA), thus highlighting the possibility that the first phasic increase in mitochondrial size during early adulthood stages preferentially depends on mitochondrial fusion events.
Figure 2.

Knockdown of mitochondrial fusion proteins suppresses the first phasic increase of mitochondrial size. A–C, Quantitative analysis of mitochondrial size (A), density (B), and total load (C) in distal ALM processes during young adulthood (1DA–4DA) of neuron-RNAi-conducive strains. All animals were grown from eggs on EV (empty vector) control RNAi plates before being grown on gene-targeted RNAi plates from 1DA to 4DA (or EV as control). Note that animals grown on EV control plates exhibited the first phasic (1DA to 4DA) upward shift in average mitochondrial size (A), density (B), and load (C) along distal neuronal processes. In contrast, RNAi treatment against fzo-1/Mfn and eat-3/Opa1, but not drp-1, abolished this phasic increase. Data were pooled from the total numbers of animals indicated in the bars in three independent trials, and are expressed as means ± SEM and compared by ANOVA with by Sidak's correction for multiple comparisons.
Long-lived genetic mutants display stably maintained mitochondrial load in ALM processes during adulthood
If the three-phasic pattern of neuronal mitochondrial maintenance is an age-related phenotype, it should be responsive to genetic manipulation of life span. To test this hypothesis, we examined ALM neuronal mitochondrial parameters in daf-2(e1370) mutants with a life span twice as long as WTs and daf-16(mu86) mutants with a slightly shortened life span. While long-lived daf-2 mutants had similar changes in mitochondrial size as WT animals, the three-phase pattern stretched over a longer period of time (Fig. 3A). daf-2 mutants also exhibited smaller amplitude of lifelong fluctuation of mitochondrial load in their ALM neuronal processes and maintained relatively constant mitochondrial density compared to WTs (Fig. 3B). As a result, neuronal mitochondrial load exhibited a more stable lifelong pattern compared to WTs: neither rapid incline nor decline was observed (Fig. 3C, Table 2). In contrast, daf-16 mutants with a slightly shortened life span (Kenyon et al., 1993; Li et al., 2007) displayed WT-like mitochondrial parameters under our nonstress experimental conditions (Fig. 3A–C). Consistently, a previous study of morphological aging in the same class of neurons showed that daf-16 loss of function held little impact on age-related neuronal restructuring, whereas daf-2 conferred demonstrative effects (Toth et al., 2012).
Figure 3.
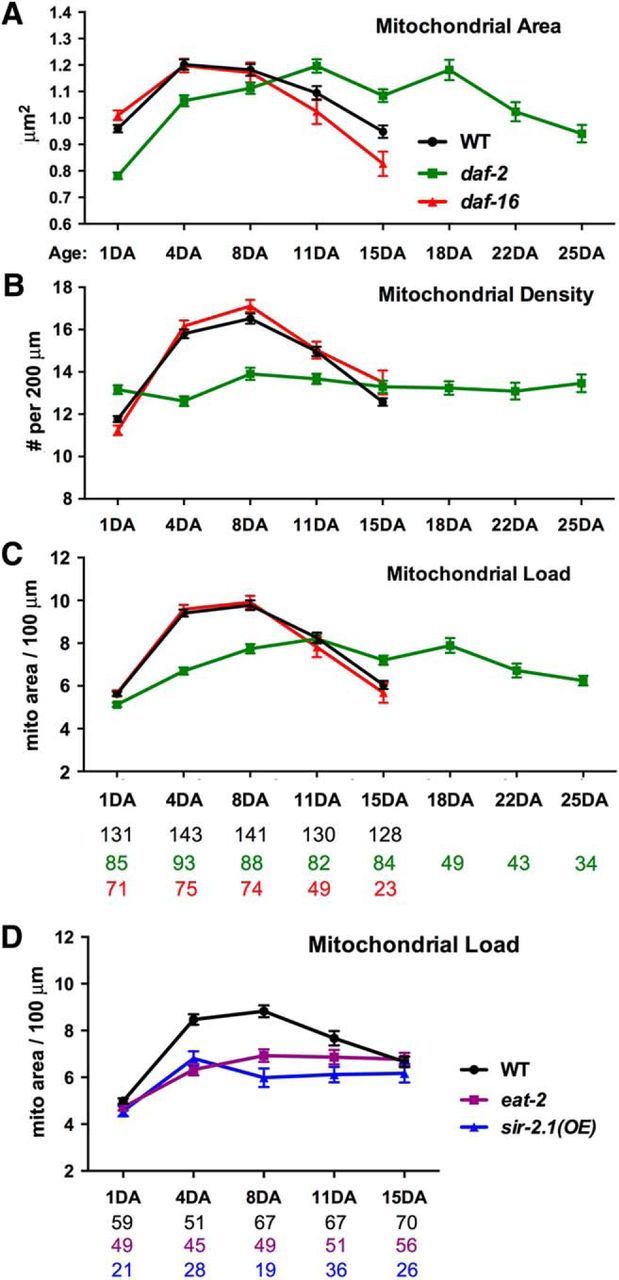
Long-lived mutants show stable mitochondrial load over animal adulthood. A–C, Quantitative analysis of mitochondrial size (A), density (B) and load (C) in distal ALM processes of WT, long-lived daf-2(e1370), and slightly short-lived daf-16(mu86) mutants over animal adulthood. Note that aging-associated triphasic changes in mitochondrial size were delayed in daf-2 mutants (A). daf-2 mutants exhibited relatively constant mitochondrial density (B) and load (C) in ALM distal processes during life: neither rapid incline nor decline was observed. All animals were age-synchronized at the egg stage (Table 2). D, Quantitative analysis of mitochondrial load in distal ALM processes of WT and long-lived eat-2(ad465) and sir-2.1OE animals. Both eat-2 and sir-2.1OE strains maintained a more stable mitochondrial load in the distal ALM processes during the 4DA to 15DA period, neither rapid incline nor decline was observed. Data were pooled from three independent trials. All animals were age synchronized at the L4 stage (due to developmental delay of eat-2). The total number of animals in each group is indicated under bar graphs. Data represent means ± SEM.
Table 2.
Age-related mitochondrial phenotype in daf-2 and daf-16 distal ALM processes
| WT |
daf-2(e1370) |
daf-16(mu86) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SEM | N | Mean | SEM | N | Mean | SEM | N | |
| Mitochondrial area (μm2) | |||||||||
| 1DA | 0.96 | 0.01 | 131 | 0.78 | 0.01 | 85 | 1.01 | 0.02 | 71 |
| 4DA | 1.20 | 0.02 | 143 | 1.06 | 0.02 | 93 | 1.20 | 0.03 | 75 |
| 8DA | 1.18 | 0.02 | 141 | 1.11 | 0.02 | 88 | 1.17 | 0.04 | 74 |
| 11DA | 1.09 | 0.03 | 130 | 1.20 | 0.03 | 82 | 1.02 | 0.05 | 49 |
| 15DA | 0.95 | 0.02 | 128 | 1.08 | 0.02 | 84 | 0.83 | 0.05 | 23 |
| 18DA | 1.18 | 0.04 | 49 | ||||||
| 22DA | 1.02 | 0.04 | 43 | ||||||
| 25DA | 0.94 | 0.03 | 34 | ||||||
| Aspect ratio | |||||||||
| 1DA | 3.4 | 0.0 | 131 | 3.6 | 0.0 | 85 | 3.5 | 0.1 | 71 |
| 4DA | 4.2 | 0.1 | 143 | 4.0 | 0.1 | 93 | 4.1 | 0.1 | 75 |
| 8DA | 4.0 | 0.1 | 141 | 4.0 | 0.1 | 88 | 3.9 | 0.1 | 74 |
| 11DA | 3.8 | 0.1 | 130 | 4.1 | 0.1 | 82 | 3.4 | 0.1 | 49 |
| 15DA | 3.3 | 0.1 | 128 | 3.7 | 0.1 | 84 | 3.0 | 0.2 | 23 |
| 18DA | 4.0 | 0.1 | 49 | ||||||
| 22DA | 3.4 | 0.1 | 43 | ||||||
| 25DA | 3.2 | 0.1 | 34 | ||||||
| IMD (μm) | |||||||||
| 1DA | 18.7 | 0.2 | 131 | 16.7 | 0.3 | 85 | 20.0 | 0.6 | 71 |
| 4DA | 13.7 | 0.2 | 143 | 17.6 | 0.3 | 93 | 13.4 | 0.3 | 75 |
| 8DA | 13.2 | 0.2 | 141 | 16.0 | 0.4 | 88 | 12.6 | 0.2 | 74 |
| 11DA | 14.6 | 0.2 | 130 | 16.1 | 0.3 | 82 | 14.7 | 0.4 | 49 |
| 15DA | 17.5 | 0.3 | 128 | 16.8 | 0.4 | 84 | 16.5 | 0.8 | 23 |
| 18DA | 16.7 | 0.4 | 49 | ||||||
| 22DA | 17.0 | 0.5 | 43 | ||||||
| 25DA | 16.6 | 0.7 | 34 | ||||||
| # mitos/200 μm | |||||||||
| 1DA | 11.8 | 0.1 | 131 | 13.2 | 0.2 | 85 | 11.2 | 0.2 | 71 |
| 4DA | 15.8 | 0.2 | 143 | 12.6 | 0.2 | 93 | 16.2 | 0.3 | 75 |
| 8DA | 16.5 | 0.2 | 141 | 13.9 | 0.3 | 88 | 17.1 | 0.3 | 74 |
| 11DA | 15.0 | 0.2 | 130 | 13.7 | 0.2 | 82 | 15.0 | 0.4 | 49 |
| 15DA | 12.6 | 0.2 | 128 | 13.3 | 0.3 | 84 | 13.5 | 0.6 | 23 |
| 18DA | 13.2 | 0.3 | 49 | ||||||
| 22DA | 13.1 | 0.4 | 43 | ||||||
| 25DA | 13.5 | 0.4 | 34 | ||||||
| Mitochondrial load (μm2/100 μm) | |||||||||
| 1DA | 5.63 | 0.10 | 131 | 5.12 | 0.10 | 85 | 5.65 | 0.14 | 71 |
| 4DA | 9.41 | 0.16 | 143 | 6.70 | 0.16 | 93 | 9.58 | 0.21 | 75 |
| 8DA | 9.77 | 0.23 | 141 | 7.74 | 0.22 | 88 | 9.91 | 0.30 | 74 |
| 11DA | 8.26 | 0.24 | 130 | 8.21 | 0.23 | 82 | 7.81 | 0.46 | 49 |
| 15DA | 6.05 | 0.19 | 128 | 7.20 | 0.21 | 84 | 5.68 | 0.47 | 23 |
| 18DA | 7.89 | 0.34 | 49 | ||||||
| 22DA | 6.72 | 0.32 | 43 | ||||||
| 25DA | 6.24 | 0.23 | 34 | ||||||
Quantitative analysis of mitochondrial size, shape (aspect ratio), intermitochondrial distance, density (total number of mitochondria per 200 μm length), and mitochondrial load (summed mitochondrial area per 100 μm process length) in distal ALM processes of WT, daf-2, and daf-16 mutants at different time points throughout the adulthood. Total number of animals in each group is indicated (N). Data represent means ± SEM.
To determine whether the stably maintained mitochondrial load is a general phenotype associated with longevity, we examined two other genetic models of life span extension: dietary restriction (eat-2; Lakowski and Hekimi 1998; Huang et al., 2004; Bansal et al., 2015) and sirtuin overexpression (sir-2.1OE; Viswanathan and Guarente 2011). Similar to daf-2 mutants, both eat-2 and sir-2.1OE animals maintained a more stable mitochondrial load in the distal ALM processes during the 4DA to 15DA period; neither rapid incline nor decline was observed (Fig. 3D). Altogether, our study provides in vivo evidence that three different mutant C. elegans strains with extended life spans share a similar mitochondrial load phenotype in the ALM processes during adulthood, thus raising the intriguing possibility that stable mitochondrial load in distal neuronal processes is associated with longevity and functionality of the nervous system.
Faster physiological aging correlates with reduced mitochondrial load in distal neuronal processes
Aging is influenced by genetic and stochastic factors. Even chronologically synchronized isogenic C. elegans population grown in the same conditions age differently; some maintain a youthful body profile, whereas others exhibit accelerated aging and deterioration (Herndon et al., 2002). To determine whether age-associated changes in neuronal mitochondrial maintenance also correlate with physiological aging, we compared mitochondria at the same chronological age (11DA) with contrasting whole-body appearance (Garigan et al., 2002; Fig. 4A,B; see Material and Methods). Previous studies showed that decrepit-looking aged animals have a shorter life expectancy (Herndon et al., 2002) and higher lipofuscin loads (Gerstbrein et al., 2005) and exhibit accelerated aging. Youthful-looking 11DA animals had higher total mitochondrial load in the distal ALM processes than decrepit-looking 11DA animals: 7.5 ± 0.5 versus 5.1 ± 0.3 μm2 of mitochondrial biomass per 100 μm length (p < 0.001; Fig. 4C), respectively. Lower mitochondrial load in decrepit 11DA animals is primarily due to smaller individual mitochondrial size, as the difference in mitochondrial density is only modestly significant (p = 0.02). Our observations of smaller neuronal mitochondria in decrepit 11DA animals suggest earlier onset of age-related mitochondrial loss in faster-aging animals. Our data raise the possibility that stochastic factors contributing to the physical deterioration may have a negative effect on mitochondrial size and total mitochondrial load in neuronal processes. Alternatively, as neurons have been shown to regulate animal life span (Bishop and Guarente, 2007; Wolkow et al., 2000; Durieux et al., 2011), age-related decline of neuronal mitochondrial load may exacerbate physiological aging of the animal by affecting neuronal signaling. In summary, our in vivo analysis illustrates correlation between naturally accelerated physical aging and the neuronal mitochondrial aging profile, which further supports observations in the three genetic manipulations that extend longevity and also feature with stable mitochondrial load in distal neuronal processes.
Figure 4.

Accelerated body aging correlates with early mitochondrial loss in distal neuronal processes. A, B, Representative images show 1DA (A) and 11DA animals (B). Whereas all 1DA animals displayed consistently good body morphology, the 11DA group ranged widely from youthful (Y11DA) to decrepit looking (D11DA). C, Quantitative analysis of mitochondrial size, density, and total load in distal ALM processes of 11DA youthful versus decrepit animals. Note that accelerated body aging correlates with reduced mitochondrial size and total load in ALM processes. All animals were raised on the same plates. Total number of animals in each group is indicated within in bars. Data represent means + SEM.
Mitochondrial trafficking in distal ALM process declines progressively with age
Mitochondrial trafficking in distal neurites enables neurons to redistribute mitochondria in response to neuronal activity and local metabolic requirements. To characterize the effect of aging on mitochondrial trafficking, we performed time-lapse imaging of mitochondria in the distal segment of ALM processes (>200 μm segment) at 1DA, 4DA, 8DA, and 11DA in WT and daf-2(e1370) animals. While the majority of mitochondria are stationary during a 5 min recording, some move along processes in a “stop and go” pattern (Fig. 5A) similar to axonal mitochondria of mammalian and fly larvae neurons (Kang et al., 2008; Mondal et al., 2011; Chen and Sheng, 2013). Consistent with an ex vivo study in mouse nerve explants (Misgeld et al., 2007), we observed that motile mitochondria were significantly shorter than the stationary ones: the average length of motile mitochondria at 1DA is 1.1 ± 0.4 μm (mean ± SEM; n = 103), compared to 2.3 ± 0.06 μm when stationary (n = 306, p < 0.01).
Given the much lower frequency of mitochondrial motility in ALM neurons in vivo compared to cultured mammalian neurons, we characterized the frequency and run length of trafficking events (Fig. 5C–E). At all ages examined, mitochondria move in the retrograde direction (toward the soma) more frequently than in the anterograde direction (toward the distal terminal). In WT animals, we observed a progressive age-related decline in both frequency and run length of motile mitochondria starting at young adulthood (1DA; Fig. 5A,C). Average anterograde trafficking frequency reduced from 1.45 to 0.56 events/5 min (linear slope, −0.094; R2 = 0.98; p = 0.009); average retrograde frequency reduced from 4.4 to 1.0 events/5 min (linear slope, −0.35; R2 = 0.98; p = 0.008) during 1DA–11DA. The slope of age-related decline in mitochondrial trafficking frequency is significantly steeper for retrograde than anterograde direction (p = 0.001). Average anterograde run length reduced from 28.2 to 7.6 μm (linear slope, −1.92; R2 = 0.92; p = 0.042); average retrograde run length reduced from 26.9 to 11.3 μm (linear slope, −1.60; R2 = 0.99; p = 0.005) during 1DA–11DA (Fig. 5C). The slope of age-related decay in anterograde mitochondrial run length was not significantly different (p = 0.49) from retrograde.
In contrast, long-lived daf-2 mutants did not exhibit any significant age-related decay in either motile mitochondria frequency or run length during the 1DA–11DA period; none of the linear regression tests gave a significant nonzero slope value (Fig. 5D). To test our hypothesis that genetically enabled life span extension is associated with mitochondrial trafficking maintenance in neurons, we measured long-lived eat-2 mutants for the same mitochondrial parameters (Fig. 5E). Similar to long-lived daf-2 animals, neither anterograde nor retrograde frequency nor run length of motile mitochondria was significantly different during 1DA–11DA age period in eat-2 animals, indicating no significant age-related decline in mitochondrial trafficking. Overall, we present in vivo evidence of a progressive age-related decline of mitochondrial trafficking at a single-neuron level starting in early adulthood in WT animals and better maintenance of neuronal mitochondrial trafficking in two long-lived mutants.
Age-associated changes in mitochondrial resistance to oxidative stress
We next investigated how organismal aging affects neuronal mitochondria ability to resist oxidative stress in vivo and whether prolongevity daf-2(e1370) mutation confers enhanced mitochondrial stress resistance. Oxidative stress resistance was defined as the ability to minimize mitochondrial oxidation following acute oxidative exposure. To measure mitochondrial redox state, we used genetically encoded redox-sensing GFP variant targeted to the mitochondrial matrix by its leader sequence (mito-ROGFP2; Hanson et al., 2004; Ghose et al., 2013). Upon oxidation, this fluorescent reporter exhibits a partial reversible shift in its peak excitation from 488 to 405 nm. The 405/488 ratio indicates the extent of ROGFP2 oxidation, thus serving as a readout of redox state in mitochondrial matrix (Hanson et al., 2004). We chose to image mitochondria in the head neurons of the previously published mito-ROGFP2 reporter strain odls111 (Ghose et al., 2013) to avoid signal interference by intestinal autofluorescence at 405 nm excitation. To quantitate the aging effect on mitochondrial oxidative stress resistance in vivo, we sampled age-synchronized WT and daf-2 populations at 1DA, 4DA, 8DA, 11DA, and 15DA and measured mito-ROGFP2 405/488 ratios in neurons of untreated animals and after incubation in 200 mm H2O2 solution for 30 min (Fig. 6A,B). H2O2-treated animals displayed an upward ratiometric shift in mito-ROGFP2 405/488 intensity ratios in both genotypes at all ages tested (Fig. 6C,D), indicating a reliable biosensor of oxidative effect on mitochondrial redox state.
Figure 6.
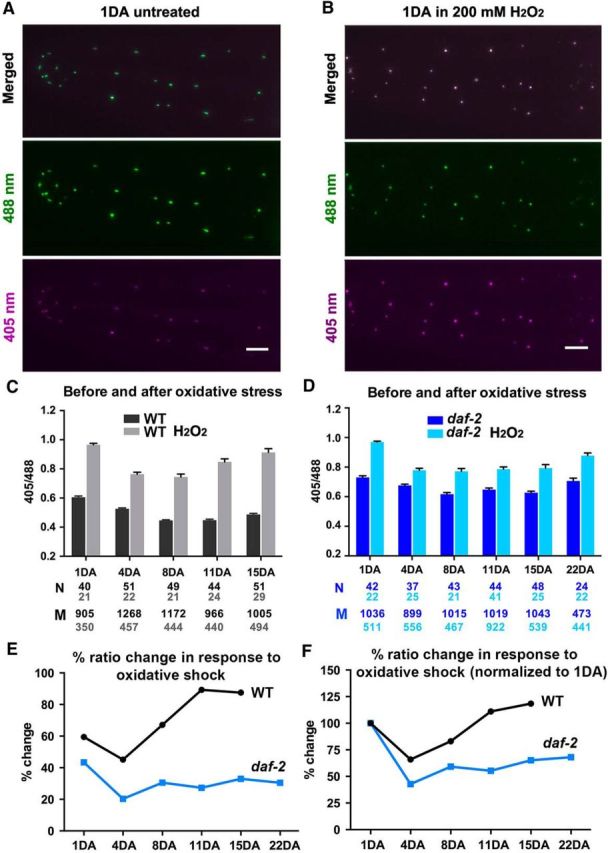
Age-associated changes in mitochondrial resistance to oxidative stress. A, B, Representative images showing mitochondria labeled with genetically encoded redox biosensor ROGFP2 in glr-1-positive head neurons of untreated animals (A) and 1DA WT animals treated with a 200 mm H2O2 solution for 30 min (B). Oxidation of ROGFP2 by H2O2 treatment concurrently decreases 488 nm-stimulated fluorescence and increases 405 nm-stimulated fluorescence (green to red shift). C, D, Relative oxidation levels of ROGFP2-labeled neuronal mitochondria in WT (C) and daf-2 (D) animals. The 405/488 nm maximum fluorescence intensity ratios of individual mitochondria in neurons of synchronized aging animals with or without 200 mm H2O2 incubation were pooled at various stages of adulthood as indicated. E, F, H2O2-triggered shifts in 405/488 ratios (from C and D) expressed as percentage changes from the same-age untreated control group (E) or as percentage changes normalized to the ratio shift of 1DA animals of each corresponding genotype (F). Larger percentage values reflect a more oxidized matrix after oxidative stress. Note that (1) WT neuronal mitochondria display more resistance to acute oxidative insult in middle adulthood (4DA–8DA) than in young (1DA) and late (15DA) adulthood, and (2) neuronal mitochondria in long-lived daf-2 mutants show higher than WT resistance to acute oxidative shock in all age groups and slower age-related decline in resistance to oxidative stress. Scale bars: 10 μm. Data in bar graphs represent means ± SEM, with ANOVA plus Tukey's post hoc analysis. The total numbers of animals (N) and mitochondria (M) analyzed are indicated below the bars.
We observed that neuronal mitochondria's ability to resist oxidative stress changes during animal life (Fig. 6C–F). H2O2 incubation triggered a 59% increase from the baseline in 405/488 ratio in 1DA WT animals (Fig. 6E). The same incubation at 4DA triggered only a 45% increase from the baseline in 405/488 ratio, indicating higher resistance to oxidation compared to 1DA animals. The same incubation at 15DA triggered an 88% increase from the baseline in 405/488 ratio, indicating lower resistance to oxidation compared to either 1DA or 4DA animals. When normalized to 1DA levels, the ratiometric shifts from oxidative shock at 4DA and 8DA were 34 and 17% lower than that at 1DA, respectively, but 18% higher at 15DA than at 1DA (Fig. 6F). We conclude that WT neuronal mitochondria display more resistance to acute oxidative insult in the early-to-middle adulthood (4DA–8DA) than in young (1DA) and late (15DA) adulthood stages. The postreproductive period of adulthood (after 4DA) is marked by progressively increasing ratiometric shift upon H2O2 exposure (Fig. 6F), indicating aging-related decline in the ability to resist acute oxidative stress.
In contrast, neuronal mitochondria in daf-2 mutants exhibited slightly elevated lifelong baseline (unstressed) mito-ROGFP2 405/488 ratios compared to WT (Fig. 6D). This finding is consistent with prior reports that whole-body mitochondria of long-lived daf-2(e1370) mutants, while having higher bioenergetic competence, also produce more H2O2 (Brys et al., 2010). When challenged with acute oxidative stress, neuronal mitochondria in daf-2 mutants showed smaller than WT ratiometric shifts at all ages tested, indicating constitutively higher resistance to acute oxidative shock (Fig. 6E,F). When normalized to 1DA levels, neuronal mitochondria in daf-2 animals exhibited a lower rate of age-related decline in oxidative stress resistance as compared to WTs, suggesting better maintenance of mitochondrial integrity through life (Fig. 6F). Elevated resistance to acute oxidative stress exhibited by neuronal mitochondria in daf-2 mutants is consistent with enhanced survival of daf-2 mutants under oxidative stress (Honda and Honda, 1999; Holzenberger et al., 2003; Bansal et al., 2015). Considering that daf-2 neuronal mitochondria are comparable in size to WT, elevated antioxidant and detoxification activity in daf-2 animals likely contributes to the enhanced resistance to oxidative stress (Vanfleteren and De Vreese, 1995; Honda and Honda, 2002; Yanase et al., 2002; McElwee et al., 2003; Murphy et al., 2003; Houthoofd et al., 2005).
Discussion
By focusing on age-associated changes in the neuronal mitochondrial maintenance in the single neuron level, we identified a triphasic pattern of mitochondrial size, density, and load in neurons in vivo during a normal animal life span. Aging is a genetically regulated process; therefore, genetic mechanisms that extend life span should also change aging-associated processes in neurons. We further modulated key longevity pathways such as insulin signaling in daf-2 mutants to show that such changes are bona fide aging-associated features. To strengthen our claim, we also include two additional genetic models of life span extension: dietary restriction and posttranslational modification associated with healthy metabolism. Similar to our documentation of daf-2 mutants, both eat-2 and sir-2.1OE populations maintained relatively constant mitochondrial load in the distal ALM processes and exhibited lower-than WT mitochondrial load at 4DA and 8DA age. A WT-type triphasic increase, maintenance, and decrease pattern of neuronal mitochondrial load was “flattened” in all three genetic models of longevity. Our data thus raise an intriguing possibility that stably maintained neuronal mitochondrial load through animal adulthood, rather than the dynamic triphasic pattern, is associated with extended longevity and functionality of the nervous system. Thus, our study serves as a foundation to elaborate the specific interactions of these genetic pathways, mitochondrial activity, and longevity.
Why do neurons increase mitochondrial size and density toward middle age? The early adulthood-associated increase may be a compensatory neuronal response to higher energy consumption and/or lower energy supply to support ATP-dependent functions required in reproduction and maintenance (Rolland et al., 2013). The latter is supported by previous findings that C. elegans exhibit a progressively steady decline in whole-body ATP concentration (Brys et al., 2010) despite having bigger mitochondria in body wall muscles at midlife compared to early adulthood (Yasuda et al., 2006). Mitochondrial size has a direct consequence on neuron activity; for example, bigger mitochondrial volume at synapses correlates with higher levels of synaptic vesicle exocytosis (Ivannikov et al., 2013). Increasing mitochondrial size and density can thus serve as a response mechanism to higher energy demands in distal neuronal processes and/or against aging-related mitochondrial stress and reduced mitochondrial respiration activity (Tondera et al., 2009; Rolland et al., 2013). Age-related phasic changes in mitochondrial size likely occur through regulating mitochondrial fusion/fission dynamics. Indeed, the perturbation of fusion genes fzo-1 and eat-3 effectively abolishes early adulthood-associated increase in mitochondrial size (Fig. 2), thus highlighting a crucial role of the fusion process in lifelong mitochondrial maintenance. The pathological relevance of altered mitochondrial size was also illustrated by the fact that mutations in genes regulating mitochondrial fission/fusion have been linked to neurodegenerative diseases (Deng et al., 2008; Chen and Dorn, 2013; DuBoff et al., 2013). We hypothesize that mitochondrial content and distribution in a neuron is achieved by a complex interplay of mitochondrial biogenesis, trafficking, fusion/fission, and degradation; each of these processes is likely to be affected by aging, quite possibly to degrees that vary among animals. Our findings establish the foundation for future investigations into determination of relative impact of each of these factors/pathways and disease- and aging-relevant changes in regulation of neuronal mitochondrial morphology and density.
Unlike long-lived daf-2 animals, short-lived daf-16(mu86) mutants did not show significant deviation from WT controls in age-related progressive decline of mitochondrial maintenance. This could be due to the fact that reduction of daf-2 function has a much more dramatic effect on life span (100% increase) than loss of daf-16 function (reduces by 15–20%). As such, it is possible that the effect of marginal reduction of life span on mitochondrial load in daf-16 ALM neurons is not readily detectable in our experimental conditions. Consistently, previous studies of the morphological aging of the same touch neurons showed that daf-16 holds little impact on age-related restructuring, whereas daf-2 confers demonstrative effects (Toth et al., 2012; Scerbak et al., 2014). Alternatively, lack of significant mitochondrial phenotype in daf-16 may indicate that other transcription factors downstream of daf-2 influence neuronal mitochondrial maintenance during adulthood.
Understanding how mitochondrial trafficking changes in response to physiological aging in WT and long-lived animals will advance our knowledge of cellular mechanisms that maintain the aging nervous system. Given the dynamic nature of neuronal activity and extremely long morphological structures between the soma and synapses, neurons face exceptional challenges in maintaining energy homeostasis throughout animal life span. We hypothesize that increasing mitochondrial load (or density) in the distal neuronal processes during young adulthood (1DA–8DA; Fig. 1C, Table 1) might be in part contributed by much faster decline of retrograde relative to anterograde mitochondrial trafficking, thus shifting the bidirectional motility balance toward accumulating mitochondria in distal ALM neurites. However, average mitochondrial run length in distal ALM processes is relatively short (Fig. 5C); thus it is possible that these trafficking events represent local reshuffling of mitochondrial material rather than net mitochondrial flux to and from the cell body. Long-lived daf-2 and eat-2 mutants exhibit much better maintenance of mitochondrial trafficking during aging compared to WTs; overall, retrograde motile frequency and run length did not show a significant decline between 1DA and 11DA ages for either daf-2 or eat-2 mutants. It is possible that these animals do exhibit age-related decline in mitochondrial trafficking, but it occurs at a much slower rate than that registered as significant within the 1DA–11DA period. The difference in age-related mitochondrial trafficking rates between WT and long-lived mutants (Fig. 5) may contribute to constant mitochondrial density in daf-2 and eat-2 distal processes throughout adulthood (Fig. 3B,D). Mitochondrial transport and distribution in mammalian neurons are in part influenced by synaptic activity (MacAskill et al., 2010; Sheng, 2014). We should note that the segments of neuronal processes imaged (Figs. 1–5) do not have synaptic connections. Thus, local synaptic activity may not be a significant contributor to the observed age-related changes in mitochondrial motility and density in main ALM process of C. elegans. Defective mitochondrial transport has been linked to the pathogenesis of several age-associated neurodegenerative diseases (Morfini et al., 2009; Court and Coleman, 2012; Sheng and Cai, 2012). Our study provides in vivo evidence that maintaining mitochondrial trafficking may be necessary for protecting against neuronal aging.
The age-associated pattern in mitochondrial stress resistance (Fig. 6F) correlates with the three phasic stages of neuronal mitochondrial size (Fig. 1C). Mitochondrial size increase by fusion has been reported as a protective mechanism against various physiological and pathological stresses (Chen et al., 2007; Tondera et al., 2009; Ghose et al., 2013; Shutt and McBride, 2013). Mitochondrial fusion protein EAT-3 (homolog of mammalian Opa1) was shown to be essential for C. elegans resistance to oxidative stress by paraquot (Kanazawa et al., 2008). Increased resistance of middle-aged mitochondria to acute oxidative stress could be due to their relatively larger size, as they may contain more antioxidant enzymes and have more anti–reactive oxygen species capacity. Although we are unable to measure neuronal antioxidant levels in vivo, whole-body transcripts of the major antioxidant enzyme catalase-1 reach maximum levels during the 2DA–5DA period (approximately four times higher than at 1DA; Lund et al., 2002). Similarly, whole-body protein levels of mitochondria-specific scavenging enzyme Mn-SOD double from young adult to middle adulthood, but decline toward the end of life to levels comparable of young adult (Yasuda et al., 2006). Our results thus illustrate the intriguing findings that elongated neuronal mitochondria in middle-aged WT animals are more resistant to acute oxidative stress than smaller mitochondria in young or old animals.
In summary, we have taken advantage of the short life span and transparent nature of C. elegans models to conduct in vivo quantitative analysis of mitochondrial maintenance during organismal aging at the single neuron level. We show that neuronal mitochondria undergo nonprogressive changes in size and density, as well as progressive decline in transport frequency and distance, throughout animal adulthood. The size and total axonal load of mitochondria can be described in three distinct stages: increase of mitochondrial content in young adulthood, maintenance of high load during midlife, and fast decline in late adulthood. These changes correlate with mitochondrial stress resistance during the aging of the organism. Genetic manipulations that extend life span altered the rate and shape of the age-related mitochondrial changes in size, load, and oxidative stress resistance. Stably maintained trafficking rates and mitochondrial load in neurons through animal adulthood, rather than a dynamic triphasic pattern, is associated with extended longevity and functionality of the nervous system. Thus, our study characterizes mitochondrial aging profile at the level of a single neuron in its native environment and provides insights into fundamental phenomena relevant to age-associated neurodegenerative diseases. We hypothesize that mitochondrial content and distribution in a neuron is achieved by a complex interplay of mitochondrial biogenesis, trafficking, fusion/fission, and degradation; each of these processes is likely to be affected by aging. Our findings build the essential foundation for future determining the relative impact of each of these factors/pathways in regulating neuronal mitochondrial maintenance in healthy aging- and disease-relevant conditions.
Footnotes
This work was supported by the Intramural Research Program of NINDS through NIH Grants NS003029 and NS002946 (Z.-H.S.), R01AG046358 (M.D.), and OD010943 (D.H.H.). We thank S. Koushika (National Centre for Biological Sciences (NCBS), Bangalore, India) for sharing the Pmec-7::mitoGFP strain and C. Rongo (Rutgers University, Piscataway, NJ) for sharing the Pglr-1::mitoROGFP2 strain. We thank L. Herndon, A. Jevince, and G. Stephney for technical help with TEM, N. R. Hall for help with statistics, and H. Ushakov for some transgenic constructions. We also thank members of the Sheng lab for constructive discussion and Marina Nogueira (NIH summer intern) for assistance in animal maintenance and imaging. Some strains were provided by the Caenorhabditis Genetics Center, funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
The authors declare no competing financial interests.
References
- Bansal A, Zhu LJ, Yen K, Tissenbaum HA. Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc Natl Acad Sci U S A. 2015;112:E277–E286. doi: 10.1073/pnas.1412192112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brys K, Castelein N, Matthijssens F, Vanfleteren JR, Braeckman BP. Disruption of insulin signalling preserves bioenergetic competence of mitochondria in ageing Caenorhabditis elegans. BMC Biol. 2010;8:91. doi: 10.1186/1741-7007-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calixto A, Chelur D, Topalidou I, Chen X, Chalfie M. Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods. 2010;7:554–559. doi: 10.1038/nmeth.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sheng ZH. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol. 2013;202:351–364. doi: 10.1083/jcb.201302040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YM, Gerwin C, Sheng ZH. Dynein light chain LC8 regulates syntaphilin-mediated mitochondrial docking in axons. J Neurosci. 2009;29:9429–9438. doi: 10.1523/JNEUROSCI.1472-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JJ, Huang C, Hughes S, Kornfeld K. The measurement and analysis of age-related changes in Caenorhabditis elegans. WormBook Jan. 2008;24:1–21. doi: 10.1895/wormbook.1.137.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Coleman MP. Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 2012;35:364–372. doi: 10.1016/j.tins.2012.04.001. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Giesbergen PC, de Rijk MC, Hofman A, Koudstaal PJ, Breteler MM. Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology. 2004;63:1240–1244. doi: 10.1212/01.WNL.0000140706.52798.BE. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBoff B, Feany M, Götz J. Why size matters - balancing mitochondrial dynamics in Alzheimer's disease. Trends Neurosci. 2013;36:325–335. doi: 10.1016/j.tins.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum Mol Genet. 2012;21:3587–3603. doi: 10.1093/hmg/dds190. [DOI] [PubMed] [Google Scholar]
- Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics. 2002;161:1101–1112. doi: 10.1093/genetics/161.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstbrein B, Stamatas G, Kollias N, Driscoll M. In vivo spectrofluorimetry reveals endogenous biomarkers that report healthspan and dietary restriction in Caenorhabditis elegans. Aging Cell. 2005;4:127–137. doi: 10.1111/j.1474-9726.2005.00153.x. [DOI] [PubMed] [Google Scholar]
- Ghose P, Park EC, Tabakin A, Salazar-Vasquez N, Rongo C. Anoxia-reoxygenation regulates mitochondrial dynamics through the hypoxia response pathway, SKN-1/Nrf, and stomatin-like protein STL-1/SLP-2. PLoS Genet. 2013;9:e1004063. doi: 10.1371/journal.pgen.1004063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MB. Mechanosensation. WormBook Jan. 2006;6:1–14. doi: 10.1895/wormbook.1.62.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH. Electron microscopy and three-dimensional image reconstruction. Methods Cell Biol. 1995;48:395–436. doi: 10.1016/S0091-679X(08)61397-7. [DOI] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13:1385–1393. [PubMed] [Google Scholar]
- Honda Y, Honda S. Life span extensions associated with upregulation of gene expression of antioxidant enzymes in Caenorhabditis elegans; studies of mutation in the age-1, PI3 kinase homologue and short-term exposure to hyperoxia. J Am Aging Assoc. 2002;25:21–28. doi: 10.1007/s11357-002-0003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houthoofd K, Fidalgo MA, Hoogewijs D, Braeckman BP, Lenaerts I, Brys K, Matthijssens F, De Vreese A, Van Eygen S, Muñoz MJ, Vanfleteren JR. Metabolism, physiology and stress defense in three aging Ins/IGF-1 mutants of the nematode Caenorhabditis elegans. Aging Cell. 2005;4:87–95. doi: 10.1111/j.1474-9726.2005.00150.x. [DOI] [PubMed] [Google Scholar]
- Huang C, Xiong C, Kornfeld K. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2004;101:8084–8089. doi: 10.1073/pnas.0400848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivannikov MV, Sugimori M, Llinás RR. Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J Mol Neurosci. 2013;49:223–230. doi: 10.1007/s12031-012-9848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa T, Zappaterra MD, Hasegawa A, Wright AP, Newman-Smith ED, Buttle KF, McDonald K, Mannella CA, van der Bliek AM. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. A conserved regulatory system for aging. Cell. 2001;105:165–168. doi: 10.1016/S0092-8674(01)00306-3. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Gao B, Lee SM, Bennett K, Fang D. RLE-1, an E3 ubiquitin ligase, regulates C. elegans aging by catalyzing DAF-16 polyubiquitination. Dev Cell. 2007;12:235–246. doi: 10.1016/j.devcel.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Lund J, Tedesco P, Duke K, Wang J, Kim SK, Johnson TE. Transcriptional profile of aging in C. elegans. Curr Biol. 2002;12:1566–1573. doi: 10.1016/S0960-9822(02)01146-6. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Atkin TA, Kittler JT. Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. Eur J Neurosci. 2010;32:231–240. doi: 10.1111/j.1460-9568.2010.07345.x. [DOI] [PubMed] [Google Scholar]
- McElwee J, Bubb K, Thomas JH. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell. 2003;2:111–121. doi: 10.1046/j.1474-9728.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Mondal S, Ahlawat S, Rau K, Venkataraman V, Koushika SP. Imaging in vivo neuronal transport in genetic model organisms using microfluidic devices. Traffic. 2011;12:372–385. doi: 10.1111/j.1600-0854.2010.01157.x. [DOI] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22:R741–R752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Pan CL, Peng CY, Chen CH, McIntire S. Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc Natl Acad Sci U S A. 2011;108:9274–9279. doi: 10.1073/pnas.1011711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland SG, Motori E, Memar N, Hench J, Frank S, Winklhofer KF, Conradt B. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proc Natl Acad Sci U S A. 2013;110:E2967–E2976. doi: 10.1073/pnas.1303872110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scerbak C, Vayndorf EM, Parker JA, Neri C, Driscoll M, Taylor BE. Insulin signaling in the aging of healthy and proteotoxically stressed mechanosensory neurons. Front Genet. 2014;5:212. doi: 10.3389/fgene.2014.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol. 2014;204:1087–1098. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrg3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt TE, McBride HM. Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta. 2013;1833:417–424. doi: 10.1016/j.bbamcr.2012.05.024. [DOI] [PubMed] [Google Scholar]
- Tank EM, Rodgers KE, Kenyon C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J Neurosci. 2011;31:9279–9288. doi: 10.1523/JNEUROSCI.6606-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M, Bartke A, Antebi A. The Endocrine Regulation of Aging by Insulin-Like Signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth ML, Melentijevic I, Shah L, Bhatia A, Lu K, Talwar A, Naji H, Ibanez-Ventoso C, Ghose P, Jevince A, Xue J, Herndon LA, Bhanot G, Rongo C, Hall DH, Driscoll M. Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J Neurosci. 2012;32:8778–8790. doi: 10.1523/JNEUROSCI.1494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanfleteren JR, De Vreese A. The gerontogenes age-1 and daf-2 determine metabolic rate potential in aging Caenorhabditis elegans. FASEB J. 1995;9:1355–1361. doi: 10.1096/fasebj.9.13.7557026. [DOI] [PubMed] [Google Scholar]
- Viswanathan M, Guarente L. Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature. 2011;477:E1–E2. doi: 10.1038/nature10440. [DOI] [PubMed] [Google Scholar]
- Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science. 2000;290:147–150. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- Yanase S, Yasuda K, Ishii N. Adaptive responses to oxidative damage in three mutants of Caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech Ageing Dev. 2002;123:1579–1587. doi: 10.1016/S0047-6374(02)00093-3. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- Yasuda K, Ishii T, Suda H, Akatsuka A, Hartman PS, Goto S, Miyazawa M, Ishii N. Age-related changes of mitochondrial structure and function in Caenorhabditis elegans. Mech Ageing Dev. 2006;127:763–770. doi: 10.1016/j.mad.2006.07.002. [DOI] [PubMed] [Google Scholar]