

Abstract
Fabry disease is a rare, progressive X-linked inborn error of the glycosphingolipid metabolic pathway. Mutations of the GLA gene result in deficiency of the lysosomal enzyme, α-galactosidase A (α-Gal A) with accumulation of glycosphingolipids, particularly globotriaosylceramide (GL3) in the vascular endothelium of various tissues. Accumulation of GL3 eventually leads to life threatening renal, cardiac and cerebrovascular complications typically in the third to fifth decades of life. The first signs and symptoms of classic Fabry disease however appear in childhood but diagnosis is often delayed. The symptoms most commonly experienced in childhood include neuropathic pain, gastrointestinal dysfunction, hyperhidrosis and heat intolerance. Timely diagnosis is important as early treatment with enzyme replacement therapy reduces GL3 accumulation, can stabilize disease progression and potentially prevent irreversible organ damage. Physicians should be familiar with the signs and symptoms of Fabry disease in childhood and be particularly vigilant for unusual or non-specific but recurrent or episodic symptoms.
Keywords: Fabry disease, paediatric, α-galactosidase, acroparaesthesia
Introduction
Fabry disease is a rare multisystem, X-linked lysosomal storage disorder of glycosphingolipid metabolism due to complete or partial deficiency of the lysosomal enzyme, α-galactosidase A (α-Gal A) caused by mutations in the GLA gene. As a result there is progressive accumulation of glycosphingolipids, particularly globotriaosylceramide (GL3), within lysosomes of a wide variety of cells throughout the body, including endothelial, renal and cardiac and dorsal root ganglion neuronal cells. Fabry disease can be caused by a variety of missense or nonsense point mutations, deletions or insertions within the GLA gene located on Xq22. More than 600 pathogenic mutations have been identified, most of which are private mutations, occurring in individual families. Disease severity is inversely related to α-Gal A activity. Mutations associated with complete loss of enzyme function result in the classic phenotype. Glycosphingolipid storage results in a cascade of structural and functional cellular changes with disruption of metabolic processes progressing to cell death, and irreversible major organ dysfunction (1).
The first signs of classic Fabry disease usually appear in childhood. The characteristic early clinical features of Fabry disease include acroparaesthesia, angiokeratoma, heat intolerance, cornea verticillata and gastrointestinal symptoms. Later complications occur with disease progression and include progressive renal failure, hypertrophic cardiomyopathy, cerebrovascular disease and reduced life expectancy. Although Fabry disease is X-linked, female heterozygotes can exhibit the same severity of symptoms but are usually more variable and tend to occur later than males and some females remain asymptomatic. Heterozygous females have α-Gal A levels than can range from deficient to normal levels. This thought to be due to X chromosome inactivation or Lyonisation.
Diagnosis of Fabry disease is often delayed for several years after the onset of symptoms and signs. The early symptoms are often non-specific and there is limited awareness of the disorder among clinicians. Early initiation of treatment with enzyme replacement therapy is effective in alleviating many of the signs and symptoms of Fabry disease, stabilise and even reverse disease progression.
Epidemiology
Fabry disease is found among all ethnic, racial, and demographic groups. In males, the estimated incidence is 1:50,000 and recent population estimates range from 1:40,000 to 1:117,000, although milder forms of the disease that present later in life may be more common (2). With increased disease awareness and screening programs it is likely that the prevalence may be higher. Newborn screening programmes in the United States have detected a high rate of later onset cases with incidences ranging from approximately 1:3,000 to 1:7,800 males. In Taiwan the frequency in newborn males may be as high as 1:1,500 (3).
Clinical manifestations
Fabry disease is a progressive multisystem disorder. The early symptoms and signs of classical Fabry disease usually present in childhood but the primary disease process starts during early fetal development. There is histopathological evidence of GL3 accumulation in renal, liver and myenteric plexus cells in affected males fetuses. Corneal whorls have been detected in a 22-week male fetus (4-6).
Males experience symptoms at an earlier age and at a higher prevalence than females. Review of the Fabry Registry data showed the median age of symptom onset in males was 6 and 9 years in females. The first clinical presentation at a median age of 7 years in males was episodic neuropathic pain presenting with burning pain of the hands and feet known as acroparaesthesia occurring in 59% of males and 41% of females at a median age of 9 years. Recurrent gastrointestinal symptoms were the second most common feature in 27% with median age of onset in males of 5 and 9.5 years in females. Hypohidrosis and heat intolerance are also common in the early stage of the disease (7). Although not life-threatening these symptoms impact on the health, quality of life and function of affected children.
The most serious complications of Fabry disease emerge in adulthood and include end stage renal failure, cerebrovascular events such as strokes, and progressive cardiomyopathy, arrhythmias and valvular disease and premature death. Despite being X-linked, Fabry disease affects females. Symptoms in female heterozygotes may be more variable, but can be of the same severity as in males (3,8). There is a wide range of inter and intrafamilial variability of clinical features.
Neurological symptoms
Neuropathic pain experienced by young patients with Fabry disease manifests as chronic acroparaesthesia superimposed by acute attacks of Fabry pain crisis. These symptoms reflect damage to small fibers in the peripheral and autonomic nervous systems as a result of accumulation of glycosphingolipids (9). Acroparaesthesia is often described by patients as a chronic, nagging, tingling, burning sensation in the hands and feet whilst a pain crisis is an episode of acute, agonizing pain, beginning in the extremities and radiating proximally. The pain is typically precipitated by rapid changes in core body temperature due to fever, illness, stress or exercise, sudden exposure to cold, changes in humidity or fatigue. Acroparaesthesia has been reported in children as young as 2 years (3).
Fabry pain crises may also manifest as acute abdominal pain. Other symptoms related to dysfunction of the autonomic nervous system observed in Fabry patients include hypo- or anhidrosis, temperature and exercise intolerance and impaired gastrointestinal motility (10).
Early cerebral involvement has been reported in an asymptomatic 8-year-old boy with white matter lesions demonstrated by brain magnetic resonance imaging (11).
Dermatological signs
Angiokeratoma appearing as small, raised, non-blanching dark-red spots are the hallmark of Fabry disease although not specific for the disorder. Angiokeratoma can be idiopathic or associated with other disorders. Diffuse angiokeratoma are typically located in the lower trunk and genital region, but may also be present on the palms, around the mouth, lips and umbilicus (2).
Abnormal autonomic nerve function and infiltration of the sweat glands is thought to result in hyperhidrosis, heat and exercise intolerance. In high temperatures patients may experience flushing, onset of neuropathic pain, headache, fatigue or heat stroke whilst in the cold they may experience pain or numbness in the extremities.
Ocular signs
Vortex keratopathy or cornea verticillata is the most frequent ocular sign in Fabry disease occurring in over 70% of males and females including children making ophthalmological examination a useful tool for early diagnosis of Fabry disease (11). It is very rare in individuals without Fabry disease. The presence of cornea verticillata has been reported in a 6-month old child and in a fetus (12). Cornea verticillata are golden brown or gray, usually bilateral, opacities that branch out from a central whorl across the inferior cornea.
Retinal vessel tortuosity and cataracts have also been reported and occur more frequently in males. The presence of vascular tortuosity has been correlated with disease severity in children and may represent a more severe phenotype (2). Ophthalmological signs may be present in children before the onset of other signs or symptoms and do not usually result in visual impairment.
Gastrointestinal symptoms
Abdominal pain, episodic nausea and vomiting, abdominal bloating, and alternating constipation and diarrhea have been reported in the paediatric Fabry population. These symptoms may be related to gastrointestinal dysmotility due to autonomic dysfunction. Gastrointestinal symptoms of altered bowel actions and abdominal pain have been reported in up to 60% of children less than 10 years of age (13).
Cardiac
The myocardium, conduction system and valves may be affected in Fabry disease. Cardiac complications become apparent in childhood with slow progression. Left ventricular hypertrophy is the most frequent cardiac manifestation with onset reported in childhood and is a major cause of morbidity and early mortality in adults. Left ventricular mass indexed to height in 20 paediatric patients was found to be above the 75th percentile of normal controls, with reduced heart rate variability. With disease progression other cardiac complications in adults include atrial fibrillation, non-sustained ventricular tachycardia, QRS broadening, short PR interval, atrioventricular block and sinus node dysfunction. In addition some patients develop mild to moderate valvular insufficiency, hypertension and coronary artery disease (14).
Renal
Renal deposition of globotriaosylceramide starts early in childhood and precedes overt clinical renal disease. Increasing albuminuria and proteinuria in children with Fabry disease is a marker of progressive vascular damage. Although more common in adults, renal disease has been reported in children. Renal failure has been reported as early as 16 years. Light and electron microscopy studies of renal biopsies in 9 symptomatic males and females aged 7 to 18 years showed podocyte inclusions, segmental foot process effacement and distal tubular inclusion in all patients with normal glomerular filtration and albuminuria in 5, confirming that deposition of globotriaosylceramide starts in early childhood before overt renal disease (15).
Other symptoms
Hearing impairment and tinnitus have also been reported in children with Fabry disease, being present in 55% of females and 39% of males from data from the Fabry Outcome Survey (13).
Diagnosis
In males, confirmation of a clinical diagnosis requires demonstration of deficient α-Gal A activity in leukocytes or dried blood spots and increased globotriaosylceramide levels in plasma or urinary sediment. In females heterozygotes α-Gal A activity and globotriaosylceramide levels may be within the normal range. Diagnostic confirmation requires identification of a mutation in the GLA gene. Confirmation of Fabry disease in an individual should lead to screening of other at risk family members and appropriate genetic counseling.
Our understanding of Fabry disease is increasing, yet misdiagnosis and diagnostic delay frequently occur (12). Contributing factors include the rarity of the disease, the non-specific, subjective nature of the early symptoms and the lack of awareness amongst clinicians. Patients are often seen by a number of specialists in different clinical fields before the diagnosis is confirmed, diagnostic delays of several years have been reported, by which time the disease may be advanced.
Importance of early diagnosis
Lack of consideration of Fabry disease as a possibility, particularly in young patients, is one of the major challenges to making a correct and timely diagnosis. Children may first present with non-specific or unusual symptoms, which, in the absence of previously diagnosed family members, are often not recognized as being associated with Fabry disease.
Recent research has shown increased psychosocial impact amongst patients whose severe clinical symptoms, such as acroparaesthesia, fatigue and heat intolerance, were either dismissed or misdiagnosed (16). Early diagnosis of Fabry disease is crucial to patient health and well-being and may help to avoid unnecessary interventions and anxiety.
Timelier diagnosis may be aided by increased awareness of this disorder and the signs and symptoms that may present in early childhood.
Table 1 provides a summary of symptoms and signs in childhood. Physicians should be particularly vigilant for persistent or recurrent symptoms. If a paediatric patient, male or female, presents with any of these symptoms, Fabry disease should be considered in the differential diagnosis.
Table 1. The diagnosis of Fabry disease should be considered in children presenting with one or more of the following signs and symptoms.
| Neurological |
| Acroparaesthesia |
| Neuropathic pain |
| Persistent “growing pains” |
| Recurrent unexplained headaches |
| Gastrointestinal tract |
| Episodic non-inflammatory/non-infectious diarrhoea |
| Unexplained recurrent abdominal pain/discomfort and or vomiting |
| Unexplained constipation |
| Ophthalmological |
| Vortex keratopathy (see Figure 1) |
| Cataract |
| Conjunctival vessel tortuosity |
| Dermatological |
| Angiokeratoma |
| Reduced sweating |
| Heat intolerance |
| Renal |
| Unexplained microalbuminuria/proteinuria |
| Cardiac |
| Left ventricular hypertrophy |
| Musculoskeletal |
| Exercise intolerance |
| Fatigue |
| Other |
| Recurrent unexplained fever |
| Depression |
Figure 1.
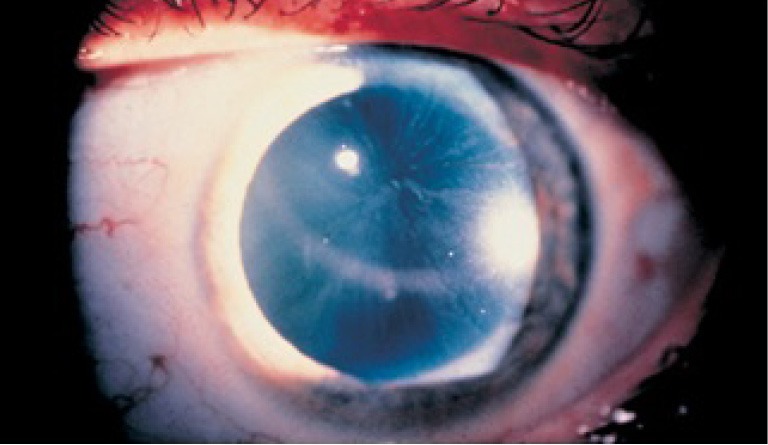
Cornea verticillata, the most frequent ocular abnormality in Fabry disease are fine, golden brown or gray, usually bilateral, opacities that branch out from a central whorl across the inferior cornea.
Early diagnosis can be significantly enhanced by taking a detailed medical and family history with emphasis on gaining insight into some of the non-specific presenting symptoms. Family history regarding renal, cardiac and neurological disease and early deaths is important when Fabry disease is suspected. Difficulties often arise in making a diagnosis when no male index case is present. Early referral to Genetic Metabolic Physician is important to circumvent the need for further unnecessary evaluations, and aid in determining a definitive diagnosis of Fabry disease. Genetic counseling and cascade testing can then be offered to at risk family members.
Management
Management of Fabry disease requires a multidisciplinary team approach often coordinated by a Genetic Metabolic Physician. Effective management involves both symptom management and the management of the disease itself. Neuropathic pain may respond to analgesics or anticonvulsants such as carbamazepine, gabapentin and pregabalin. Factors that are identified to trigger acute episodes of pain such as extremes of heat, physical exertion should be avoided. Gastrointestinal symptoms may require a change in diet such as small, frequent meals, and effective control of constipation.
Treatment
Specific treatment with intravenous enzyme replacement therapy is available for Fabry disease. Enzyme replacement therapy reduces levels of GL3 in plasma, urine and tissues and can reduce symptom severity, improve pain-related quality of life, gastrointestinal symptoms and slow disease progression (17). The overall goal of enzyme replacement therapy is to prevent further disease progression and, potentially, reverse underlying pathologic abnormalities. However, delayed diagnosis may lead to irreversible organ damage and reduced treatment efficacy (18). A definitive diagnosis ensures patients have early access to optimal monitoring, supportive management and appropriate treatment in order to prevent irreversible life threatening complications.
Fabry disease is a progressive disorder; in younger patients, the use of enzyme replacement therapy therefore focuses on disease prevention where it has the potential to provide greater long-term benefits than in adults. Progressive podocyte injury has been associated with the development of renal complications in children with Fabry disease (19). Recent data demonstrate a significant correlation between dose of enzyme replacement therapy and reduction in globotriaoscylceramide podocyte inclusions after long-term treatment in young patients (20). The same study showed complete clearance in mesangial and glomerular endothelial cells in all patients after 5 years of enzyme replacement therapy, irrespective of dose.
Clinical trials with up to 4 years follow-up have established that the use of enzyme replacement therapy in children is well-tolerated, reduces pain and improves pain-related quality of life (19-23). However, due to its high cost, the timing of access to enzyme replacement therapy continues to be guided predominantly by country-specific treatment criteria. In Australia there are eligibility criteria for males and females, with treatment initiated only after demonstration of the onset of clinically significant signs or symptoms. Earlier access, before irreversible organ dysfunction, should be considered. However, longer-term follow-up studies are needed to define when initiation of enzyme replacement therapy during childhood can prevent future organ damage (18).
Conclusions
Fabry disease is rare and presents with diverse symptoms in childhood and diagnosis is often delayed. Physicians should be familiar with early signs and symptoms of Fabry disease, which should be considered in the differential diagnosis when a young patient presents with one or more unusual or non-specific but recurrent or episodic symptoms, particularly unexplained neuropathic pain, gastrointestinal symptoms, exercise intolerance, fatigue and hypohidrosis. Early recognition of Fabry related symptoms and initiation of enzyme replacement therapy prior to irreversible organ damage is crucial.
Acknowledgements
None.
Editor’s note: “Rare Diseases Column” is chaired by Dr. Zhanhe Wu from The Children’s Hospital at Westmead, Australia, featuring articles related to rare diseases mostly genetic based, presented in early life disease, with chronic phase but frequently progressive, disabling and life threatening diseases. Article types of original articles, review articles, case reports, perspectives, etc. are welcomed to be submitted to the column.
Footnotes
Conflicts of Interest: The author has no conflicts of interest to declare.
References
- 1.El-Abassi R, Singhal D, England JD. Fabry's disease. J Neurol Sci 2014;344:5-19. [DOI] [PubMed] [Google Scholar]
- 2.Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM 2010;103:641-59. [DOI] [PubMed] [Google Scholar]
- 3.Laney DA, Peck DS, Atherton AM, et al. Fabry disease in infancy and early childhood: a systematic literature review. Genet Med 2015;17:323-30. [DOI] [PubMed] [Google Scholar]
- 4.Desnick RJ, Raman MK, Bendel RP, et al. Prenatal diagnosis of glycosphingolipidoses: Sandhoff’s and Fabry’s disease. J Pediatr 1973;83:149-50. [Google Scholar]
- 5.Vedder AC, Strijland A, vd Bergh Weerman MA, et al. Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis 2006;29:106-11. [DOI] [PubMed] [Google Scholar]
- 6.Tsutsumi A, Uchida Y, Kanai T, et al. Corneal findings in a fetus with Fabry’s disease. Acta Ophthalmol (Copenh) 1984;62:923-31. [DOI] [PubMed] [Google Scholar]
- 7.Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr Res 2008;64:550-5. [DOI] [PubMed] [Google Scholar]
- 8.Alfadhel M, Sirrs S. Enzyme replacement therapy for Fabry disease: some answers but more questions. Ther Clin Risk Manag 2011;7:69-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burlina AP, Sims KB, Politei JM, et al. Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. BMC Neurol 2011;11:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolodny EH, Pastores GM. Anderson-Fabry disease: extrarenal, neurologic manifestations. J Am Soc Nephrol 2002;13 Suppl 2:S150-3. [PubMed] [Google Scholar]
- 11.Cabrera-Salazar MA, O'Rourke E, Charria-Ortiz G, et al. Radiological evidence of early cerebral microvascular disease in young children with Fabry disease. J Pediatr 2005;147:102-5. [DOI] [PubMed] [Google Scholar]
- 12.Sodi A, Ioannidis AS, Mehta A, et al. Ocular manifestations of Fabry's disease: data from the Fabry Outcome Survey. Br J Ophthalmol 2007;91:210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr 2006;95:86-92. [DOI] [PubMed] [Google Scholar]
- 14.Kampmann C, Weithoff CM, Whybra C, et al. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr 2008;97:463-9. [DOI] [PubMed] [Google Scholar]
- 15.Tøndel C, Bostad L, Hirth A, et al. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis 2008;51:767-76. [DOI] [PubMed] [Google Scholar]
- 16.Bouwman MG, de Ru MH, Linthorst GE, et al. Fabry patients' experiences with the timing of diagnosis relevant for the discussion on newborn screening. Mol Genet Metab 2013;109:201-7. [DOI] [PubMed] [Google Scholar]
- 17.Alegra T, Vairo F, de Souza MV, et al. Enzyme replacement therapy for Fabry disease: A systematic review and meta-analysis. Genet Mol Biol 2012;35:947-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007;146:77-86. [DOI] [PubMed] [Google Scholar]
- 19.Najafian B, Mauer M, Hopkin RJ, et al. Renal complications of Fabry disease in children. Pediatr Nephrol 2013;28:679-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tøndel C, Bostad L, Larsen KK, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol 2013;24:137-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borgwardt L, Feldt-Rasmussen U, Rasmussen AK, et al. Fabry disease in children: agalsidase-beta enzyme replacement therapy. Clin Genet 2013;83:432-8. [DOI] [PubMed] [Google Scholar]
- 22.Ramaswami U. Update on role of agalsidase alfa in management of Fabry disease. Drug Des Devel Ther 2011;5:155-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramaswami U, Parini R, Pintos-Morell G, et al. Fabry disease in children and response to enzyme replacement therapy: results from the Fabry Outcome Survey. Clin Genet 2012;81:485-90. [DOI] [PubMed] [Google Scholar]