

Abstract
Background:
The ε4 allele of Apolipoprotein E is involved in lipid metabolism. Oxidative stress produces an increase in lipid peroxidation that has been implicated in the pathogenic cascade in Alzheimer’s disease. This study estimated the effect of the ε4 allele, malondialdehyde and lipid levels on the risk for Alzheimer’s disease.
Methods:
A total of 41 control subjects and 73 patients with Alzheimer’s disease were recruited. The Apolipoprotein E genotype was determined by amplification of exon 4 of the Apolipoprotein E by polymerase chain reaction (PCR); malondialdehyde concentration was determined by high-pressure liquid chromatography, and serum lipids were measured by routine photometric techniques.
Results:
Malondialdehyde levels were significantly higher in Alzheimer’s disease patients independent of the Apolipoprotein E genotype and ε4 allele. The ε4 allele increases the risk of Alzheimer’s disease by 5.114 times and elevated malondialdehyde levels increase the risk by 9.342.
Conclusion:
The presence of ε4 allele and elevated malondialdehyde levels are independent risk factors for Alzheimer’s disease. These findings support the hypothesis that lipid peroxidation and ε4 allele contribute to the pathogenic cascade in Alzheimer’s disease by different pathways.
Keywords: Alzheimer’s disease, Apolipoprotein E, lipids, malondialdehyde, lipid peroxidation
Background
The Apolipoprotein E (APOE) ε4 allele is considered the most prevalent and strongest genetic risk factor described in cases of Alzheimer’s disease (AD) of late or sporadic onset.1–4 The relative risk of suffering sporadic AD has been shown to be different depending on the genetic load of ε4 alleles, and the disease appears earlier in homozygotic individuals with this allele (APOE ε4/ε4).5,6 The mechanism by which the APOE genotype has an impact on AD is not fully understood, although there are proposed mechanisms through which APOE4 increases AD risk, including both amyloid-β (Aβ)-dependent effects, as modulation of Aβ levels, aggregation, neurotoxicity and neuroinflammation, and Aβ-independent effects, as neuronal development, glucose metabolism, brain activity and lipid metabolism.7 Potentially, the major impact may be related to its function as a lipid transporter. The human brain consists of 25% lipids, yet the role they play in the course of the disease is still a matter of research debate.8–11 APOE is an important modulator of many stages of lipoprotein metabolism and abnormal lipid metabolism is strongly related to the pathogenesis of AD. Traditionally, the increased risk was attributed to lower efficient of APOE4 in delivering cholesterol and essential lipids for the maintenance of synaptic integrity and plasticity.7,12–17
Furthermore, recent evidence demonstrates the multifunctional nature of the APOE protein. Of the three alleles of APOE, ε4 exhibits the least anti-oxidizing activity (ε2 > ε3 > ε4) and has been associated with other cerebral metabolic alterations such as oxidative stress (OS).18–24 The process of lipid peroxidation results in a range of intermediates and end products including lipid hydroperoxides, aldehydes and malondialdehyde (MDA).25,26 This lipid peroxidation produces a loss of membrane lipids resulting in an alteration of the structure, fluidity and permeability of the neuronal membranes. There is evidence to suggest that OS may also contribute to the pathogenic cascade in AD20,25,27–32 and this could potentially be the decisive step setting off a series of processes that take place in the disease.
The relation between plasma levels of lipid peroxidation, particularly MDA, and AD has been examined in many studies, of which have demonstrated increased levels of MDA in patients with AD.22,30,31,33 However, these studies did not adjust for APOE genotype and the associations of elevated level of plasma MDA and APOE polymorphism with AD is unknown. The aim of this study was to examine the effects of ε4 allele and plasma levels of MDA on the risk of AD. Also, we aimed to investigate the association between APOE genotypes and ε4 allele with MDA and lipid levels in AD.
Methods
Selection of patients and controls
The participants of this study were recruited from the Neurological Service Outpatients ’clinic at the General University Hospital, Elche, Spain. Patients and controls were free of known diseases related to oxidative metabolism: cardiovascular, respiratory, hepatic or neurological diseases, diabetes mellitus, neoplasic syndrome, severe arthrosis or alcoholism, and malnutrition. Furthermore, they had not taken antioxidants for at least 3 months.
Patients with probable AD according to McKhann et al.’s34 1984 criteria were included in the study. The standard study protocol (general laboratory tests, luetic serology, Thyroid stimulating hormone (TSH), vitamin B12 and folic acid, cerebral computed tomography) was applied to rule out other secondary dementias. Control subjects were free from any memory deficits (Mini Mental State Examination (MMSE) ⩾ 24 (20 in illiterate)), central neurological disease or symptoms (corroborated by an informant).
The study was conducted according to the provisions of the Helsinki Declaration, and Ethics Committee for Clinical Research of the General University Hospital of Elche approved the study. All participants or their families signed their consent for the participation in this study.
A total of 73 patients with probable AD (41 females/32 males; aged: 79.0 (65.0–89.0) years) and 41 control subjects (24 females/17 males; aged: 74.0 (62.0–84.0) years) were included for this study.
Blood samples were collected from all subjects in vacutainer tubes (Becton, Dickinson and Company (BD)). A whole blood sample was held for 30 min at room temperature to allow clotting. The sample was subsequently centrifuged at 3000 g for 10 min at 4°C. A fully automatic biochemistry analyzer (Olympus AU 5400 Automatic Analyzer) was used to detect levels of total cholesterol (TC), triglyceride (TG), high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C).
Ethylenediaminetetraacetic acid (EDTA) plasma was obtained for determining MDA and total blood with EDTA for determining APOE genotype. All the tubes were centrifuged immediately for 10 min at 2500 r/min, and plasma and buffy coat were transferred into separate tubes and stored at −80°C.
Laboratory measurements
MDA methodology
MDA concentration was determined by high-pressure liquid chromatography (HPLC) in the isocratic phase, in the Agilent 1100 series instrument, with fluorescent detection (Waters 474), using a kit from Chromsystems Instruments & Chemicals GmbH (ref. 67000, Munich, Germany). Sample preparation was based on an efficient protein precipitation; this procedure ensured proteins were efficiently removed (and consequently protein–MDA complex) along with any other interfering substances, followed by step of derivatization for MDA formation. The resulting fluorophore was specific and detectable at very low levels. The supernatant was injected in the HPLC system and fluorescence detection occurred with excitation at 515 nm and emission at 553 nm. The concentration of MDA was calculated from the area, on the basis of a calibration chromatogram performed with a standard solution of MDA. The samples were sheltered from the light throughout the full process of conservation and analysis.
Lipids methods
Lipids were measured by colorimetric assay: TC (cholesterol oxidase/esterase/peroxidase), HDL-C (selective immunological precipitation of B-lipoproteins and cholesterol oxidase/esterase/peroxidase), LDL-C (direct method, cholesterol oxidase/esterase/peroxidase) and TG (lipase/glycerol kinase).
APOE genotyping
APOE genotype was determined in the total blood collected in a tube with EDTA K3 as anticoagulant. The samples were centrifuged and the buffy coat was collected and DNA extracted. DNA was extracted automatically in the Bio Robot EZ1 kit from Qiagen which purifies genomic DNA from magnetic particles, thereby ensuring the production of a pure, high-quality product. The DNA sample to be amplified by PCR is introduced in a reagent mixture containing an excess of deoxynucleoside 5′-triphosphates (dNTPs), biotinylated primers and thermostable DNA polymerase. By heating, the two strands of the DNA helix are separated (denaturation) in order to expose the target sequences to the primers. These primers are complementary to the region flanking the target sequence. Therefore, upon cooling to a specific temperature, the primers will bind to their target region (annealing). At another temperature, and utilizing the dNTPs, the thermostable DNA polymerase will extend the annealed primers along the target template (extension). This way, two exact biotinylated copies of the template sequence are produced after one cycle of denaturation, annealing and extension. After 30 cycles, a multi-amplified biotinylated target sequence is obtained. The sample was amplified in a thermocycler Gene Amp PCR System 9700 from Applied Biosystems using the amplification kit from Innogenetics INNO-LIPA APOE, Innogenetics, Germany. Amplification products are subsequently hybridized using one typing strip on which four sequence-specific DNA probe lines and one control line are fixed. Reverse hybridization was performed with colorimetric detection of the different variants of APOE (in an Auto-LiPA-48 analyzer).
Statistical methods
The results of each group were analyzed using the statistical software package SPSS 22.0. The Kolmogorov–Smirnov test was applied in all continuous variables to define the presence of normality. Quantitative data were expressed as median and ranges (variables were non-normally distributed). Differences between control and AD groups were analyzed by the Mann–Whitney U-test. Comparisons between groups with different APOE genotypes were analyzed by chi-squared and Kruskal–Wallis tests for categorical and continuous variables, respectively. For evaluation of association between age and lipids with MDA, non-parametric Spearman’s correlation test was used.
Receiver operating characteristic (ROC) curve and the area under curve (AUC) were calculated for MDA levels. The effect of each variable was evaluated independently by logistic regression analyses. The MDA levels were categorized as the value of highest sensitivity and specificity obtained from the ROC curve. Adjusted odds ratio (OR) was calculated with multivariable logistic regression, including all variables that were significantly associated with the AD with a p-value of less than 0.10 (age, ε4 allele and higher MDA levels). In addition, OR and 95% confidence intervals (CIs) were calculated. p-values < 0.05 were accepted as statistically significant.
Results
The comparisons of age and sex across different groups are shown in Table 1. AD patients were significantly older than the control group (79.0 (65.0–89.0) vs 74.0 (62.0–84.0) years; p < 0.001). The frequencies for men and women did not differ significantly either in patients or controls (p = 0.806). As expected from the demographic distribution in the elderly population, there were more women than men in both groups. AD patients performed significantly worse on the MMSE than controls subjects (20.7 (14.0–26.0) vs 28.3 (25.0–30.0) years; p < 0.001).
Table 1.
Distribution of age, gender, APOE genotypes and alleles in AD and control subjects.
| AD (n = 73) | Controls (n = 41) | p | |
|---|---|---|---|
| Age (years) | 79.0 (65.0–89.0) | 74.0 (62.0–84.0) | <0.001* |
| Sex, n (%) | |||
| Male | 32 (43.8) | 17 (41.5) | 0.806 |
| Female | 41 (56.2) | 24 (58.5) | |
| MMSE | 20.7 (14.0–26.0) | 28.3 (25.0–30.0) | <0.001* |
| Alleles APOE, n (%) | |||
| ε2 | 5 (3.4) | 6 (7.3) | 0.091 |
| ε3 | 96 (65.8) | 66 (80.5) | 0.018* |
| ε4 | 45 (30.8) | 10 (12.2) | <0.001* |
| Genotypes APOE, n (%) | |||
| ε2/ε3 | 4 (5.5) | 6 (14.6) | 0.527 |
| ε3/ε3 | 34 (46.6) | 28 (68.3) | 0.370 |
| ε3/ε4 | 24 (32.9) | 4 (9.8) | <0.001* |
| ε4/ε4 | 10 (13.7) | 3 (7.3) | 0.132 |
| ε2/ε4 | 1 (1.4) | 0 | – |
| ε4 Allele | |||
| Non-carriers | 38 (52.1) | 34 (82.9) | 0.637 |
| Carriers | 35 (47.9) | 7 (17.1) | <0.001* |
APOE: Apolipoprotein E; AD: Alzheimer’s disease.
p < 0.05.
The APOE genotypes and allele frequencies observed in each group are presented in Table 1. The ε3 allele was the most abundant in our sample, with a frequency of 65.8% and 80.5% in AD patients and control participants, respectively. This frequency was significantly higher in the control group compared to AD patients (p = 0.018). We also found that the ε3/ε3 genotype was the most frequent in both groups (46.6% of AD patients and 68.3% of controls). The ε4 allele was the second most frequent, 30.8% in AD patients and 12.2% in control subjects. As expected, the proportion of ε4 allele was significantly higher in the AD patients (p < 0.001). The ε2 allele was less frequent in both groups (3.4% in AD patients and 7.3% in control subjects). The ε3/ε4 genotype was significantly higher in AD patients than the control group (32.9% vs 9.8%; p < 0.001). The frequency of patients ε4 allele carriers was significantly higher in AD patients than controls (47.9% vs 17.1%; p < 0.001).
Table 2 shows the MDA and lipid levels in plasma of AD and control groups, stratified according to the APOE genotype, and classified as APOE ε4 carriers and non-carriers. There was a significant increase in MDA in AD patients when compared to the control group (21.3 (10.2–45.6) and 12.4 (3.4–29.7) µg/L, respectively; p < 0.001). No significant differences were found between MDA levels in AD or control groups, stratified according to the APOE genotype, although we found the highest levels in the group with ε4/ε4 genotype (23.8 (14.0–33.2) µg/L for AD patients and 19.2 (8.8–23.9) µg/L for control subjects). Also, we observed that all AD subgroups showed higher plasma MDA levels compared to their corresponding control subgroups, although significant differences were found only in the ε3/ε3 and ε3/ε4 subgroups (p < 0.001 and p = 0.037, respectively). No statistically significant differences were found in the MDA levels of AD patients or control subjects between ε4 carriers and non-carriers. The MDA levels in AD subgroups with and without the ε4 allele were significantly higher than those observed in the control subgroup (24.5 (14.0–37.2) vs 12.4 (8.8–23.9) µg/L; p < 0.001 and 21.8 (10.2–30.0) vs 13.3 (3.41–29.7) µg/L; p = 0.09, respectively). A correlation study was performed to examine whether MDA levels were influenced by age (Table 3). No correlation was found in the total patients or in each group of patients (p > 0.05).
Table 2.
MDA and lipid levels of AD and control groups stratified according to the APOE genotype and the presence or absence of the ε4 allele.
| Group | All subjects | Genotype |
ε4 Allele |
||||
|---|---|---|---|---|---|---|---|
| ε2/ε3 (n = 10) | ε3/ε3 (n = 62) | ε3/ε4 (n = 28) | ε4/ε4 (n = 13) | non-carriers (n = 72) | carriers (n = 42) AD | ||
| AD | |||||||
| Total cholesterol | 209.0 (118.0–325.0) | 211.5 (179.0–311.0) | 205.0 (128.0–325.0) | 197.0 (118.0–290.0) | 251.0 (211.0–293.0) | 223.0 (128.0–311.0) | 199.5 (133.0–264.0) |
| HDL-C | 54.2 (26.5–85.5) | 51.1 (43.0–71.6) | 50.8* (36.2–85.5) | 54.9 (26.5–83.0) | 63.2 (50.5–84.4) | 47.4 (36.2–85.5) | 57.2 (26.5–70.3) |
| LDL-C | 131.0 (71.0–219.0) | 148.5 (109.0–202.0) | 130.0 (71.0–219.0) | 125.0 (76.0–190.0) | 161.0 (141.0–183.0) | 151.0 (71.0–202.0) | 119.0 (76.0–161.0) |
| Triglycerides | 105.0 (47.0–511.0) | 136.0 (101.0–511.0) | 108.5 (47.0–260.0) | 85.5 (57.0–375.0) | 105.5 (70.0–172.0) | 159.0 (47.0–511.0) | 85.5 (57.0–375.0) |
| MDA | 21.3* (10.2–45.6) | 19.4 (14.5–28.4) | 21.4* (10.2–45.6) | 18.5* (10.6–37.2) | 23.8 (14.0–33.2) | 21.8* (10.2–30.0) | 24.5* (14.0–37.2) |
| Controls | |||||||
| Total cholesterol | 214.0 (149.0–281.0) | 216.0 (157.0–259.0) | 200.0 (149.0–281.0) | 225.0 (164.0–259.0) | 263.0 (201.0–278.0) | 207.0 (149.0–281.0) | 242.0 (164.0–278.0) |
| HDL-C | 53.9 (35.7–167.0) | 59.9 (47.5–128.0) | 56.4* (39.0–167.0) | 47.6 (45.9–72.0) | 50.2 (47.2–52.4) | 56.4 (39.0–167.0) | 47.7 (45.9–72.0) |
| LDL-C | 136.0 (95.0–191.0) | 138.0 (96.0–179.0) | 133.0 (95.0–174.0) | 143.0 (109.0–158.0) | 175.0 (132.0–191.0) | 134.0 (95.0–179.0) | 154.0 (109.0–191.0) |
| Triglycerides | 112.0 (46.4–294.0) | 92.0 (47.7–123.0) | 112.0 (46.4–212.0) | 124.0 (52.0–160.0) | 160.0 (122.0–172.0) | 102.0 (46.4–212.0) | 156.0 (52.0–172.0) |
| MDA | 12.4* (3.4–29.7) | 13.2 (10.3–21.7) | 13.3* (3.4–29.7) | 10.8* (9.3–18.5) | 19.2 (8.8–23.9) | 13.3* (3.41–29.7) | 12.4* (8.8–23.9) |
MDA: malondialdehyde; AD: Alzheimer’s disease; APOE: Apolipoprotein E; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol.
The results of lipids are expressed in mg/dL; the results of MDA are expressed in µg/L.
Mann–Whitney U-test, p < 0.05.
Table 3.
Correlation between MDA and age and lipids levels in the groups studied.
| All subjects |
AD |
Controls |
||||
|---|---|---|---|---|---|---|
| MDA (µg/L) |
||||||
| r | p-value | r | p-value | r | p-value | |
| Age | 0.067 | 0.146 | −0.045 | 0.723 | −0.256 | 0.509 |
| Total cholesterol | 0.035 | 0.744 | −0.015 | 0.916 | 0.069 | 0.674 |
| HDL-C | −0.179 | 0.095 | −0.272 | 0.056 | 0.031 | 0.851 |
| LDL-C | 0.037 | 0.729 | 0.016 | 0.913 | 0.067 | 0.691 |
| Triglycerides | 0.135 | 0.204 | 0.081 | 0.572 | 0.044 | 0.792 |
MDA: malondialdehyde; AD: Alzheimer’s disease; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol.
Relative to lipid levels, no significant differences were found in serum levels of total TC, TG, HDL-C and LDL-C of AD patients compared to the control group (Table 2). When compared the other subgroups, the only difference was found between HDL-C levels between AD and controls that have the genotype ε3/ε3 (p = 0.04). Spearman’s correlation coefficient was performed to find any correlation between the plasma MDA concentration and the level of lipids. No correlation was found in any of the groups studied. Neither correlation was found considering all patients (Table 3). To analyze the differences between the levels of MDA as well as lipid levels, use an analysis of covariance (ANCOVA) to eliminate the influence that age may have on our results. We obtained the same results obtained by the Kruskal–Wallis and Mann–Whitney U-test by means of ANCOVA analysis, so that not-found significance with the age variable and our results are not influenced by the age.
The ROC curves for MDA levels are presented in Figure 1. The ROC analysis gives an AUC of 0.770 (p < 0.001, CI: 0.677–0.863). MDA level of 12.45 µg/L has a sensitivity of 90.8% and a specificity of 48.7% for detecting AD. Table 4 shows crude and adjusted OR with regard to the factors studied and AD. All significant factors, age, ε4 allele and MDA levels, were then analyzed together using multivariate logistic regression. Carrying the ε4 allele was a significant risk factor for AD (OR: 5.114; 95% CI: 1.808–14.468); after adjustment for confounder of age and MDA levels, this association was unchanged (less than 10%) (OR: 4.850; 95% CI: 1.588–14.806). The correlation between MDA levels and the risk of AD was analyzed. OR: 1.170 and 95% CI: 1085–1262 were obtained (data not shown). To see that the increase in a unit of MDA levels elevates 1.17 times the risk of AD, we proceeded to categorize the MDA levels selecting as a cutoff point the MDA levels obtained from the ROC curve “⩽12.45” or “>12.45.” MDA levels >12.45 µg/L was significant risk factor for AD (9.342; 95% CI: 3.274–26.658). This association was unchanged (less than 10%) after adjustment for age and the ε4 allele (9.433; 95% CI: 3.293–27.568). There was no relationship between the ε4 allele and higher MDA levels. We also explored the potential interaction between these variables and lipid levels. No interaction effect was found (Table 4).
Figure 1.
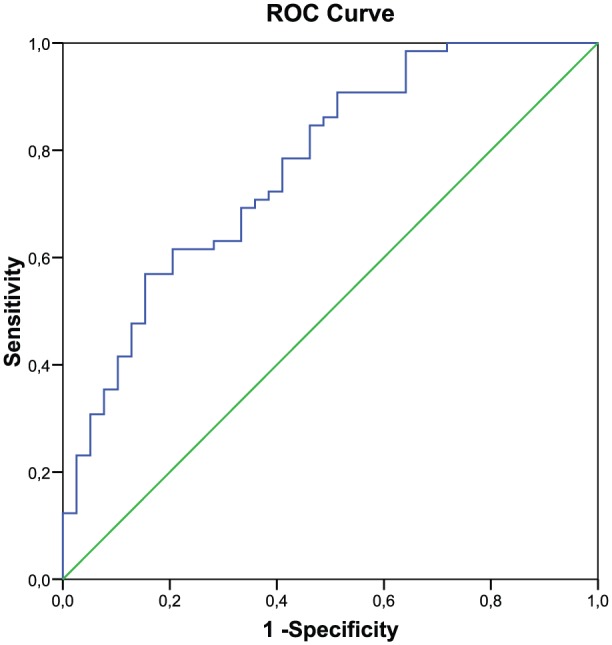
ROC curves of MDA.
Table 4.
Factors associated with AD.
| ORa | 95% CI | Adjusted ORb | 95% CI | |
|---|---|---|---|---|
| ε4 Allele | ||||
| Presence | 5.114 | (1.808–14.468) | 4.850 | (1.588–14.806) |
| MDA levels | ||||
| >12.45 | 9.342 | (3.274–26.658) | 9.443 | (3.293–27.568) |
| Total cholesterol | 1.000 | (0.990–1.010) | 1.014 | (0.969–1.060) |
| HDL-C | 0.979 | (0.956–1.002) | 0.992 | (0.961–1.024) |
| LDL-C | 0.998 | (0.985–1.011) | 0.971 | (0.916–1.029) |
| Triglycerides | 1.006 | (0.998–1.013) | 1.007 | (0.995–1.018) |
AD: Alzheimer’s disease; OR: odds ratio; CI: confidence interval; MDA: malondialdehyde; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol.
Crude OR.
ORs adjusted for age, ε4 allele and higher MDA levels.
Discussion
This study has shown that MDA levels, product of lipid peroxidation, were significantly higher in AD patients than in control subjects. Many studies have shown increased lipid peroxidation in the brains of AD patients and some of the products of lipid peroxidation, 4-hydroxynonenal, MDA, are found in high concentrations in AD patients.20,22,25,27–32 This result supports evidence that lipid peroxidation is elevated in AD and contributes to the pathogenic cascade in AD.
In addition, we have found that high MDA levels do not differ in AD depending on APOE genotype or the presence of the ε4 allele, suggesting that APOE genotype or ε4 allele does not influence the intensity of lipid peroxidation. There are many mechanisms by which APOE ε4 could affect many biological processes and increase the risk of AD. It has been recognized that antioxidant properties of APOE are altered by genotype. Studies with AD patients have revealed that APOE ε4 is associated with increased lipid peroxidation in post-mortem brains33 and with elevated hydroxyl radical levels in blood.21,23 However, as shown in this study and another study,21 the increased lipid peroxidation associated with APOE ε4 is not reflected when measured MDA in blood. This result suggests that significantly higher levels of MDA found in AD patients were independent of the ε4 allele. It is noteworthy that the levels of MDA were independent of the lipid levels as they were not dependent on the APOE genotype or presence of ε4 allele.
Extensive epidemiological data are available which demonstrates an association between the ε4 allele and risk of AD, but there was no information available about the association between MDA level and risk AD. This is the first study that investigated the risk associated with the presence of ε4 allele and MDA, together, with AD. OR analyses were in agreement with the hypothesis of an interaction between the ε4 allele and elevated lipid peroxidation for the development of AD. The ε4 allele, by itself, increased the risk of AD by 5.114 times and elevated MDA levels increased the risk by 9.342. In our study, the association between the ε4 allele and AD was not altered after adjustment for elevated MDA and lipid levels (OR: 4.850). Similarly, the association of elevated MDA levels and AD was not altered after adjustment for the ε4 allele and lipid levels (OR: 9.443). These findings indicate that the ε4 allele and high plasma value for MDA are independent risk factors for AD and could be used for identifying those patients with a high risk for AD. These results could be applied in clinical practice, especially in patients with mild cognitive impairment (MCI) who have a cognitive impairment but not having AD to assess the possible risk for AD and may be reviewed periodically and even treated earlier.
However, some studies have reported that the ε4 allele is associated with increased peripheral lipid levels.12,16 A review has suggested that this polymorphism accounts for approximately 10% of the variation in serum TC levels in the general population.12 Our result showed that lipid levels did not significantly differ between AD patients and control subjects as reported by some authors.11 In order to exclude the presence of high levels of lipid peroxidation due to high lipid levels, these were also measured and correlated with MDA levels. The results showed that the increment in plasma MDA described could not be ascribed to the presence of higher lipids levels.
Our study is limited by its small sample size; this is because the subjects of this study were free of know diseases that strongly affect the levels of OS. However, despite the small sample size, we were able to detect significant increases in MDA levels in patients with AD.
In conclusion, AD patients have higher lipid peroxidation independent to the presence of the allele ε4 and lipid levels; it is likely that other factors besides APOE are involved in OS in AD. The presence of allele ε4 and elevated MDA levels are independent risk factors for AD. This study could not establish the association between the presence of ε4 allele and lipid peroxidation, supporting the idea that this factors contribute to the pathogenic cascade in AD by different pathways.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical approval: Ethical approval for this study was obtained from the local Ethics Committee of the General University Hospital of Elche Foundation (Spain) with the code FIBElx08/07.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study received financial support from the General University Hospital of Elche Foundation (Spain) (FIBElx06/01 and FIBElx08/07).
Informed consent: Written informed consent was obtained from all subjects before the study.
References
- 1. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993; 261(5123): 921–923. [DOI] [PubMed] [Google Scholar]
- 2. Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993; 43(8): 1467–1472. [DOI] [PubMed] [Google Scholar]
- 3. Van Duijn CM, de Knijff P, Cruts M, et al. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat Genet 1994; 7(1): 74–78. [DOI] [PubMed] [Google Scholar]
- 4. Kivipelto M, Rovio S, Ngandu T, et al. Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med 2008; 12(6B): 2762–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997; 278(16): 1349–1356. [PubMed] [Google Scholar]
- 6. Cruz-Sanchez FF, Durany N, Thome J, et al. Correlation between apolipoprotein-E polymorphism and Alzheimer’s disease pathology. J Alzheimers Dis 2000; 2(3–4): 223–229. [DOI] [PubMed] [Google Scholar]
- 7. Liu CC, Kanekiyo T, Xu H, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 2013; 9(2): 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stefani M, Liguri G. Cholesterol in Alzheimer’s disease: unresolved questions. Curr Alzheimer Res 2009; 6(1): 15–29. [DOI] [PubMed] [Google Scholar]
- 9. Vance JE, Hayashi H, Karten B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol 2005; 16(2): 193–212. [DOI] [PubMed] [Google Scholar]
- 10. Kamino K, Tanaka T, Kida T, et al. The role of lipid metabolism in Alzheimer’s disease. Nihon Shinkei Seishin Yakurigaku Zasshi 2002; 22(4): 103–110. [PubMed] [Google Scholar]
- 11. Wood WG, Li L, Muller WE, et al. Cholesterol as a causative factor in Alzheimer’s disease: a debatable hypothesis. J Neurochem 2014; 129(4): 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lane RM, Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J Lipid Res 2005. May; 46(5): 949–968. [DOI] [PubMed] [Google Scholar]
- 13. Wehr H, Parnowski T, Puzynski S, et al. Apolipoprotein E genotype and lipid and lipoprotein levels in dementia. Dement Geriatr Cogn Disord 2000; 11(2): 70–73. [DOI] [PubMed] [Google Scholar]
- 14. Isbir T, Agachan B, Yilmaz H, et al. Apolipoprotein-E gene polymorphism and lipid profiles in Alzheimer’s disease. Am J Alzheimers Dis Other Demen 2001; 16(2): 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harris JR, Milton NG. Cholesterol in Alzheimer’s disease and other amyloidogenic disorders. Subcell Biochem 2010; 51: 47–75. [DOI] [PubMed] [Google Scholar]
- 16. De Chaves EP, Narayanaswami V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol 2008; 3(5): 505–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mahley RW, Rall SC., Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 2000; 1: 507–537. [DOI] [PubMed] [Google Scholar]
- 18. Kharrazi H, Vaisi-Raygani A, Rahimi Z, et al. Association between enzymatic and non-enzymatic antioxidant defense mechanism with apolipoprotein E genotypes in Alzheimer disease. Clin Biochem 2008; 41(12): 932–936. [DOI] [PubMed] [Google Scholar]
- 19. Jofre-Monseny L, Minihane AM, Rimbach G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res 2008; 52(1): 131–145. [DOI] [PubMed] [Google Scholar]
- 20. Su B, Wang X, Nunomura A, et al. Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res 2008; 5(6): 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramassamy C, Averill D, Beffert U, et al. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol Dis 2000; 7(1): 23–37. [DOI] [PubMed] [Google Scholar]
- 22. Aybek H, Ercan F, Aslan D, et al. Determination of malondialdehyde, reduced glutathione levels and APOE4 allele frequency in late-onset Alzheimer’s disease in Denizli, Turkey. Clin Biochem 2007; 40(3–4): 172–176. [DOI] [PubMed] [Google Scholar]
- 23. Tamaoka A, Miyatake F, Matsuno S, et al. Apolipoprotein E allele-dependent antioxidant activity in brains with Alzheimer’s disease. Neurology 2000; 54(12): 2319–2321. [DOI] [PubMed] [Google Scholar]
- 24. Ferguson S, Mouzon B, Kayihan G, et al. Apolipoprotein E genotype and oxidative stress response to traumatic brain injury. Neuroscience 2010; 168(3): 811–819. [DOI] [PubMed] [Google Scholar]
- 25. Chang YT, Chang WN, Tsai NW, et al. The roles of biomarkers of oxidative stress and antioxidant in Alzheimer’s disease: a systematic review. Biomed Res Int 2014; 2014: 182303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014; 2014: 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sinem F, Dildar K, Gokhan E, et al. The serum protein and lipid oxidation marker levels in Alzheimer’s disease and effects of cholinesterase inhibitors and antipsychotic drugs therapy. Curr Alzheimer Res 2010; 7(5): 463–469. [DOI] [PubMed] [Google Scholar]
- 28. Pulido R, Jimenez-Escrig A, Orensanz L, et al. Study of plasma antioxidant status in Alzheimer’s disease. Eur J Neurol 2005; 12(7): 531–535. [DOI] [PubMed] [Google Scholar]
- 29. Galbusera C, Facheris M, Magni F, et al. Increased susceptibility to plasma lipid peroxidation in Alzheimer disease patients. Curr Alzheimer Res 2004; 1(2): 103–109. [DOI] [PubMed] [Google Scholar]
- 30. Gustaw-Rothenberg K, Kowalczuk K, Stryjecka-Zimmer M. Lipids’ peroxidation markers in Alzheimer’s disease and vascular dementia. Geriatr Gerontol Int 2010; 10(2): 161–166. [DOI] [PubMed] [Google Scholar]
- 31. Shichiri M. The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr 2014; 54(3): 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGrath LT, McGleenon BM, Brennan S, et al. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM 2001; 94(9): 485–490. [DOI] [PubMed] [Google Scholar]
- 33. Mazur-Kolecka B, Kowal D, Sukontasup T, et al. The effect of oxidative stress on accumulation of apolipoprotein E3 and E4 in a cell culture model of beta-amyloid angiopathy (CAA). Brain Res 2003; 983(1–2): 48–57. [DOI] [PubMed] [Google Scholar]
- 34. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984; 34(7): 939–944. [DOI] [PubMed] [Google Scholar]