

Abstract
Growing evidence links abnormal epigenetic control to the development of hematological malignancies. Accordingly, inhibition of epigenetic regulators is emerging as a promising therapeutic strategy. The acetylation status of lysine residues in histone tails is one of a number of epigenetic post-translational modifications that alter DNA-templated processes, such as transcription, to facilitate malignant transformation. Although histone deacetylases are already being clinically targeted, the role of histone lysine acetyltransferases (KAT) in malignancy is less well characterized. We chose to study this question in the context of acute myeloid leukemia (AML), where, using in vitro and in vivo genetic ablation and knockdown experiments in murine models, we demonstrate a role for the epigenetic regulators CBP and p300 in the induction and maintenance of AML. Furthermore, using selective small molecule inhibitors of their lysine acetyltransferase activity, we validate CBP/p300 as therapeutic targets in vitro across a wide range of human AML subtypes. We proceed to show that growth retardation occurs through the induction of transcriptional changes that induce apoptosis and cell-cycle arrest in leukemia cells and finally demonstrate the efficacy of the KAT inhibitors in decreasing clonogenic growth of primary AML patient samples. Taken together, these data suggest that CBP/p300 are promising therapeutic targets across multiple subtypes in AML.
INTRODUCTION
Acute myeloid leukemia (AML) is an often fatal hematological malignancy1 characterized by abnormal transcriptional programs and driven by a plethora of heterogeneous mutations.2 A central and recurrent theme is mutation of epigenetic regulators.3 Among these are the transcriptional co-activators CREB (cyclic-AMP response element binding protein)-binding protein (CREBBP or KAT3A, hereafter referred to as CBP) and its paralogue EP300 (KAT3B, hereafter referred to as p300). CBP and p300 modulate locus-specific transcription via a number of separate mechanisms.4 These include direct lysine acetyltransferase (KAT) catalytic activity, where CBP and p300 can acetylate both histone and non-histone proteins,5 as well as through multiple protein–protein interactions between CBP or p300 and transcription factors, chromatin remodelling complexes and the basal transcriptional machinery.6 Cbp and p300 are required during development for the generation and function of normal hematopoietic stem cells7 and we have recently shown that Cbp is also required for adult hematopoietic stem cell maintenance and function.8 Recently, inactivating mutations in CBP and p300 have been described in a number of hematological malignancies9–11 and this, together with the description of germline mutations of CBP in the cancer predisposition syndrome Rubinstein-Taybi syndrome12 and of hematological malignancies in Cbp-deficient mice,8,13 identify CBP and p300 as tumor suppressor genes. However, in AML, there have been reports of rearrangements of both CBP and p300 with the MLL (mixed lineage leukemia) gene and of CBP with the MOZ (MYST3) gene, all leading to the generation of leukemogenic fusion proteins.14,15 Additionally, many oncogenes have been demonstrated to interact with CBP, including the oncogenic fusions MOZ-TIF2 and NUP98-HOXA9, MLL-AFX and TCF3-PBX.16–19 Furthermore, structure function analysis of these fusions has demonstrated that removal of the protein domains that interact with CBP abrogates transformation.16–17 These apparently counterintuitive data suggest that CBP and p300 may also function as oncogenes, as has been described for other tumor suppressor genes,20 and facilitate transformation in certain types of AML.
AML remains a disease with an overall dismal outlook, with over 70% of patients eventually succumbing to the disease.21 In addition, the mainstays of AML therapy are unchanged for the last 20 years.1 Therefore, novel therapeutics are urgently required to improve the long-term outlook in this aggressive disease. To this end, agents targeting epigenetic regulators, particularly those with catalytic function, such as DNA methyltransferases and histone deacetylase inhibitors, have recently been used with some success in AML and other hematological malignancies.22 In addition, we and others have recently targeted so-called ‘epigenetic readers’,3 through the inhibition of protein–protein interactions between acetylated histone tails and the BET protein family of transcriptional activators.23 However, the role of histone acetyltransferase activity has not been systematically investigated, nor targeted in AML. In this report, we support this strategy by demonstrating that Cbp and p300 are genetically required for efficient leukemogenesis. Moreover, we demonstrate that pharmacologically targeting the catalytic activity of the lysine acetyltransferases (KAT) CBP and p300 has pre-clinical efficacy in many subtypes of AML. This occurs via the induction of cell-cycle arrest and apoptosis, while sparing normal hematopoietic progenitors in similar assays. Mechanistically, cell-cycle arrest and apoptosis appear to be mediated through alteration of a transcriptional program associated with genomic integrity. Finally we demonstrate a significant decrement of clonogenic growth in AML patient samples following CBP/p300 KAT inhibition. Taken together, these data suggest targeting CBP/p300 activity as a promising clinical strategy in AML.
RESULTS
Cbp is required for efficient immortalization in vitro and induction and maintenance of AML in vivo
To assess the requirement for Cbp during transformation, we retrovirally transduced c-kit+ bone marrow (BM) cells from Cbpfl/fl; Mx1-Cre− mice (hereafter Cbp wt) or Cbpfl/fl;Mx1-Cre+ mice following administration of poly-I poly-C (pIpC) (hereafter Cbp−/−), with AML-associated oncogenes MOZ-TIF2 (MT2) or NUP98-HOXA9 (NHA9), both of which are known to interact with CBP. Transformation was assessed in standard serial replating and growth in liquid culture assays.24 No differences in colony numbers or growth were demonstrated between MT2 and NHA9 Cbp wt or Cbp−/− progenitor cells (Figure 1a), suggesting that in vitro immortalization by MT2 and NHA9 is not absolutely dependent on Cbp expression, and may proceed in its absence. We next examined whether Cbp is required for continued in vitro self-renewal in cell lines expressing MT2 and NHA9. Cbpfl/fl c-kit+ progenitor cells were first transduced with either MT2 or NHA9 and serially replated in methylcellulose. Similar cells expressing MLL-ENL (ME), a fully transforming fusion protein not documented to interact with CBP, were included as a control. Following the third round of plating, cells were transduced with pBabe-Cre-puro retrovirus to excise Cbp in vitro, or with a puro-empty vector as a control, before further serial plating under initial puromycin selection. Although transduction with Cre recombinase did not significantly alter the self-renewal potential in any cell line at the population level (Figure 1b and data not shown), genotyping of the cells revealed preferential growth by cells that lacked recombination and therefore still expressed Cbp, demonstrating its involvement in transformation. Importantly, by the third and fourth round of plating, almost all cells immortalized by NHA9 and MT2 demonstrated the non-recombined allele, suggesting a selective advantage for cells that retained Cbp (Figure 1c). In contrast, cells expressing ME demonstrated efficient and sustained excision of Cbp (Figure 1c and data not shown). Taken together, these strongly suggest that loss of Cbp may affect the self-renewal programs maintained by oncogenes that interact with it, including MT2 and NHA9, but not by those that do not interact with Cbp, as exemplified by ME.
Figure 1.

Cbp−/− cells are rapidly outcompeted by Cbp wt cells, under selective in vitro conditions, in MT2- and NHA9-driven AML. (a) Serial replating assays of MT2- and NHA9-driven leukemias demonstrate no difference in colony number or serial replating activity between transduced Cbp wt and Cbp−/− progenitor cells. (b) Similar replating assays demonstrate no differences in the in vitro self-renewal potential of MT2 and NHA9 AML murine cell lines generated from Cbpfl/fl progenitors following expression of either Cre-puro or an empty puro vector, as both cell lines retained serial replating potential post-Cbp excision. (c) Genotyping of pooled colonies at the end of each round of replating revealed serial re-emergence of the un-excised Cbp allele, in the NHA9 and MT2, but not in the ME immortalized murine cell lines. *P < 0.05.
We next assessed the requirement for Cbp during the initiation and maintenance of leukemia in vivo, using the short latency MT2 AML model.16,24 To test the requirement for Cbp during leukemia initiation, c-kit+ BM cells from previously pIpC-treated Cbp wt or Cbp−/− mice were transduced with MSCV-MT2-IRES-GFP (Figure 2a). The transduction efficiency between Cbp wt and Cbp−/− cells (as measured by the percentage of GFP+ cells) was similar and genotyping of the transduced cells revealed excision of the Cbp allele (Figure 2a) before transplantation. All MT2 mice succumbed to disease within 2–4 months after transplantation, with a similar macroscopic and histological AML phenotype (Figure 2a; Supplementary Figure 1). However, similarly to the findings in vitro, genotyping from various heavily infiltrated tissues of the leukemic mice from the Cbp−/− group demonstrated that the majority of cells actually retained the non-recombined Cbpfl/fl allele and therefore expressed Cbp. Indeed, when these populations were enriched for leukemic cells, by sorting for GFP, all cells lacked recombination at the Cbp allele (Figure 2a). Thus, in agreement with our in vitro studies, there is a significant growth advantage during leukemia induction for cells that continue to express Cbp.
Figure 2.
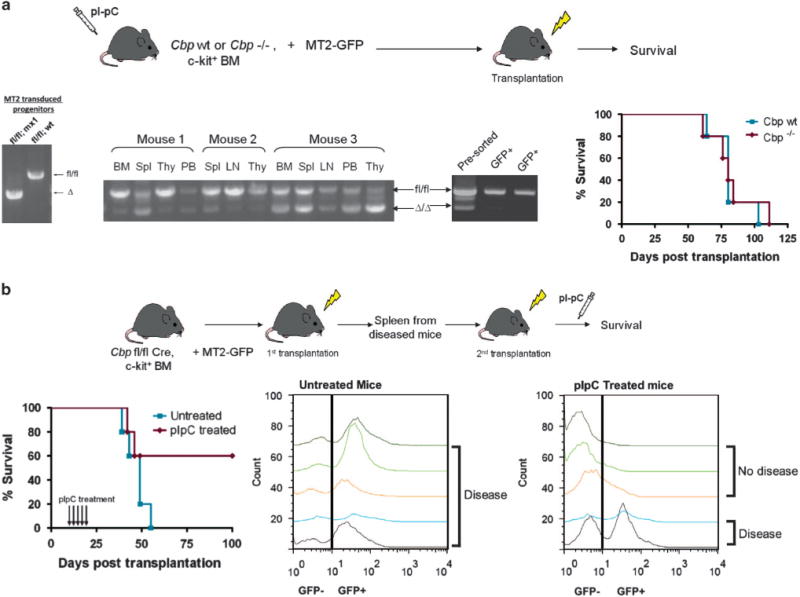
Cbp confers a selective advantage during initiation/progression of MT2-driven AML, under selective in vivo conditions. (a) Cbp wt and Cbp−/− progenitor cells were transduced with MT2 and transplanted into lethally irradiated recipients. Genotyping of the Cbp wt and Cbp−/− progenitor cells post-transduction confirmed almost complete excision of the Cbp allele. However, genotyping of leukemic cells revealed reemergence and clonal expansion of the un-excised Cbp allele. Furthermore, enrichment of leukemic cells via GFP sorting demonstrated only non-recombined cells, suggesting that Cbp loss confers a significant growth disadvantage during leukemia induction. All recipient mice succumbed to AML within 4 months post-transplantation. (b) c-kit+ Cbpfl/fl;Mx1-Cre+ BM cells were transduced with MT2 and transplanted into lethally irradiated recipients. The leukemias that arose in the primary recipients were transplanted into secondary animals. Excision of the Cbp allele in the secondary recipient animals resulted in decreased penetration of disease (only 40% of animals developed AML). Representative flow cytometry demonstrates that no GFP+ cells could be detected in the peripheral blood of non-diseased animals at the end of the experiment (d120).
We next examined whether Cbp is required for maintenance of leukemia. c-kit+ BM cells were isolated from Cbpfl/fl;Mx1-Cre+ mice (not previously treated with pIpC). These cells were transduced with MSCV-MT2-IRES-GFP and transplanted into lethally irradiated recipient mice (Figure 2b). Leukemic cells were harvested from primary mice and further transplanted into sub-lethally irradiated secondary recipients. Five doses of pIpC were administered to half of the recipient mice, starting at 10 days post-transplantation to allow for proper engraftment, with control mice receiving no treatment. Mice from the untreated group succumbed to disease between 19 and 35 days later (Figure 2b). By contrast, only 40% of the mice from the pIpC-treated group died from disease, although they did so with a similar latency. In the remaining pIpC-treated mice, no GFP-positive cells could be detected in the peripheral blood on serial testing, nor were Cbpfl/fl or recombined alleles detected by genotyping and no disease was evident at the termination of the experiment on day 120 (Figure 2b and data not shown). Thus, similarly to our in vitro replating assays, loss of Cbp in vivo compromises effective induction and maintenance of MT2-associated AML.
Functional redundancy exists between Cbp and p300 during myeloid transformation
Cbp and its closely related paralogue p300 have similar, but also unique functions.7 We hypothesized that p300 may partially compensate for Cbp loss and explain why Cbp was not an absolute requirement for immortalization in vitro. To test this hypothesis, we knocked down p300, using two lentiviral shRNAs (sh747 and sh1945) in Cbp wt and Cbp−/− cells immortalized by MT2 or NHA9. Modest knockdown (up to 60% of mRNA, sh747, Figure 3a) of p300 in Cbp wt MT2 expressing cells decreased the numbers of colonies in methylcellulose culture, in comparison with cells expressing a control shRNA construct that targets luciferase (Figure 3b). This decrease was even more marked in Cbp−/− cells (Figure 3b), with similar results also demonstrated in the NHA9 cell line (Figure 3b). Taken together, our data suggest functional redundancy between Cbp and p300, which cooperate to facilitate the maintenance of self-renewal programs in cells immortalized by the AML-associated fusion proteins MT2 and NHA9.
Figure 3.
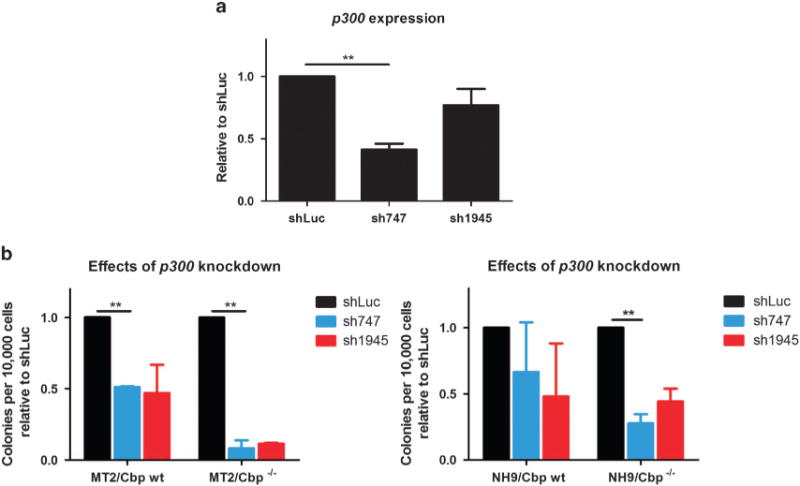
Functional redundancy of Cbp and p300 during myeloid transformation. (a) p300 expression following lentiviral transduction of Cbp−/− cells, using two different shRNAs. Expression was assessed by q-PCR using beta-actin as a reference gene. (b) sh-mediated knockdown of p300 decreases clonogenic potential of MT2 and NHA9 AML cell lines, particularly on a Cbp−/− background. **P < 0.01.
Inhibition of the KAT activity of CBP/p300 suppresses the growth of multiple AML subtypes through the induction of cell-cycle arrest and apoptosis
To bypass the functional redundancy between Cbp and p300 and to determine whether other subtypes of AML also require CBP/p300, we employed a pharmacological strategy that utilized a highly selective small molecule inhibitor of CBP and p300 KAT activity, C646 (Supplementary Figure 2A).25 Initially, all experiments were conducted with both an inactive structural analog of C646, C37 and vehicle control (DMSO), but as responses to C37 and DMSO were identical, we continued only with DMSO as a control (Supplementary Figure 2B and data not shown). We first demonstrated that C646 treatment was not associated with any significant toxicity to normal murine hematopoietic stem and progenitor cells in colony formation, colony composition and in serial replating assays up to a dosage of 30 μM (Figure 4a and data not shown). All subsequent assays were therefore performed at a dosage of 20 μM. At this dosage, we demonstrated significant inhibition of growth for MT2 and NHA9 immortalized cell lines in liquid culture and in methylcellulose colony assays (Figure 4b and Supplementary Figure 3A). We extended this analysis to a panel of human AML cell lines representative of common oncogenic driver mutations (Figure 4c and Supplementary Table 1). Growth rates for 10 separate cell lines were assessed in liquid culture assays (DMSO vs C646). As demonstrated in Figure 4c, 8 cell lines demonstrated a significant reduction in growth, with 7/10 cell lines demonstrating a greater than 50% reduction in growth in liquid culture following C646 treatment. Similarly, in methylcellulose assays, colony growth was significantly reduced in 8/10 and by greater than 50% in 5/10 cell lines (Figure 4d). Taken together, these results demonstrate significant efficacy for CBP/p300 KAT inhibition across a number of AML subtypes.
Figure 4.

Pharmacological inhibition of CBP and P300 suppresses the growth and decreases clonogenic potential of multiple AML cell lines in vitro. (a) C646 treatment of normal murine BM cells (n = 3) does not lead to significant changes of the number, or the types of colonies produced in serial replating assays. (b) C646 suppresses the growth of the NHA9 immortalized murine cell line in liquid culture and in methylcellulose assays. (c) Ten different human AML cell lines were tested against C646, over a period of 12 days in liquid culture conditions. The majority of these were responsive to C646 treatment (e.g., Kasumi-1), with 7/10 cell lines demonstrating a >50% decrease in cell numbers on day 12, compared with DMSO control. Two cell lines showed no response to CBP/p300 KAT inhibition (K562 and MOLM13). (d) Similarly, methylcellulose assays revealed a significant decrease in clonogenic potential in 8/10 human AML cell lines treated with C646. (e) Treatment with C646 induces apoptosis in the sensitive (Kasumi-1) but not in the resistant (K562) cell lines, as measured by Annexin/7-AAD staining. (f) Induction of apoptosis in the sensitive cell lines is accompanied by a modest G1 cell-cycle arrest (assessed by PI staining), however this is lacking in the resistant K562 cell line. *P < 0.05; **P < 0.01; ***P < 0.0001.
To further determine the cellular consequences of KAT inhibition, we performed apoptosis and cell-cycle analysis post-C646 treatment. Inhibition with C646 was associated with a variable induction of apoptosis at 72 h (Figure 4e and Supplementary Figure 3C). There was no induction of apoptosis in normal human CD34+ cells in liquid culture at similar dosage (Figure 6b). A modest alteration of cell cycle was observed with induction of G1/G0 arrest in the responsive, but not in the resistant cell lines (Figure 4f).
Figure 6.
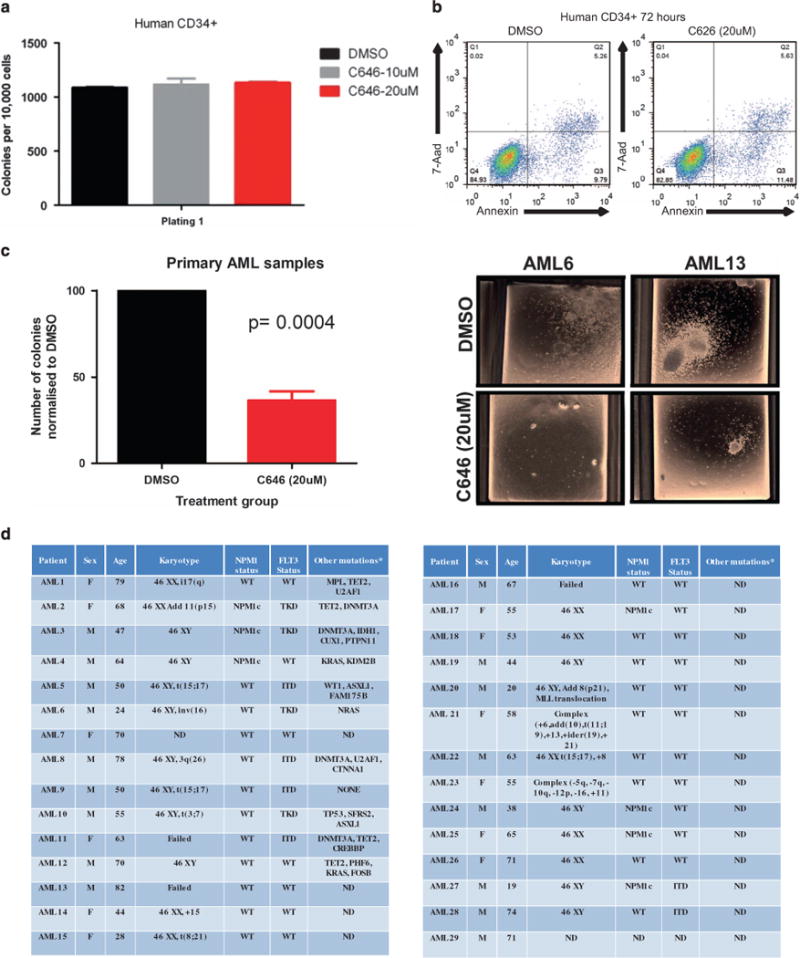
C646 treatment decreases growth of primary human AML samples. (a) C646 treatment of normal human CD43+cells (n = 2) did not alter their clonogenic potential. (b) No induction of apoptosis was observed following treatment of normal CD34+ cells with C646, as measured by Annexin/7-AAD staining. (c) By contrast, a significant reduction in colony numbers was seen in 19/29 primary AML samples exposed to the C646 compound. (d) A number of primary AML samples (n = 29), covering a range of molecular subtypes, with variable karyotypic mutational and prognostic status, as shown in this table were assessed for sensitivity to C646 (see text and Supplementary Figure 6).
Inhibition of CBP/p300 lysine acetyltransferase (KAT) activity in human leukemia cells alters a transcriptional program associated with genomic integrity
To determine the critical transcriptional programs altered in AML cells that mediate cell-cycle arrest and cell death, we analyzed global gene expression patterns following treatment with C646 or DMSO control, in two sensitive (Kasumi-1 and KG-1) and one resistant (K562) cell lines. Gene expression was assessed 24 h post treatment, a time point before the induction of significant apoptosis or cell-cycle alteration (Supplementary Figure 4). Significant differential gene expression changes (false discovery rate < 5%; fold change: ± 1.5-fold (Log2 scale)) were noted for 348 and 231 genes in the sensitive cells lines KG-1 and Kasumi-1, respectively, and for 47 genes for the resistant cell line K562 (Figure 5a). There was a high correlation between the deregulated genes in the two sensitive cell lines (KG-1 and Kasumi-1, (P < 2.2− 16) (Figure 5b), suggesting that KAT inhibition generates specific rather than general transcriptional changes. Ninety-eight genes were commonly differentially expressed in the two sensitive cell lines. To further prioritize gene expression changes associated with sensitivity to C646, 11 genes that were also differentially expressed in the resistant cell line K562 were subtracted from this gene list (Figure 5c). When the GO terms for the remaining 87 genes (57 genes downregulated and 30 genes upregulated, Figure 5d and Supplementary Table 2) were examined, there was significant enrichment for genes involved in DNA replication, DNA repair, the control of mitosis and the cell cycle (Figure 5d). Moreover, 43% (38/87) of these genes have been previously identified as direct p300 targets (p300 chromatin immunoprecipitation (ChIP) data from the ENCODE project,26 Supplementary Table 2). In agreement with the histone acetyltransferase function of CBP/p300, transcriptional repression was accompanied by reduced H3K18Ac levels in the promoter region of selected candidate genes, as validated by ChIP-PCR (Figure 5e) 24 h post KAT inhibition. The differential (and sensitive cell line-specific) expression pattern fits well with the cellular consequences observed following HAT inhibition. These findings are also in agreement with previous observations of a cell-cycle defect upon Cbp deletion in normal hematopoietic stem and progenitor cells.8 Gene set enrichment analysis corroborated that DNA replication and cell-cycle progression are affected upon KAT inhibition, as the treated vs untreated profiles were significantly enriched for genes involved in those processes (Supplementary Figure 5).
Figure 5.
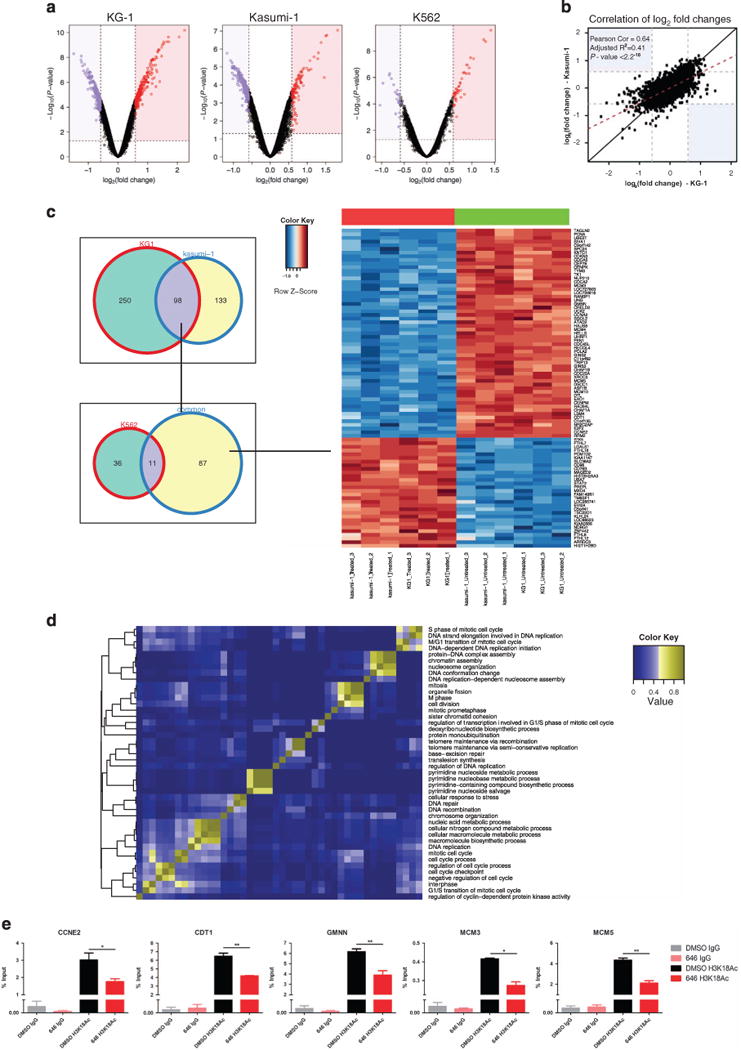
C646 treatment results in distinct transcriptional and chromatin acetylation changes associated with genomic integrity in sensitive AML cell lines. (a) Volcano plots for C646 vs DMSO treated samples (KG-1, Kasumi-1 and K562), showing fold-change (log2) and P-value significance level (log10) for all genes. A larger number of genes are differentially regulated in the sensitive (KG-1 and Kasumi-1) versus the resistant (K562) cell line. (b) Log fold change correlation plot for all genes between the sensitive KG-1 and Kasumi-1 following treatment, demonstrating a high degree of similarity in the transcriptional changes induced by C646. (c) Venn diagram of genes significantly differentially regulated (1.5 FC; < 0.05 adj. P-value) upon C646 treatment, between the different cell lines (left panel). Heatmap for the common list of genes (87 genes) found to be deregulated upon treatment in KG-1 and Kasumi-1 but not in K562 cells (right panel). (d) The ’common’ gene list was subjected to a Gene Ontology (Molecular Function) over-representation analysis. The significant results were displayed using a heatmap to highlight the percentage of shared gene between the categories, and demonstrates enrichment for processes including cell-cycle control, mitosis and DNA replication and repair. (e) ChIP-PCR analysis of H3K18 acetylation levels (24 h post-treatment) in the promoter regions of selected candidates from the list of the 87 downregulated genes. *P < 0.05; **P < 0.01.
CBP/p300 KAT inhibitors decrease growth of primary human AML samples
Finally, the efficacy of KAT inhibition was tested in primary human AML samples. Importantly, no evidence of C646 toxicity to normal CD34+ cells was seen in methylcellulose or apoptosis assays (Figures 6a and b). However, in primary blasts from a large set of AML patients a markedly different pattern was demonstrated. C646 was tested on 29 AML cases harboring a number of differing molecular and cytogenetic abnormalities. Similar to the effects seen in AML cell lines, a significant reduction in clonogenic potential was demonstrated in the majority of patient samples (P = 0.0004, Figure 6c). Indeed, 65% of samples (19/29) across a number of AML molecular subtypes, including those associated with a poor prognosis, demonstrated a significant (>50%) reduction in colony number (Figure 6d and Supplementary Figure 6).
DISCUSSION
Survival rates for AML patients are dismal, with over 70% of patients eventually succumbing to the disease. This statistic has not been helped by the recent lack of novel therapeutics developed for AML. Agents with efficacy against specific molecular subtypes, including ATRA for acute promyelocytic leukemia,23 the toxin-conjugated anti-CD33 antibody Myelotarg for core-binding factor mutated AML27 and FLT3 inhibitors for patients with activating FLT3 mutations28 have shown promising effects in specific subtypes of AML. However, agents with effects against a wider number of AML subtypes have failed to emerge. Recently, inhibitors of epigenetic regulators have demonstrated promise in hematopoietic malignancies. In this report, we present data supporting the role of the epigenetic regulators CBP/p300 in AML pathogenesis and, using pharmacological inhibition of their KAT activity in preclinical studies identify these proteins as therapeutic targets across a wide range of AML subtypes.
Our genetic experiments using in vitro and in vivo murine models of leukemia were strongly suggestive of a Cbp role in AML induction and maintenance. This was further supported by our shRNA assays, which suggested and addressed the compensatory role of p300 following Cbp loss. To bypass the limitations of these genetic approaches and therapeutically validate these results, we utilized a small molecule inhibitor that specifically targets both CBP and p300 KAT activity, thus allowing dissection of the relative roles of their catalytic activity and their non-catalytic protein scaffold functions in leukemogenesis. We encountered no evidence of toxicity to normal hematopoiesis (murine and human cells) in our in vitro assays. This is in keeping with the mild hematopoietic phenotype we observed in vivo under homeostatic conditions following acute Cbp deletion.8 Of note, the hematopoietic phenotype of mice carrying a KAT defective mutant of p300 is minimal, whereas mice with a p300 KIX-domain mutant allele, predicted to abrogate multiple protein-protein interactions, demonstrate a marked hematopoietic phenotype.29
Also supportive of a lack of general toxicity were the specific gene expression changes observed following treatment. Pharmacological inhibition of CBP and p300 affected specific and partially overlapping programs in two sensitive cell lines. Many of these differentially expressed genes are involved in DNA integrity and cell-cycle control. These alterations are in keeping with the proposed role of CBP and p300 in other DNA-templated processes, including repair, replication and recombination.30 In particular, multiple members of the minichromosome maintenance pre-replication complex (MCM3, MCM4 and MCM5), as well as its interacting proteins (MCM10) and loading/regulatory factors for replication origin licensing (CDT1 and GMNN) were downregulated in sensitive, but not in resistant cell lines. These data would also, at least partially, explain the observed altered cellular phenotype of cell-cycle arrest and apoptosis.
Although the exact mechanism(s) whereby CBP and p300 inhibition alter transcription in AML remains unknown, a number of non-mutually exclusive mechanisms could be envisaged. CBP and p300 are known to interact with multiple hematopoietic transcription factors and also with known oncogenes, therefore inhibition of direct co-activator function for driver oncogenes may underlie effects in certain AML subtypes. Our genetic experiments in MOZ-TIF2 and NUP98-HOXA9 support this, as do reports of the efficacy in acute leukemias of small molecule inhibitors that directly target the interaction between CBP and/or p300 and β-Catenin31 and the observation that nearly half of the differentially expressed genes following CBP/p300 KAT inhibition are direct targets of p300, as shown by ChIP-Seq data deposited in the ENCODE database. Another possibility is that these inhibitors target CBP and p300 co-activator activity at critical transcriptional mediators downstream of oncogenic mutations. Finally, lysine acetylation of non-histone proteins mediated by CBP and/or p300 has been suggested to facilitate leukemogenesis. In support of this concept, targeting KAT activity in AML1-ETO positive leukemia cells led to decreased leukemia growth associated with decreased site-specific AML1-ETO acetylation.32 Furthermore, AML1-ETO proteins mutated to prevent site-specific acetylation were poorly transforming in vivo and AML1-ETO positive patient samples and cell lines are sensitive to CBP/p300 inhibition.33 Thus, acetylation of both histone and non-histone proteins by CBP and p300 may facilitate leukemogenesis. Further investigations are warranted, therefore, to address the specific role of these mechanisms in transformation in individual AML subtypes.
Unfortunately, the C646 HAT inhibitor lacks sufficient potency and shows unfavorable pharmacokinetic properties (unpublished data) to allow effective dosing in vivo. However, Cbp ablation in vivo significantly impeded leukemic growth. In addition, the majority of AML cell lines and most AML patient samples, including those with a number of poor risk genetic characteristics (including FLT3-ITD, TP53, DNMT3A, TET2 and ASXL1 mutations and EVI-1 and TP53 rearrangements, as documented by directed NGS in some patients, Figure 6d), demonstrated a significant decrease in clonogenic growth upon inhibitor treatment in vitro. Importantly, no significant toxicity was seen in normal murine and human hematopoietic cells treated with the same dose of inhibitor. Taken together, our in vivo genetic data and pharmacological in vitro data suggest promising efficacy in targeting CBP and p300 function in AML. Therefore, novel potent and specific agents, optimized for in vivo use, are urgently required to validate the promising efficacy and lack of toxicity of CBP/p300 KAT inhibition, and to move this potential treatment paradigm toward clinical testing in AML. However, as a note of caution, if efficacy is demonstrated, due to the association of CBP and p300 loss with tumor induction and a chemo-resistant phenotype,9–11 caution will be warranted as to how CBP/p300 inhibitors can best be integrated into combination therapies for AML.
MATERIALS AND METHODS
Retroviral transduction assays
Retroviral transduction protocols and constructs (MSCV-NUP98-HOXA9-neo, MSCV-NUP98-HOXA9-IRES-GFP, MSCV-MOZ-TIFF2-neo and MSCV-MOX-TIFF2-IRES-GFP) have been previously described.10 Cre cDNA was subcloned into a pBabe-puro vector. BM cells were harvested from 6- to 8-week-old Cbpfl/fl;wt(wt) or Cbpfl/fl;Mx(Mx) mice that had been treated with five doses of pIpC (Sigma-Aldrich Ltd, Dorset, UK; 300 μg/dose). c-kit+ BM cells were selected using CD117MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). For the serial replating assays, 104 transduced cells were plated in duplicate in methylcellulose. Suspension culture was established by growing cells from the third plating in RPMI with 10% FBS (Sigma-Aldrich Ltd) and 10 ng/μl of mIL3 (Peprotech EC Ltd, London, UK). To excise Cbp in vitro, cells with Cbpfl/fl;wt background from the third plating were collected and transduced with pBabe-puro or pBabe-Cre-puro. Serial replating assays were set up as described above with 2 μg/ml puromycin.
Mouse disease model
The Cbp;Mx1-Cre mouse model, transplantation procedures and tissue processing have been previously described.8,34 To examine the requirement of Cbp for leukemia initiation, 1 × 106 Cbp-wt or Cbp-excised myeloid progenitor cells, which had been transduced with MSCV-MT2-IRES-GFP, were injected intravenously into lethally irradiated (2 × 550rads) C57BL/6 recipients. To examine the role of Cbp in the maintenance of leukemia, we first established MT2 induced AML by transplanting Cbpfl/fl;Mx+ (with no pIpC treatment) cells that had been transduced with MSCV-MT2-IRES-GFP into lethally irradiated recipients. When mice demonstrated illness, spleen cells were collected and 106 cells were transplanted into sub-lethally irradiated (550 rads) C57BL/6 recipients. To excise Cbp in vivo, 10 days after transplantation, five doses of pIpC were injected intraperitoneally. GFP+ cells were sorted using a MoFlo Cell Sorter (Beckman Coulter Ltd, High Wycombe, UK). PCR primers for Cbp genotyping were GGGGAAATTTTGGTCTGGTAAG (Forward), and CTGCTCTACCTAAATTCCC AG (Reverse). All mice were housed in a pathogen-free animal facility. Experiments were conducted under UK Home Office regulations.
RNAi experiments
p300 shRNA in pLKO.1 lentiviral vector and control vector targeting the Luciferase sequence (SHC007) were obtained from Sigma-Aldrich Ltd. Lentiviral supernatants were produced in 293T cells with packaging plasmids psPAX2 and pMD2G (Sigma-Aldrich Ltd) using TransIT-LT1 reagent. Transduction of leukemia cell lines was performed using standard protocols24 with puromycin (2 μg/ml final concentration) as a selection marker. 104 transduced cells were plated in methylcellulose and suspension cultures were set up. To assess p300 expression, total mRNA was prepared using Trizol reagent. cDNA was synthesized using Super-Script cDNA synthesis kit (Life Technologies Ltd, Paisley, UK). Quantitative RT-PCR was carried out with SYBR Green PCR mastermix using the ABI Prism 7000 system (Life Technologies Ltd). RNA expression levels were normalized to beta-actin.
Cell culture and methylcellulose assays
Kasumi-1, KG-1, ME-1, HEL, U937, SKM-1, Nomo-1, MOLM-13, OCI-AML3 and K562 cells were grown in RPMI-1640 medium supplemented with FBS (10–20% final concentration, Sigma-Aldrich Ltd). Human CD34+ cells were maintained in liquid culture using StemSpan Serum-free media and CC100 cytokine cocktail (STEMCELL Technologies, Vancouver, BC, Canada). All growth media also contained 1% penicillin/streptomycin. Unless otherwise stated, mouse BM, MT2 and NHA9 mouse immortalized progenitors, leukemic cell lines and human control CD34+ and AML patient primary cells were plated at a concentration of 10 000–20 000 cells/plate (in duplicate) using the MethoCult H4531, MethoCult H4435 Enriched (for human cells) or MethoCult GF M3434 (for mouse cells) (STEMCELL Technologies). Colonies were scored at 7–12 days.
CBP/p300 KAT inhibitor (C646)
Mouse and human cells were treated with the CBP/p300 HAT inhibitor C646 (whose properties are described in detail in Bowers et al.25), at various concentrations. The inactive form C37 and DMSO vehicle were included as controls. For the liquid growth experiments, cells were re-suspended at a concentration of 0.5 × 106 cells/ml, and the appropriate amount of drug was added. At the appropriate time points, wells were counted, washed in PBS and re-suspended at the same concentration, with fresh drug added each time.
Western blotting
Western blotting was performed using standard protocols (15% SDS-PAGE gels). Antibodies used were Histone 4 (17036, Abcam, Cambridge, UK) and Anti-acetyl-Histone H3 (06-599, Millipore Limited, Nottingham, UK). IRDye 680RD and IRDye 800CW were used as secondary antibodies (LI-COR Biosciences Ltd, Cambridge, UK). Immunoblots were scanned using an Odyssey Infrared Imaging system (LI-COR Biosciences Ltd).
Flow-cytometry assays
The apoptosis assays were performed using the BD Pharmingen Apoptosis Detection kit (Becton Dickinson Ltd, Oxford, UK). Cell-cycle experiments were performed on 80% EtOH-fixed cells, using Propidium Iodide (Sigma-Aldrich Ltd). Flow cytometry was performed on a CyAn ADP Flow-Cytometer (Dako UK Ltd, Ely, UK) or a BD LSRFortessa cell analyser (Becton Dickinson Ltd) and all data were analyzed with FloJo software (Tree Star, Inc., Ashland, OR, USA).
ChIP and ChIP-PCR assays
ChIP was performed on KG-1 cells harvested 24 h post treatment as previously described.34 IgG (I5006, Sigma-Aldrich Ltd) or H3K18Ac (ab1191, Abcam) antibodies were used (2.5 mg/reaction). ChIP-PCR was carried out with SYBR Green PCR mastermix using the ABI Prism 7000 system (Applied Biosystems). The following primers were used: CCNE2: CCTTGCTTCCTCTCTT CTCCA (Forward)/GTGGTGGCGATCTTTCTTCC (Reverse), CDT1: TCGCTACGA GGATTGAGCG (Forward)/CCTGCAGCTGTCAAAGTAGG (Reverse), GMNN: TC GCTACGAGGATTGAGCG (Forward)/CCTGCAGCTGTCAAAGTAGG (Reverse), MCM3: GTGGACCGGATCTGTTTGG (Forward)/GGTGCCGGGAAGTTTAAGTC (Reverse), MCM5: GGTTCTTGTCTCCCCTGGTT (Forward)/CCGGACTCCAACCC CAGT (Reverse).
Patient material
Control CD34+ human cells, patient BM or peripheral blood cells (480% blasts) were obtained following donor/patient consent and under full ethical approval at each involved institute. Mutations were determined by solution capture hybridization followed by deep sequencing with baits and mutation calling as defined in Papaemmanuil et al.35
Statistical analysis
Unless otherwise stated, all statistical analyses used Student’s t-test on raw data. Values equal to, or less than 0.05 were considered as statistically significant. Survival curves were constructed using the Kaplan–Meier method. Error bars represent s.e.m. Symbols: *P < 0.05; **P < 0.01; ***P < 0.0001.
ENCODE mining, GEP and bioinformatic analysis
Illumina human whole genome 6 V2 gene expression data from all three cell lines were normalized using RMA algorithm and analyzed in R.36 P300 peaks for CREBBP in the K562 AML cell line were downloaded from the ENCODE project (http://genome.ucsc.edu/ENCODE/).
Gene set enrichment analysis and leading edge analysis
Gene set enrichment analysis tools were obtained from http://www.broadinstitute.org/gsea/index.jsp. Default settings and all curated gene sets (c2.All.v3.0.symbols.gmt) were used for the analysis.
Supplementary Material
Acknowledgments
Funding in the Huntly laboratory comes from Cancer Research UK, Leukemia Lymphoma Research, the Kay Kendal Leukemia Fund, the Leukemia lymphoma Society of America, the Wellcome Trust, The Medical Research Council and an NIHR Cambridge Biomedical Research Centre grant. Patient samples were processed in the Cambridge Blood and Stem Cell Biobank.
Footnotes
AUTHOR CONTRIBUTIONS
BH, GG and W-IC designed the experiments, GG, W-IC, SJH, PG, AF and EP performed experiments. GG, WI-C, DR, EP, PC, BG and BH analyzed data. CC, JMVD and PAC provided critical reagents. BH oversaw the study. BH, GG and W-IC wrote and all authors reviewed the manuscript.
CONFLICT OF INTEREST
PA Cole is a cofounder, equity holder and paid consultant for Acylin Therapeutics which is developing p300 HAT inhibitors.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367:647–657. doi: 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- 4.Blobel GA. CREB-binding protein and p300: molecular integrators of hematopoietic transcription. Blood. 2000;95:745–755. [PubMed] [Google Scholar]
- 5.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 6.Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics. 2010;5:9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci USA. 2002;99:14789–14794. doi: 10.1073/pnas.232568499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan WI, Hannah RL, Dawson MA, Pridans C, Foster D, Joshi A, et al. The transcriptional coactivator Cbp regulates self-renewal and differentiation in adult hematopoietic stem cells. Mol Cell Biol. 2011;31:5046–5060. doi: 10.1128/MCB.05830-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007;9:1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- 13.Kung AL, Rebel VI, Bronson RT, Ch’ng LE, Sieff CA, Livingston DM, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–277. [PMC free article] [PubMed] [Google Scholar]
- 14.Taki T, Sako M, Tsuchida M, Hayashi Y. The t(11;16)(q23;p13) translocation in myelodysplastic syndrome fuses the MLL gene to the CBP gene. Blood. 1997;89:3945–3950. [PubMed] [Google Scholar]
- 15.Ida K, Kitabayashi I, Taki T, Taniwaki M, Noro K, Yamamoto M, et al. Adenoviral E1A-associated protein p300 is involved in acute myeloid leukemia with t(11;22)(q23; q13) Blood. 1997;90:4699–4704. [PubMed] [Google Scholar]
- 16.Deguchi K, Ayton P, Carapeti M, Kutok J, Snyder C, Williams I, et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. 2003;3:259–271. doi: 10.1016/s1535-6108(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 17.Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999;19:764–776. doi: 10.1128/mcb.19.1.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.So CW, Cleary ML. MLL-AFX requires the transcriptional effector domains of AFX to transform myeloid progenitors and transdominantly interfere with forkhead protein function. Mol Cell Biol. 2002;22:6542–6552. doi: 10.1128/MCB.22.18.6542-6552.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayly R, Chuen L, Currie RA, Hyndman BD, Casselman R, Blobel GA, et al. E2A-PBX1 interacts directly with the KIX domain of CBP/p300 in the induction of proliferation in primary hematopoietic cells. J Biol Chem. 2004;279:55362–55371. doi: 10.1074/jbc.M408654200. [DOI] [PubMed] [Google Scholar]
- 20.Payne SR, Kemp CJ. Tumor suppressor genetics. Carcinogenesis. 2005;26:2031–2045. doi: 10.1093/carcin/bgi223. [DOI] [PubMed] [Google Scholar]
- 21.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 22.Oki Y, Issa JP. Epigenetic mechanisms in AML - a target for therapy. Cancer Treat Res. 2010;145:19–40. doi: 10.1007/978-0-387-69259-3_2. [DOI] [PubMed] [Google Scholar]
- 23.Baljevic M, Park JH, Stein E, Douer D, Altman JK, Tallman MS. Curing all patients with acute promyelocytic leukemia: are we there yet? Hematol Oncol Clin North Am. 2011;25:1215–1233 viii. doi: 10.1016/j.hoc.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 25.Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119:6198–6208. doi: 10.1182/blood-2011-11-325050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grunwald MR, Levis MJ. FLT3 inhibitors for acute myeloid leukemia: a review of their efficacy and mechanisms of resistance. Int J Hematol. 2013;97:683–694. doi: 10.1007/s12185-013-1334-8. [DOI] [PubMed] [Google Scholar]
- 29.Kimbrel EA, Lemieux ME, Xia X, Davis TN, Rebel VI, Kung AL. Systematic in vivo structure-function analysis of p300 in hematopoiesis. Blood. 2009;114:4804–4812. doi: 10.1182/blood-2009-04-217794. [DOI] [PubMed] [Google Scholar]
- 30.Allard S, Masson JY, Cote J. Chromatin remodeling and the maintenance of genome integrity. Biochim Biophys Acta. 2004;1677:158–164. doi: 10.1016/j.bbaexp.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 31.Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2013;33:2169–2178. doi: 10.1038/onc.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science. 2011;333:765–769. doi: 10.1126/science.1201662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao XN, Lin J, Ning QY, Gao L, Yao YS, Zhou JH, et al. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLoS One. 2013;8:e55481. doi: 10.1371/journal.pone.0055481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–3627. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.