

Abstract
Objective
To test the hypothesis that autoantigen modifications by peptidylarginine deiminase type 4 (PAD-4) increase immunoreactivity.
Methods
We assembled sera from patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Felty’s syndrome (FS), and antineutrophil cytoplasmic antibody–associated vasculitides (AAVs), as well as sera from control subjects without autoimmune diseases. The sera were tested for binding to activated neutrophils, deiminated histones, and neutrophil extracellular chromatin traps (NETs). IgG binding to lipopolysaccharide-activated neutrophils was assessed with confocal microscopy, and binding to in vitro–deiminated histones was measured using enzyme-linked immunosorbent assay (ELISA) and Western blotting. In addition, we quantitated histone deimination in freshly isolated neutrophils from the blood of patients and control subjects.
Results
Increased IgG reactivity with activated neutrophils, particularly binding to NETs, was paralleled by preferential binding to deiminated histones over nondeiminated histones by ELISA in a majority of sera from FS patients but only in a minority of sera from SLE and RA patients. Immunoblotting revealed autoantibody preference for deiminated histones H3, H4, and H2A in most FS patients and in a subset of SLE and RA patients. In patients with AAVs, serum IgG preferentially bound nondeiminated histones over deiminated histones. Increased levels of deiminated histones were detected in neutrophils from RA patients.
Conclusion
Circulating autoantibodies in FS are preferentially directed against PAD-4–deiminated histones and bind to activated neutrophils and NETs. Thus, increased reactivity with modified autoantigens in FS implies a direct contribution of neutrophil activation and the production of NET-associated nuclear autoantigens in the initiation or progression of FS.
Autoimmune disorders such as systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) may progress slowly and follow a chronic path with gradual worsening of disease manifestations (1,2). In other individuals, sudden flares of more intense disease manifestations may interrupt lengthy periods of symptom quiescence. One notable example of worsening in a chronically progressing disorder is provided by Felty’s syndrome (FS), a variant of RA that is defined by arthritis involving axial joints, enlargement of the spleen, and a decline in neutrophil numbers (3). The decrease in neutrophil numbers is thought to be due to excessive activation of mature neutrophils and their clearance in the patient’s spleen. FS occur in 1–3% of RA patients, usually after 10–15 years of fairly typical symptoms (3). An alternative viewpoint is that FS does not arise as chronic progression of RA, but instead may be closely related to a T cell form of large granular lymphocyte leukemia with which it shares its defining clinical features and an oligoclonal CD8+ T cell expansion (4). Due to the neutropenia, FS patients experience an elevated risk of infections. The factors determining the course of disease in any given patient are largely unknown. A prevalent finding is that autoimmune disorders may worsen in parallel with various types of infections (5), although it is difficult to separate environmental effects from underlying genetic and stochastic contributions.
It has not been established just how infections may affect autoimmune reactivity and potentially lead to sudden flares in the presentation of autoimmune disorders. One possibility is that, as a result of infection, the number of apoptotic cells could transiently rise because diverse pathogens induce apoptosis in infected cells (6). The increased numbers of apoptotic cells may exceed the clearance capacity of tissue-resident scavenger cells and lead to the stimulation of the immune system with antigens from the apoptotic cells (7,8). This proposed mechanism is consistent with the increased risk of autoimmunity arising from genetic defects in serum factors that recognize and bind apoptotic cells, or with defects in phagocyte receptors that function in uptake and clearance of apoptotic cells (8). Whether inefficient clearance of cells that die from nonapoptotic death also increases the risk of autoimmunity has been less thoroughly tested.
An alternative form of cell death that is induced during an infection is “NETosis.” NETosis received its name from neutrophil extracellular chromatin traps (NETs) that are released in response to infectious agents ranging from bacteria to fungi (9). Once at the site of an infection, neutrophils deploy extracellular chromatin that is studded with additional bactericidal granule components and may serve to immobilize and destroy microbes (10). The release of NETs is induced by a wide range of inflammatory stimuli (11) and depends on signals from the cell surface and the participation of the cytoskeleton (12). Autoantibodies to NET components, including elastase, myeloperoxidase (MPO), cathepsin G, and proteinase 3 (PR3), arise in autoimmune disorders (13–15), suggesting that NETs should be viewed as possible stimuli for such antibodies. Nevertheless, conclusive evidence linking NETs to the induction of autoantibodies remains elusive.
Core histones in NETs contain arginines that are converted to citrullines (11) by peptidylarginine deiminase type 4 (PAD-4), a posttranslational modification that is essential for NET release (16). PAD-4 acts on various autoantigens (17–21), such that antibody reactivity against citrulline-containing peptides has proven useful in the diagnosis of autoimmunity. In RA, autoantibodies react with a peptide of filaggrin, provided it contains citrulline (22), and anti–cyclic citrullinated peptide (anti-CCP) autoreactivity constitutes a reliable marker for this condition (23).
Because histones are the major substrates of PAD-4 (24), and because antihistone antibodies are produced in SLE (25), RA (26), and FS (27), we hypothesized that, if NETs are the source of the autoantigens, autoantibodies from patients with these disorders might bind deiminated histones preferentially over nondeiminated histones. Our results with FS patient sera support this hypothesis; by immunofluorescence, enzyme-linked immunosorbent assay (ELISA), and Western blotting, we found preferential binding to deiminated histones H3, H4, H2A, or H1. In comparison, only few RA and SLE patient sera showed preferential binding to deiminated histones. A control group of patients with anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitides (AAVs) expressed antibodies with preference for nondeiminated histones over deiminated histones. We propose that activated neutrophils contribute, as sources of deiminated autoantigens, to the pathogenesis of FS.
PATIENTS AND METHODS
Patient sera
Sera were collected at the University of Lübeck, the Hospital for Special Surgery, and the University of Tennessee Health Science Center and from the Veterans Administration RA (VARA) serum repository (28) and used in accordance with Institutional Review Board approvals from each of the participating institutions. Patient data are shown in Table 1. All patients fulfilled the American College of Rheumatology (ACR) 1987 revised classification criteria for RA (29), the ACR 1982 revised classification criteria for SLE (30), or the Chapel Hill Consensus Conference nomenclature/criteria for vasculitis (31). FS was clinically defined by the coexistence of RA, chronic neutropenia (<2000/μl), and splenomegaly (3), except that patients with sera obtained from VARA were classified as having FS based on the determination of an International Classification of Diseases, Ninth Revision code corresponding to this disorder (714.1).
Table 1.
Characteristics of the patients and controls*
| SLE (n = 32) |
RA (n = 37) |
FS (n = 23) |
GPA (n = 25) |
MPA (n = 15) |
CSS (n = 15) |
Controls (n = 10) |
|
|---|---|---|---|---|---|---|---|
| Males | 3 | 18 | 17 | 16 | 7 | 9 | 8 |
| Females | 29 | 19 | 6 | 9 | 8 | 6 | 2 |
| Age, mean years | 35.2 | 64.9 | 70.3 | 54.7 | 67.4 | 60.7 | 36.2 |
| Active disease | 10 | 31 | 21 | 25† | 15 | 15 | – |
| pANCA positive | 1 | 1 | – | 2 | 14 | – | – |
| cANCA positive | – | 1 | 1 | 23 | – | – | – |
| ACPA positive | 2 | 22 | 17 | 1 | – | – | – |
| Azathioprine | 9 | – | – | 2 | 3 | 1 | – |
| Prednisolone | 26 | 24 | 8 | 24 | 12 | 14 | – |
| Methotrexate | 1 | 25 | 12 | 9 | 3 | 2 | – |
| Cyclophosphamide | 2 | – | – | 9 | 5 | 8 | – |
| Hydroxychloroquine | 17 | – | – | – | – | – | – |
| Mycophenolate | 9 | – | – | – | – | – | – |
| Anti-TNF | – | 4 | 6 | – | – | – | – |
Except where indicated otherwise, values are the number of patients. SLE = systemic lupus erythematosus; RA = rheumatoid arthritis; FS = Felty’s syndrome; MPA = microscopic polyangiitis; CSS = Churg-Strauss syndrome; pANCA = perinuclear antineutrophil cytoplasmic antibody; cANCA = cytoplasmic ANCA; ACPA = anti–citrullinated protein antibody; anti-TNF = anti–tumor necrosis factor.
Sera from the patients with granulomatosis with polyangiitis (Wegener’s) (GPA) were also tested in the inactive phase of disease.
Neutrophil isolation and stimulation
Neutrophils were isolated from healthy donor blood (Key Biologics) or from patients with RA or FS, as described (11). Briefly, neutrophils were enriched using dextran sedimentation centrifugation, recovered by isolymph density-gradient centrifugation (Gallard-Schlesinger), and suspended in phosphate buffered saline (PBS; without Ca++/Mg++) with 0.1% glucose and 0.5% heat-inactivated human serum. Neutrophils were stimulated with 100 ng of lipopolysaccharide (LPS)/ml of Hanks’ balanced salt solution (HBSS) or PBS with 2 mM Ca++ for 2 hours and lysed in 2% sodium dodecyl sulfate (SDS), 5% 2-mercaptoethanol, and 10% glycerol in 62.5 mM Tris, pH 6.8. Alternatively, autoimmune or control sera were diluted to 2 μg/ml of IgG and used in place of LPS.
Confocal microscopy
Purified neutrophils were allowed to settle onto poly-L-lysine-coated glass coverslips and examined as described (11). Cells were treated with LPS (100 ng/ml) for 1 hour at 37°C, rinsed with ice-cold HBSS, and fixed with 4% paraformaldehyde in HBSS. Coverslips were incubated with patient serum (10 μg/ml IgG) or rabbit anti–citrullinated histone H3 antibodies (catalog no. ab5103; Abcam) at 1:100 dilution for 1 hour. Antibody binding was detected with Alexa Fluor 647–conjugated goat anti-human IgG or Alexa Fluor 488–conjugated goat anti-rabbit IgG and Sytox orange (all from Invitrogen) and analyzed using confocal microscopy.
ELISA
Calf thymus histones or recombinant human histone H3.3 (Millipore) were incubated with 0.2 μM recombinant PAD-4 to completion of reaction as described (32). Flat-bottomed, 96-well microtiter plates (4HBX; Immulon) were coated with 5 μg/ml of deiminated or nondeiminated histones. As a control, recombinant PAD-4 was added to histones in the presence of EDTA to test whether binding to PAD-4 confounded our ELISA readings. Blank values were determined from plates incubated with coating buffer. Plates were blocked with 2.5% bovine serum albumin (BSA) in PBS, and sera were serially diluted from 20 μg/ml of IgG and incubated for 2 hours. Binding was quantified using alkaline phosphatase–conjugated goat anti-human IgG (Southern-Biotech) after addition of phosphatase substrate (Sigma) and measurement of optical density. ANCAs were detected using immunofluorescence or capture ELISA, as described (33). Anti–citrullinated protein antibodies (ACPAs) were measured by anti–citrullinated vimentin ELISA (Orgentec) or anti-CCP ELISA (Inova Diagnostics), according to the manufacturers’ instructions.
Western blotting
Proteins were resolved by 15% SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose. Membranes were blocked with 5% BSA, 0.1% Tween 20 in Tris buffered saline (TBS) and incubated for 2 hours with patient sera at 5 μg/ml of IgG in TBS containing 2.5% BSA, 1% Nonidet P40, and 0.1% SDS. Horseradish peroxidase–conjugated anti-human IgG was used for chemiluminescence development with “Western Lightning” substrate (PerkinElmer).
Statistical analysis
We used an unpaired 1-tailed t-test to evaluate whether FS patient IgG had greater preference for deiminated histones than IgG from other experimental groups. Chemiluminescence images of Western blots were scanned, and the differences in binding between different sera were evaluated by paired permutation test (further information is available at http://www.uthsc.edu/research/research_data/RadicM/ar11-0195RevFinalSuppl.pdf). The relative abundance of deiminated histones in neutrophils ex vivo was determined by dividing the intensity of binding to deiminated histone H3 by the total histone H3 binding intensity. The statistical differences between the groups were computed by Wilcoxon’s 2-sample rank sum test.
RESULTS
Characteristics of the patient populations
To analyze potential mechanisms that induce autoreactivity, we collected sera from 32 patients with SLE, 37 with RA, 23 with FS, and 55 with small-vessel vasculitides, along with sera from 10 healthy controls (Table 1). Despite the fact that SLE, RA, and FS primarily affect women, the total patient group included 77 females and 70 males with ages ranging from 13 to 88 years (mean 62 years). The large number of male patients is because 34 of the FS and RA patient samples were from the VARA repository, which contains mostly sera from male patients. Disease was active in 52 of 60 RA and FS patients, whereas 22 of the SLE patients had inactive disease. In addition, our study included 32 males and 23 females with active vasculitis, including 25 patients with granulomatosis with polyangiitis (Wegener’s) (GPA), 15 with microscopic polyangiitis (MPA), and 15 with Churg-Strauss syndrome (CSS). Most patients were treated with combinations of azathioprine, prednisolone, methotrexate, and cyclophosphamide, and 10 RA or FS patients were receiving anti–tumor necrosis factor therapy (Table 1). Clinical assays were performed to assess the levels of ACPAs or anti-CCP antibodies. Most FS patients (17 of 23) and RA patients (22 of 37) had antibodies to citrullinated antigens, an indication of more rapidly progressing arthritis. In contrast, 23 GPA patients and 14 MPA patients had cytoplasmic ANCA or perinuclear ANCA, respectively (Table 1). The clinical usefulness of these assays was recently evaluated (33,34).
Neutrophil activation increases autoantibody reactivity
Neutrophils respond to bacterial breakdown products, such as LPS, and other proinflammatory stimuli by releasing granule enzymes and casting chromatin NETs (10). These complex morphologic and enzymatic reactions could potentially affect autoantibody binding. To test whether neutrophil activation increases autoantibody reactivity, we prepared neutrophils from healthy donors, exposed them to LPS or left them untreated, and measured binding of IgG from SLE, RA, and FS patients. Bound IgG was detected with fluorescent rabbit anti-human IgG antibodies and visualized relative to DNA (Figure 1). Control serum IgG did not detectably bind to unstimulated neutrophils or to activated neutrophils (Figures 1A and B). In contrast, IgG from FS, SLE, and RA patients reacted strongly with LPS-activated neutrophils (Figures 1D, F, H, J, L, and N), yet binding to unstimulated neutrophils remained low.
Figure 1.
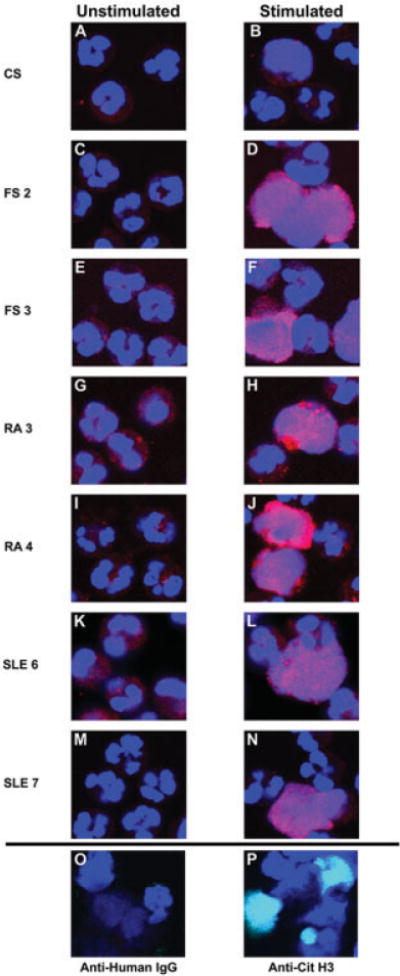
Binding of autoimmune sera to activated neutrophils. A–N, Unstimulated and lipopolysaccharide (LPS)–stimulated neutrophils were tested for binding by autoimmune and control serum (CS) IgG. Representative sera are shown. A and B, Control serum. C and D, Serum from Felty’s syndrome (FS) patient 2. E and F, Serum from FS patient 3. G and H, Serum from rheumatoid arthritis (RA) patient 3. I and J, Serum from RA patient 4. K and L, Serum from systemic lupus erythematosus (SLE) patient 6. M and N, Serum from SLE patient 7. Stimulated neutrophils, particularly those that had released neutrophil extracellular chromatin traps, reacted more strongly with autoimmune IgG. O, As a negative control, anti-human IgG alone was used on LPS–stimulated neutrophils. P, LPS-treated neutrophils were used to detect deiminated histone H3. anti–citrullinated histone H3 (Anti-Cit H3) was detected with green fluorescence. IgG binding is shown in red; DNA is shown in blue. The overlap of green and blue appears as aqua. Original magnification × 400.
Binding of patient IgG to NETs was particularly striking. Binding of IgG from FS patient 2 largely overlapped with NET chromatin (Figure 1D), and IgG binding closely matched the lace-like distribution of dispersed nuclear DNA. A similar pattern of IgG binding was observed with serum from FS patient 3 (Figure 1F). The binding of IgG from RA patients 3 and 4 was focused into domains that mostly coincided with the location of DNA, although certain patches of chromatin remained unbound (Figures 1H and J). Binding of IgG from SLE patient 6 was observed in close association with NETs, and it overlapped extensively with the extracellular chromatin (Figure 1L), as did IgG from SLE patient 7, the binding of which closely matched the distribution of NET DNA (Figure 1N). The pattern of patient IgG binding resembled, in most instances, the distribution of deiminated histone H3 (Figure 1P). The congruent distributions of IgG immunoreactivity and of deiminated histone H3 suggested that, at least in part, immunoreactivity may be enhanced by histone deimination.
Preferential antibody binding to deiminated histones in ELISA
Previous reports have indicated that neutrophils efficiently deiminate core histones (24). Our own data confirmed that core histones are the major substrates of PAD-4 deimination in activated neutrophils (further information is availableat http://www.uthsc.edu/research/research_data/RadicM/ar11-0195RevFinalSuppl.pdf), although a number of other proteins also are PAD-4 substrates. To directly assess whether deiminated histones are preferred targets for FS, RA, or SLE patient autoantibodies, we deiminated histones in vitro and measured IgG binding by ELISA.
Binding curves were determined for sera from 23 FS, 37 RA, and 32 SLE patients. Figures 2A–D show representative curves. Five of 6 FS patient sera showed IgG binding to histones on ELISA, and, for each of those 5 sera, binding to deiminated histones exceeded binding to nondeiminated histones by 2–10-fold (Figure 2A). The sixth sample showed negligible binding. In contrast, RA sera bound less avidly to histones and, except for 1 sample, the sera showed little to no preference for deiminated histones (Figure 2B). Histone binding was most pronounced with SLE sera, yet except for 1 sample (triangles in Figure 2C), the binding to deiminated and nondeiminated histones was comparable. In the exceptional serum sample, IgG binding to deiminated histones was >30-fold stronger than to nondeiminated histones. Control sera showed very little to no binding to either type of histone (Figure 2D). LG2-2, a mouse anti–histone H2B monoclonal antibody (35), bound equally to either substrate, thus indicating equal coating of plates with deiminated and nondeiminated histones (Figure 2E).
Figure 2.
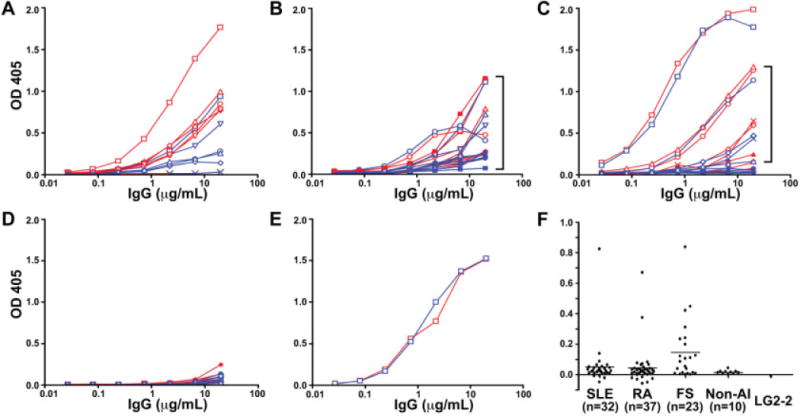
Serum IgG binding to deiminated or nondeiminated histones. A–D, Sera from FS patients (A), RA patients (B), or SLE patients (C) and from control subjects without autoimmune diseases (Non-AI) (D) were tested for binding to deiminated histones (red curves) and nondeiminated histones (blue curves) by enzyme-linked immunosorbent assay. Optical density (OD) values are plotted along the y-axes. E, A mouse anti–histone H2B monoclonal antibody (LG2-2) was used as a control. F, Differences in binding to deiminated and nondeiminated histones were computed for each serum at 6.6 μg/ml of total IgG (in the linear range of titration curves). Horizontal bars show the mean. Significant differences between sera from FS patients and sera from SLE patients, RA patients, and control subjects without autoimmune disease were observed (P = 0.028, P = 0.017, and P = 0.002, respectively). Different symbols in A–E represent different patient or control serum samples. Brackets in B and C indicate sera with the greatest preference for deiminated histones. See Figure 1 for other definitions.
We subtracted the binding to nondeiminated histones from the binding to deiminated histones at 6.6 μg/ml IgG, in order to determine the relative binding preference for deiminated histones (Figure 2F). This measure indicated that FS patient sera were unique among the sera tested, in that binding to deiminated histones was generally higher than binding to nondeiminated histones. The SLE group had 1 serum sample with greatly enhanced preference for deiminated histones, and other sera that bound with only low relative preference to deiminated histones. IgG from RA patient sera showed only slightly higher binding to histones than did control sera, and only 2 sera bound preferentially to deiminated histones on ELISA. These data indicate that FS patients differ significantly from RA and SLE patients in preferential binding of their antibodies to deiminated histones.
Autoantibodies distinguish between different deiminated histones
The ELISA with total histones revealed binding preferences that were robust and directed at native antigens. However, subtle preferences directed at individual histones may not have been detected with the ELISA. To determine which histones are preferentially bound by autoantibodies, we separated deiminated and nondeiminated histones by SDS-PAGE and probed them with patient and control sera (Figure 3). As shown by Coomassie blue staining, both types of histones were present in equivalent amounts. Moreover, the faster mobility of the deiminated histones and their immunochemical reaction pattern indicated that, as expected, 3 core histones contained citrullines.
Figure 3.
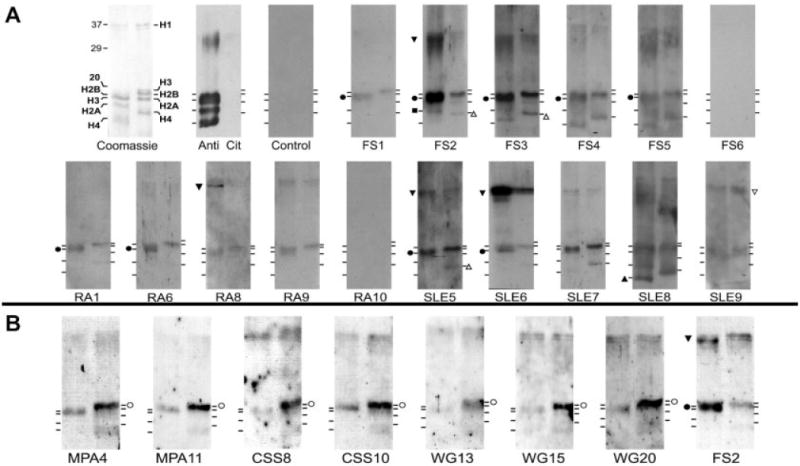
Autoantibodies distinguish between different histones. A, Representative SLE, RA, and FS patient sera exhibit different preferences for individual deiminated or nondeiminated histones. Equal amounts of in vitro–deiminated histones (left lanes) or nondeiminated histones (right lanes) were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and stained with Coomassie blue or reacted with an anticitrulline antibody (Anti Cit) on a Western blot. Binding of IgG from healthy individuals (Control) or from FS, RA, or SLE patients to deiminated and nondeiminated histones was detected on Western blots. B, Sera from patients with vasculitis preferentially recognize nondeiminated histones. Western blotting was performed with sera from patients with microscopic polyangiitis (MPA), Churg-Strauss syndrome (CSS), and granulomatosis with polyangiitis (Wegener’s) (WG) that bound histones on enzyme-linked immunosorbent assay. Serum from FS patient 2 was used as a positive control for deiminated histone binding. Increased binding to individual histones is marked by symbols. Solid symbols indicate preferential binding to deiminated histones; open symbols indicate preferential binding to nondeiminated histones. Circles represent histone H3; squares represent histone H2A; upright triangles represent histone H4; inverted triangles represent histone H1. See Figure 1 for other definitions.
The majority of FS patient sera and a minority of the RA and SLE patient sera (some quite weakly, e.g., serum from RA patient 1) bound with preference to one or more deiminated histones (Figure 3A). Most of the samples that scored positive for antibodies to deiminated histones bound deiminated histone H3, although preferential reactivities with deiminated histone H2A (serum from FS patient 2) or deiminated histone H4 (serum from SLE patient 8) were also observed. Preferential binding to citrulline-containing epitopes from different core histones demonstrated that flanking sequences contribute to antibody binding. Preferential binding to deiminated over nondeiminated histone H3 was confirmed by probing multiple gels and quantifying the statistical significance of binding (further information is available at http://www.uthsc.edu/research/research_data/RadicM/ar11-0195RevFinalSuppl.pdf).
Interestingly, 4 sera (from FS patient 2, RA patient 8, and SLE patients 5 and 6) preferentially bound to a deiminated protein migrating near 35 kd. Analyses using anti–modified citrulline antibody revealed a band migrating near 35 kd, the approximate size of linker histone H1, suggesting that histone H1 is recognized by a variety of patient sera. Although it will be necessary to demonstrate conclusively that histone H1 is a substrate for PAD-4, the possibility is intriguing because histone H1 controls the higher-order structure of chromatin. The analysis of patient sera by Western blotting confirmed the striking preference of FS patient antibodies for deiminated histones and identified histone epitopes that may be particularly autoreactive.
Autoantibodies from patients with AAVs preferentially bind to nondeiminated histones
As a control, we analyzed antihistone autoantibodies in patients with autoimmune disorders characterized by antineutrophil immunoreactivity. GPA, MPA, and CSS constitute a group of vasculitic disorders associated with ANCAs (36). The characteristics of 25 GPA patients, 15 MPA patients, and 15 CSS patients with vasculitis, including their autoantibody profiles and therapy, are shown in Table 1. By ELISA, only 7 AAV patient sera bound to histones (data not shown). When those sera were tested by Western blot analysis, all 7 preferentially bound nondeiminated histones over deiminated histones (Figure 3B). As before, serum from FS patient 2 preferentially bound deiminated histones. These results showed that AAV patients overall have a low incidence of antihistone autoantibodies and that binding of those antibodies is inhibited by deimination.
LPS stimulation of neutrophils generates preferred histone autoepitopes
The above-described experiments demonstrated that in vitro deimination of histones with PAD-4 affects autoreactive IgG binding. To test whether neutrophil activation also generates autoepitopes that mediate autoreactive IgG binding, we exposed neutrophils to LPS for 2 hours and probed blots of neutrophil lysates with serum from FS patient 2 and a control antiserum (Figure 4A). Indeed, the antibodies from FS patient 2 showed preferential binding to a protein migrating with the expected mobility of deiminated histone H3, providing evidence that the relevant FS patient 2 IgG histone autoepitopes were generated by neutrophil exposure to inflammatory stimuli. In addition, other proteins were immunoreactive, and LPS stimulation broadly increased the binding of FS patient 2 IgG to cell extracts (Figure 4A). These results were consistent with the induction of immunoreactive epitopes by PAD-4 activation in neutrophils.
Figure 4.
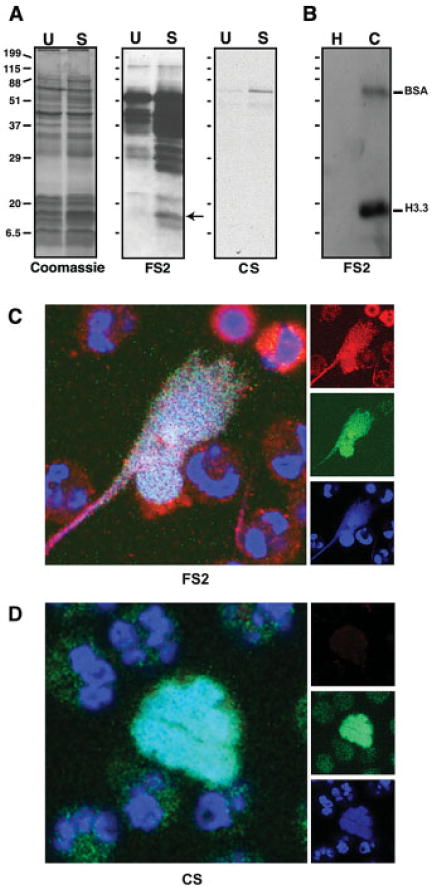
Serum from FS patient 2 preferentially binds histones from activated neutrophils, deiminated recombinant histone H3.3, and neutrophil extracellular chromatin traps. A, Equal amounts of cellular proteins from unstimulated (U) or LPS-stimulated (S) neutrophils were stained with Coomassie blue or probed with serum from FS patient 2 or control serum. Protein of appropriate mobility for histone H3 (arrow) was more reactive with FS patient 2 IgG following LPS treatment, consistent with increased binding to the deiminated histone H3. Control serum did not bind. B, Equal amounts of nondeiminated (H) or deiminated (C) proteins (histone H3.3 and bovine serum albumin [BSA]) were probed by Western blotting using FS patient 2 IgG. C and D, LPS-stimulated neutrophils were tested for binding to serum from FS patient 2 (C) and control serum (D). Human IgG binding is shown in red; DNA is shown in blue; rabbit anti–citrullinated histone H3 is shown in green. The red, green, and blue channel images that yielded the composite images are shown along the right. Original magnification × 700. See Figure 1 for other definitions.
Deimination is responsible for preferential histone binding
Histones accept numerous posttranslational modifications that, in various combinations, constitute a “histone code” (37). In order to exclude possible contributions of other posttranslational modifications to the preferential binding of autoantibodies, we used histone H3.3 that was produced in Escherichia coli and was thus devoid of eukaryotic posttranslational modifications. We deiminated histone H3.3 with PAD-4 in vitro. In parallel, we deiminated BSA to test IgG cross-reactivity with citrullines in an unrelated polypeptide.
By Western blotting, FS patient 2 IgG bound with strong preference to the deiminated histone H3.3 over the nondeiminated histone. This result indicated that citrulline is a major determinant of FS patient 2 IgG binding. Furthermore, a low level of binding to deiminated BSA was also observed (Figure 4B), suggesting that binding can accommodate some degree of heterogeneity in flanking sequences.
To localize FS patient 2 IgG binding relative to deiminated histone H3, we stimulated neutrophils with LPS on coverslips and incubated them with a dilution of FS patient 2 serum and rabbit anti–deiminated histone H3. Binding was visualized with anti-human IgG and anti-rabbit IgG antibodies (red versus green in Figure 4C). The FS patient 2 IgG and rabbit IgG antibodies were detected along NET chromatin that was visualized using Sytox orange. The extent of colocalization between the antibodies and DNA was nearly complete (Figure 4C). This confirmed that FS patient 2 serum contained antibodies that recognize deiminated histone H3 and other, more discontinuously arranged NET components. FS patient 2 IgG also bound neutrophils prior to the release of nuclear chromatin, suggesting that autoantigen posttranslational modifications occur in cytoplasmic foci of LPS-activated neutrophils.
Circulating neutrophils from RA patients show spontaneous activation
To assess histone deimination in neutrophils circulating in sera from RA and FS patients and control subjects, we isolated neutrophils and probed them for deiminated histone H3 by Western blotting. Deiminated histone H3 signal intensities were normalized relative to total histone H3. The relative intensities showed a 2.9-fold higher level of histone H3 deimination in RA patient neutrophils compared with control subject neutrophils (P < 0.008) (Figure 5A). The increased levels of deiminated histones in circulating neutrophils from RA patients suggest a greater degree of PAD-4 activation. In contrast, we observed no significant increase in histone deimination in FS patient neutrophils compared with control subject neutrophils (P > 0.1).
Figure 5.
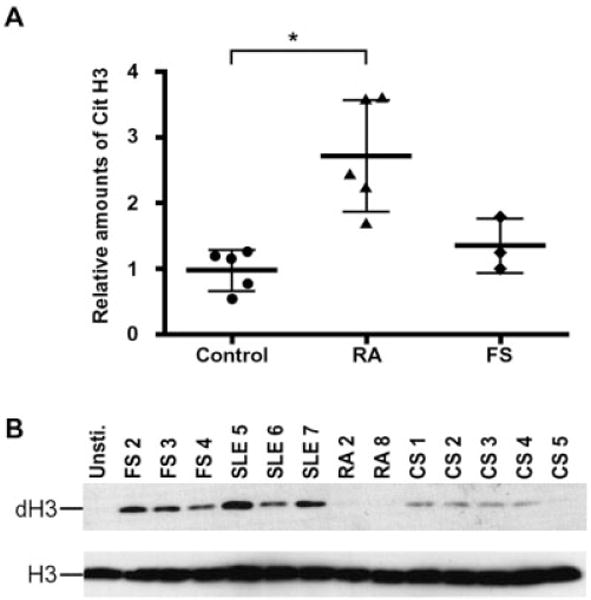
Deiminated histone H3 levels in circulating neutrophils, and stimulation of neutrophils by autoimmune sera. A, Neutrophil lysates were prepared from freshly isolated neutrophils from RA and FS patients and control subjects. Histone H3 deimination was determined by rabbit anti–deiminated histone H3 Western blotting and normalized relative to total histone H3. The normalized amounts of deiminated histone H3 were plotted relative to the control group, the mean value of which was set to 1. Values are the mean ± SD. The difference between the RA patient and control group sera was statistically significant (* = P < 0.008), but the difference between the FS patient and control group sera was not (P > 0.1). B, Human neutrophils were stimulated with autoimmune sera diluted to 2 μg/ml of IgG for 2 hours and analyzed for the presence of deiminated histone H3 (dH3). The blot was stripped and reprobed with rabbit anti–total histone H3. Cit H3 = citrullinated histone H3; Unsti. = unstimulated (see Figure 1 for other definitions).
To explore whether sera from FS, RA, or SLE patients contain substances that activate neutrophils, we incubated neutrophils from healthy donors with 2 μg/ml of patient sera and observed histone H3 deimination after 2 hours of incubation. Both SLE and FS patient sera induced histone H3 deimination, suggesting the presence of immune complexes or cytokines that activate neutrophils, whereas RA patient sera were inactive as inducers of PAD-4 in neutrophils (Figure 5B). Control subject sera showed low or undetectable induction of histone H3 deimination.
DISCUSSION
Autoantibodies target diverse posttranslational modifications in autoantigens (38–40), yet few posttranslational modifications are uniquely associated with infections and inflammation. The exception is histone deimination by PAD-4. A variety of inflammatory stimuli induce histone deimination in neutrophils, whereas apoptosis suppresses deimination (11). In the current study, we have demonstrated that autoantibodies to deiminated histones are prevalent in patients with FS. In contrast, autoantibodies to deiminated histones are rare in RA and SLE patients and absent from patients with AAVs. Our results thus suggest that inflammatory activation of neutrophils plays a central role in the induction of FS.
Our observations match findings of others that NET components are antigens in autoimmunity. Autoantibodies from patients with systemic autoimmunity recognize innate immune defense proteins that associate with NETs. This is the case for the granule proteases elastase, cathepsin G, and PR3 (13–15), as well as for various bactericidal peptides that are stored in granules and copurify with NETs, such as neutrophil defensins and the LL37 peptide (14,41). PAD-4, the enzyme responsible for histone deimination, is an autoantigen in RA patients (42), and anti–PAD-4 antibodies precede development of clinical disease and predict its severity (43). In turn, a growing number of PAD-4 substrates have been recognized as targets of autoantibodies. These include deiminated vimentin (20) and proteins of the extracellular matrix (17,19,21), suggesting that PAD-4 is active both inside and outside of cells.
How PAD-4 may act on extracellular substrates became clear when it was discovered that PAD-4 activation is part of the innate immune response to infections (11). Neutrophils release nuclear chromatin and associated enzymes in response to an encounter with pathogens (9,10). The externalized chromatin entangles pathogens and thus becomes linked to potent adjuvants that may stimulate adaptive immunity. Clearly, uptake and presentation of “NET-ted” pathogens juxtaposes microbial antigens and NET autoantigens, including DNA and histones. Upon pathogen uptake and processing, self and foreign antigens may be presented to T cells. The identification of autoantibodies to deiminated histones strongly suggests that NETs provide a source of autoantigens in vivo. Identification of autoantibodies to deiminated histones and other NET components (13,14) implicates NETs as the stimulus for autoantibody production and suggests a link between infections or inflammation and the development of autoreactivity.
To directly examine the availability of autoantigens in vivo, we quantified deiminated histone H3 in freshly isolated blood neutrophils from RA and FS patients and control subjects by Western blot analysis. We observed nearly 3-fold higher levels of deiminated histones in RA patients relative to controls (Figure 5A), suggesting ongoing spontaneous activation of circulating neutrophils. Deiminated histones were not significantly elevated in FS patients relative to controls. Because FS exhibits spontaneous activation and accelerated clearance of circulating neutrophils, our observation may be skewed by the fact that an increased proportion of circulating neutrophils in FS consists of immature (PAD-4–negative) neutrophils (44). Thus, the deiminated histones that were detected in FS patient neutrophils may have been derived from a small fraction of total neutrophils that expressed sufficiently high levels of PAD-4. The observation of potent neutrophilstimulating activity in FS and SLE patient sera (Figure 5B) supports this conclusion. Together, these results suggest depletion of mature neutrophils from FS patients along with the chronic generation of neutrophil-derived autoantigens in patients with systemic autoimmune disorders.
In certain forms of vasculitis, neutrophils are hyperactivated and serum levels of neutrophil granule components are elevated in parallel with more advanced disease (45). In addition, the severity of lupus correlates with levels of autoantibodies to the NETs’ structural scaffold, DNA (46). Other studies directly point to neutrophil activation as a key factor in the stimulation of autoantibodies. Notably, SLE patient neutrophils show an exacerbated response to autoantibody complexes and are more prone to NET release (47). One interesting aspect of these findings and those of our studies is that reactivities with different NET antigens correlate with different disease manifestations. Just as PR3 ANCAs predominantly occur in GPA and MPO ANCAs in MPA (48), the anti–deiminated histone autoantibodies associate selectively with FS. We interpret the rare development of anti–deiminated histone antibodies in RA as an indication that tolerance to histones remains active and resilient even after development of clinical disease. Tolerance to deiminated histones may only be broken after strong stimulation of adaptive immunity by unique epitopes that drive both B and T cell expansion.
In conclusion, we propose that anti–deiminated histone autoantibodies are markers deserving particular attention in patients with severe inflammation and chronic activation of neutrophils who may be at risk of progression to more severe disease. Chronic presentation of deiminated antigens may, over time, lead to the development of antigen-specific responses that target the modified antigen determinants and contribute to the development of other clinical features of FS. The data presented in this report allow us to propose a model of FS pathogenesis that is based on the biologic properties of neutrophils, the disease manifestations of FS, and our data on the high incidence of autoantibodies to deiminated histones. In FS, preexisting arthritis and an increased incidence of infections may stimulate a neutrophil response that includes histone deimination and the expulsion of chromatin from the cell. NETs containing deiminated histones, in complex with bacterial adjuvants, are the most likely antigenic trigger to account for the production of autoantibodies to the modified histones. These autoantibodies or their immune complexes may further stimulate neutrophils, thus completing a self-sustaining cycle that drives depletion of mature neutrophils.
Acknowledgments
The authors thank Dr. Srinivasa K. Puranam for help with statistical analysis of the data and Mr. Tim Higgins, senior illustrator, for expert assistance in the preparation of figures.
Supported by the Lupus Research Institute and the Dana Foundation Program in Human Immunology (research grants to Dr. Radic).
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Radic had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Dwivedi, Upadhyay, Knuckley, Thompson, Radic.
Acquisition of data. Dwivedi, Upadhyay, Neeli, Khan, Pattanaik, Myers, Kirou, Knuckley, Thompson, Crow, Mikuls, Csernok, Radic.
Analysis and interpretation of data. Dwivedi, Upadhyay, Neeli, Khan, Pattanaik, Hellmich, Knuckley, Thompson, Radic.
References
- 1.Barr SG, Zonana-Nacach A, Magder LS, Petri M. Patterns of disease activity in systemic lupus erythematosus. Arthritis Rheum. 1999;42:2682–8. doi: 10.1002/1529-0131(199912)42:12<2682::AID-ANR26>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Aletaha D, Breedveld FC, Smolen JS. The need for new classification criteria for rheumatoid arthritis. Arthritis Rheum. 2005;52:3333–6. doi: 10.1002/art.21410. [DOI] [PubMed] [Google Scholar]
- 3.Balint GP, Balint PV. Felty’s syndrome [review] Best Pract Res Clin Rheumatol. 2004;18:631–45. doi: 10.1016/j.berh.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Loughran TP., Jr The spectrum of large granular lymphocyte leukemia and Felty’s syndrome [review] Curr Opin Hematol. 2011;18:254–9. doi: 10.1097/MOH.0b013e32834760fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rose NR. The role of infection in the pathogenesis of autoimmune disease. Semin Immunol. 1998;10:5–13. doi: 10.1006/smim.1997.0100. [DOI] [PubMed] [Google Scholar]
- 6.Grassme H, Jendrossek V, Gulbins E. Molecular mechanisms of bacteria induced apoptosis. Apoptosis. 2001;6:441–5. doi: 10.1023/a:1012485506972. [DOI] [PubMed] [Google Scholar]
- 7.Cline AM, Radic MZ. Apoptosis, subcellular particles, and autoimmunity [review] Clin Immunol. 2004;112:175–82. doi: 10.1016/j.clim.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells [review] Cell. 2010;140:619–30. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 10.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180:1895–902. doi: 10.4049/jimmunol.180.3.1895. [DOI] [PubMed] [Google Scholar]
- 12.Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun. 2009;1:194–201. doi: 10.1159/000206974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manolova I, Dancheva M, Halacheva K. Antineutrophil cytoplasmic antibodies in patients with systemic lupus erythematosus: prevalence, antigen specificity, and clinical associations. Rheumatol Int. 2001;20:197–204. doi: 10.1007/s002960100108. [DOI] [PubMed] [Google Scholar]
- 14.Tamiya H, Tani K, Miyata J, Sato K, Urata T, Lkhagvaa B, et al. Defensins- and cathepsin G-ANCA in systemic lupus erythematosus. Rheumatol Int. 2006;27:147–52. doi: 10.1007/s00296-006-0173-9. [DOI] [PubMed] [Google Scholar]
- 15.Schnabel A, Csernok E, Isenberg DA, Mrowka C, Gross WL. Antineutrophil cytoplasmic antibodies in systemic lupus erythematosus: prevalence, specificities, and clinical significance. Arthritis Rheum. 1995;38:633–7. doi: 10.1002/art.1780380509. [DOI] [PubMed] [Google Scholar]
- 16.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–62. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senshu T, Kan S, Ogawa H, Manabe M, Asaga H. Preferential deimination of keratin K1 and filaggrin during the terminal differentiation of human epidermis. Biochem Biophys Res Commun. 1996;225:712–9. doi: 10.1006/bbrc.1996.1240. [DOI] [PubMed] [Google Scholar]
- 18.Pritzker LB, Joshi S, Gowan JJ, Harauz G, Moscarello MA. Deimination of myelin basic protein. 1. Effect of deimination of arginyl residues of myelin basic protein on its structure and susceptibility to digestion by cathepsin D. Biochemistry. 2000;39:5374–81. doi: 10.1021/bi9925569. [DOI] [PubMed] [Google Scholar]
- 19.Masson-Bessiere C, Sebbag M, Girbal-Neuhauser E, Nogueira L, Vincent C, Senshu T, et al. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the α- and β-chains of fibrin. J Immunol. 2001;166:4177–84. doi: 10.4049/jimmunol.166.6.4177. [DOI] [PubMed] [Google Scholar]
- 20.Vossenaar ER, Despres N, Lapointe E, van der Heijden A, Lora M, Senshu T, et al. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res Ther. 2004;6:R142–50. doi: 10.1186/ar1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uysal H, Bockermann R, Nandakumar KS, Sehnert B, Bajtner E, Engstrom A, et al. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J Exp Med. 2009;206:449–62. doi: 10.1084/jem.20081862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–81. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–63. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Nakashima K, Hagiwara T, Yamada M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J Biol Chem. 2002;277:49562–8. doi: 10.1074/jbc.M208795200. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Burlingame RW, Casiano CA, Boey ML, Rubin RL. Antihistone antibodies in systemic lupus erythematosus: assay dependency and effects of ubiquitination and serum DNA. J Rheumatol. 1994;21:1081–91. [PubMed] [Google Scholar]
- 26.Tuaillon N, Muller S, Pasquali JL, Bordigoni P, Youinou P, van Regenmortel MH. Antibodies from patients with rheumatoid arthritis and juvenile chronic arthritis analyzed with core histone synthetic peptides. Int Arch Allergy Appl Immunol. 1990;91:297–305. doi: 10.1159/000235131. [DOI] [PubMed] [Google Scholar]
- 27.Cohen MG, Webb J. Antihistone antibodies in rheumatoid arthritis and Felty’s syndrome. Arthritis Rheum. 1989;32:1319–24. doi: 10.1002/anr.1780321020. [DOI] [PubMed] [Google Scholar]
- 28.Miriovsky BJ, Michaud K, Thiele GM, O’Dell JR, Cannon GW, Kerr G, et al. Anti-CCP antibody and rheumatoid factor concentrations predict greater disease activity in men with rheumatoid arthritis. Ann Rheum Dis. 2010;69:1292–7. doi: 10.1136/ard.2009.122739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 30.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 31.Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 32.Kearney PL, Bhatia M, Jones NG, Yuan L, Glascock MC, Catchings KL, et al. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry. 2005;44:10570–82. doi: 10.1021/bi050292m. [DOI] [PubMed] [Google Scholar]
- 33.Csernok E, Holle J, Hellmich B, Willem J, Tervaert C, Kallenberg CG, et al. Evaluation of capture ELISA for detection of anti-neutrophil cytoplasmic antibodies directed against proteinase 3 in Wegener’s granulomatosis: first results from a multicentre study. Rheumatology (Oxford) 2004;43:174–80. doi: 10.1093/rheumatology/keh028. [DOI] [PubMed] [Google Scholar]
- 34.Mathsson L, Mullazehi M, Wick MC, Sjoberg O, van Vollenhoven R, Klareskog L, et al. Antibodies against citrullinated vimentin in rheumatoid arthritis: higher sensitivity and extended prognostic value concerning future radiographic progression as compared with antibodies against cyclic citrullinated peptides. Arthritis Rheum. 2008;58:36–45. doi: 10.1002/art.23188. [DOI] [PubMed] [Google Scholar]
- 35.Losman MJ, Fasy TM, Novick KE, Monestier M. Relationships among antinuclear antibodies from autoimmune MRL mice reacting with histone H2A-H2B dimers and DNA. Int Immunol. 1993;5:513–23. doi: 10.1093/intimm/5.5.513. [DOI] [PubMed] [Google Scholar]
- 36.Csernok E. Anti-neutrophil cytoplasmic antibodies and pathogenesis of small vessel vasculitides [review] Autoimmun Rev. 2003;2:158–64. doi: 10.1016/s1568-9972(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 37.Jenuwein T, Allis CD. Translating the histone code [review] Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 38.Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999;190:815–26. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utz PJ, Gensler TJ, Anderson P. Death, autoantigen modifications, and tolerance [review] Arthritis Res. 2000;2:101–14. doi: 10.1186/ar75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dieker J, Muller S. Epigenetic histone code and autoimmunity [review] Clin Rev Allergy Immunol. 2010;39:78–84. doi: 10.1007/s12016-009-8173-7. [DOI] [PubMed] [Google Scholar]
- 41.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andrade F, Darrah E, Gucek M, Cole RN, Rosen A, Zhu X. Autocitrullination of human peptidyl arginine deiminase type 4 regulates protein citrullination during cell activation. Arthritis Rheum. 2010;62:1630–40. doi: 10.1002/art.27439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolfenbach JR, Deane KD, Derber LA, O’Donnell CI, Gilliland WR, Edison JD, et al. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum. 2010;62:2633–9. doi: 10.1002/art.27570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dancey JT, Brubaker LH. Neutrophil marrow profiles in patients with rheumatoid arthritis and neutropenia. Br J Haematol. 1979;43:607–17. doi: 10.1111/j.1365-2141.1979.tb03793.x. [DOI] [PubMed] [Google Scholar]
- 45.Holle JU, Wu QJ, Moosig F, Gross WL, Csernok E. Membrane proteinase 3 (mPR3) expression on neutrophils is not increased in localized Wegener’s granulomatosis (WG) and Churg-Strauss syndrome (CSS) Clin Exp Rheumatol. 2010;28:46–50. [PubMed] [Google Scholar]
- 46.Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107:9813–8. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Dong Y, Zeng X, Li Y, Tang F. Clinical and pathological manifestations of patients with antineutrophil cytoplasmic autoantibodies directed against proteinase 3 or myeloperoxidase. Chin Med Sci J. 2002;17:32–5. [PubMed] [Google Scholar]