

Abstract
The biological functions and molecular mechanisms of miR-223 action in liver cells and liver diseases remain unclear. We therefore determined the effect and mechanism of action of miR-233 in Fas-induced hepatocyte apoptosis and liver injury. Wild-type (WT) and miR-223 knockout (KO) mice were treated i.p. with 0.5 μg/g body weight anti-Fas antibody Jo2, and the animals were monitored for survival and the extent of liver injury. Although WT mice died 4 to 6 hours after Jo2 injection (n = 6), all of the miR-223 KO mice (n = 6) survived. In comparison to WT mice, the miR-223 KO mice showed resistance to Fas-induced liver injury, as indicated by less tissue damage under histopathological examination, fewer apoptotic hepatocytes under caspase-3 immunostaining, and less elevation of serum transaminases. miR-223 KO livers showed less caspase-3, caspase-8, and caspase-9 activation and less poly (ADP-ribose) polymerase cleavage compared with WT livers (P < 0.05). Furthermore, tail vein injection of miR-223 lentiviral vector to miR-223 KO mice restored Jo2-induced liver injury. Transfection of miR-223 KO hepatocytes with miR-223 mimic enhanced Jo2-induced activation of caspase-3, caspase-8, and caspase-9, whereas transfection of WT hepatocytes with the miR-223 inhibitor attenuated Jo2-induced apoptosis. These findings demonstrate that miR-223 deficiency protects against Fas-induced hepatocyte apoptosis and liver injury. Further in vitro and in vivo data indicate that miR-223 regulates Fas-induced hepatocyte apoptosis and liver injury by targeting the insulin-like growth factor 1 receptor.
miRNAs are short noncoding RNA molecules that negatively regulate gene expression predominantly by binding to 3′-untranslated regions (UTRs) of the target genes. miR-223 was first implicated in the development and homeostasis of the immune system.1, 2, 3 Subsequently, it has been recognized that miR-223 also plays important roles in various physiological and pathological processes, including inflammation, carcinogenesis, autoimmune diseases, and several other diseases.4, 5 A recent study reports that miR-223 also coordinates cholesterol homeostasis.6 In the liver, the expression of miR-223 is increased during ischemic and reperfusion injury.7 miR-223 in hepatic macrophages (Kupffer cells) has been shown to inhibit IL-1β production and thus suppress proinflammatory response during concanavalin A–induced acute liver injury.8 A separate study shows that miR-223 in hepatocytes down-regulates the expression of cytochrome b5, thereby modulating cytochrome P450 activities in the liver.9 Although miR-223 is commonly repressed in hepatocellular carcinoma,10 miR-223 transfer from human macrophages was found to inhibit hepatocellular cancer cell proliferation.11 In addition, elevated serum levels of miR-223 are found in patients with hepatocellular carcinoma or chronic hepatitis B virus and have the potential to serve as a biomarker for liver injury.12 These findings suggest a potential role of miR-223 in liver pathobiology, although detailed biological functions and molecular mechanisms of miR-223 action in liver cells and liver diseases remain to be further defined.
Fas-induced hepatocyte apoptosis and liver injury are implicated in a wide range of liver diseases, such as viral hepatitis, alcoholic and nonalcoholic steatohepatitis, ischemia/reperfusion injury, and fulminant hepatic liver failure.13, 14, 15, 16, 17, 18, 19, 20 The current study was designed to investigate the effect and mechanisms of miR-223 in Fas-induced hepatocyte apoptosis and liver injury by using mice and hepatocytes with or without miR-223 deletion. Our findings provide novel evidence that miR-223 regulates Fas-induced hepatocyte apoptosis and liver injury by targeting the insulin-like growth factor receptor 1 (IGF1R) signaling pathway.
Materials and Methods
Animal Experiments
C57BL/6 wild-type (WT) mice and miR-223 knockout (KO) mice, obtained from the Jackson Laboratory (Bar Harbor, ME), were maintained and propagated at the Tulane University (New Orleans, LA) Health Sciences Center Animal Facility. Eight-week-old male C57BL/6 WT mice and miR-223 KO mice were used for experiments in this study. All experimental animals used in this study were treated according to the protocol approved by the Institutional Animal Care and Use Committee of Tulane University.
Liver injury induction was performed by i.p. injection with 0.5 μg/g body weight of the anti-Fas antibody Jo2 [dissolved in sterile phosphate-buffered saline (PBS); obtained from BD Bioscience, Franklin Lakes, NJ]. After Jo2 injection, the mice were sacrificed at indicated time points to assess the extent of liver injury. The liver tissues were excised, and the specimens were fixed in 10% formalin fixation for histological evaluation, as we previously described.21, 22 Portions of the liver samples were frozen in −80°C for protein extractions. On sacrifice, blood samples were collected from the orbital sinus cavity of the mice, as previously described.23 The blood samples were centrifuged at 450 × g for 15 minutes to separate serum from red blood cells. The sera were kept in −80°C until alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured using an automatic analyzer at the Department of Clinical Chemistry, Tulane University Hospital.
For experiments using IGF1R inhibitor treatment, the mice were injected i.p. with either dimethyl sulfoxide (Calbiochem, EMD Millipore, Billerica, MA) or NVP-AEW541 hydrochloride (25 mg/kg body weight; Cayman Chemical, Ann Arbor, MI) 30 minutes before Jo2 administration (0.5 μg/g of body weight), and the animals were sacrificed 4 hours after Jo2 injection.
In selected experiments, we used pre–miR-223 lentiviral vector to restore miR-223 expression in miR-223 KO mice. Specifically, 8-week-old miR-223 KO mice received a tail vein injection of 200 μL of lentiviral particle solution containing either scrambled control miRNA (L/control) or pre–miR-223 (L/miR-223) (purchased from Genecopoeia, Rockville, MD). Seven days after lentiviral injections, the mice were i.p. injected with Jo2 (0.5 μg/g of body weight). Four hours after Jo2 injection, the animals were sacrificed and the livers were harvested for further analysis.
In separate experiments, pBabe-bleo-Igf1r expression plasmid (Addgene, Cambridge, MA) was injected into 8-week-old WT mice via hydrodynamic tail vein injection. For this purpose, a total volume of 2 mL of 20 μg/mL sterile saline plasmid solution was administered into the tail vein of each animal. After 72 hours, the mice were i.p. injected with 0.5 μg/g of body weight Jo2; 4 hours after Jo2 injection, the animals were sacrificed and the livers were harvested for further analysis.
H&E Staining
Liver samples were harvested and fixed in 10% buffered formalin for paraffin embedding. Tissue sections (4 μm thick) were processed for hematoxylin and eosin (H&E) staining. Light microscopy was performed to evaluate the extent of liver tissue damage.
Caspase Activity Assays
Cytosolic extracts from liver tissues were prepared as previously studied.24 Caspase-3/7, caspase-8, and caspase-9 activities were analyzed using the Caspase-Glo Assay Kit (Promega Corp., Madison, WI).
Hepatocyte Isolation and Culture
Hepatocytes were isolated from WT and miR-223 KO mice by an adaptation of the two-step collagenase perfusion technique, as previously described.25 Cells were plated onto 6-well collagen-coated plates (BD Biosciences, San Jose, CA) and plated at 1 × 106 cells per well. Hepatocytes were cultured in Williams' medium E (Invitrogen, Grand Island, NY) that was supplemented with Hepatocyte Maintenance Supplement Pack (Invitrogen, Carlsbad, CA) with 10% fetal calf serum (Sigma, St. Louis, MO), 2 mmol/L l-glutamine (Invitrogen), and antibiotic-antimycotic (Invitrogen). The cells were allowed to adhere to the surface of the collagen-coated plates for at least 2 hours before proceeding forward with transfection or Jo2 treatment procedures. Cell lysates were obtained for Western blot analysis and caspase activity measurement.
Hepatocyte Transfection
Hepatocytes were harvested from both WT and miR-223 KO mice and plated onto collagen-coated 6-well plates (BD Biosciences, San Jose, CA) at 1 × 106 per well. After 2 hours of cell attachment, WT hepatocytes were transfected with control miRNA (Qiagen, Valencia, CA) or miR-223 inhibitor (Qiagen). miR-223 KO mice were transfected with control miRNA (Qiagen) or miR-223 mimic (Qiagen). These transfection processes were used with Targefect F2 plus Virofect enhancer (Targeting Systems, El Cajon, CA). In separate experiments, WT and miR-223 KO hepatocytes were transfected with IGF1Rα/β siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) using Targefect F2 plus Virofect enhancer. The cells were then incubated with 0.2 μg/mL Jo2 for 4 hours. Cell lysates were obtained for Western blot analysis.
Luciferase Reporter Assay
The 3′-UTR of mouse IGF1R was cloned downstream of the firefly luciferase reporter gene in pMIR-REP-dCMV vector (Flemington Lab, Tulane Medical Center, New Orleans, LA). Mutated IGF1R plasmid was constructed using the Quick Change II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). The primers for the cloning were as follows: forward, 5′-TATACGCGTAGAGACAGCCTTACGCTCAC-3′ (MluI); reverse, 5′-GTGAAGCTTCAATTCTCGAGTGTCCTGTTGC-3′ (HindIII). The mutagenic primers were as follows: forward, 5′-CTTTACCCGAACCCTTGACCGACATGGGCCTCTGC-3′; reverse, 5′-GCAGAGGCCCATGTCGGTCAAGGGTTCGGGTAAAG-3′. Hepatocytes were isolated from WT mice and plated onto collagen-coated 6-well plates at a density of 1 × 106 cells per well. After 2 hours of cell attachment, primary hepatocytes were transfected with miR-223 mimic (Qiagen) or control miRNA (Qiagen) using Targefect F2 plus Virofect enhancer (Targeting Systems). Twelve hours later, the hepatocytes were transfected with the 3′-UTR-IGF1R and mutants using Targefect-Hepatocyte reagent (Targeting Systems). Hepatocytes were harvested 24 hours after plasmid transfection. Luciferase activity was measured by centro xs3 lb 960 microplate luminometer (Berthold Technologies, Bad Wildbad, Germany) using a dual-luciferase reporter assay system (Promega, Madison, WI).
Quantitative Real-Time RT-PCR
Total RNA was extracted from the liver tissues with TRIzol (Invitrogen) and RNA reverse transcribed using Qiagen miScript RT kit (Qiagen). Qiagen miScript SYBR Green PCR kit and miR-223 miScript Primer Assay were used on the Bio-Rad (Hercules, CA) C1000 Thermal Cycler to determine the expression level of miR-223.
Western Blot Analysis
Proteins were isolated from liver tissue by lysing with NP-40 lysis buffer with one tablet of protease inhibitor cocktail (Roche Life Science, Indianapolis, IN) and one tablet of phosphatase inhibitor cocktail (Roche Life Science) in 10 mL lysis buffer. The homogenates were spun at 12,000 × g for 15 to 20 minutes at 4°C, where the supernatants were collected to measure protein concentration. After boiling for 10 minutes in the presence of 10% β-mercaptoethanol, the samples were separated by SDS-PAGE, transferred onto a nitrocellulose membrane (Bio-Rad), and then blocked in 10% dry nonfat milk/PBS with Tween 20 (PBS with 0.1% Tween 20) for 1 hour at room temperature. The blots were incubated with primary antibody in PBS with Tween 20 (1:1000 dilution) overnight at 4°C. After three washes, membranes were then incubated with secondary antibody (IRDye 680LT/IRDye 800CW secondary antibodies; LI-COR, Lincoln, NE) in PBS with Tween 20 (1:10,000 dilution) for 60 minutes at room temperature or 4°C overnight. Signals were visualized by ODYSSEY infrared imaging system (LI-COR Biosciences, Lincoln, NE). The primary antibodies used include IGF1R, phospho-IGF1R, phospho-Akt, poly (ADP-ribose) polymerase, caspase-3, cleaved caspase-8, and caspase-9 (Cell Signaling Technology, Beverly, MA); goat anti-rabbit or goat anti-mouse secondary antibodies were purchased from LI-COR Biosciences.
Statistical Analysis
Statistical analyses were performed using SPSS version 13.0 (SPSS Inc, Chicago, IL) and Microsoft Excel (Redmond, WA). Comparison between two groups was analyzed using paired t-test. To measure mortality rates, Kaplein-Meier analysis (log-rank) was used. P value of 0.05 or less was considered statistically significant.
Results
miR-223 Deletion Protects against Fas-Induced Liver Injury
To investigate the potential role of miR-223 in Fas-induced liver injury, miR-223 KO mice and age-/sex-matched C57BL/6 WT mice were injected i.p. with a single dose of 0.5 μg/g of body weight anti-Fas antibody Jo2, and the animals were observed hourly to assess mortality. Although all of the WT mice died within 8 hours after Jo2 injection, all of the miR-223 KO mice survived beyond the 72-hour time point (P < 0.01, Kaplan-Meier analysis and log-rank test) (Figure 1A).
Figure 1.

Deletion of miR-223 prevents Fas-induced liver injury. A: Wild-type (WT) and miR-223 knockout (KO) mice were i.p. injected with 0.5 μg/g Jo2 per 1 g body weight, and the animal survival was recorded at hourly intervals. B: Gross images of livers from WT and miR-223 KO mice 4 and 6 hours after Jo2 injection. C: Serum transaminase levels [alanine aminotransferase (ALT) and aspartate aminotransferase (AST)] from WT and miR-223 KO mice (4 and 6 hours after Jo2 treatment). Jo2-treated miR-223 KO mice show lower serum ALT and AST levels compared with Jo2-treated WT mice. D: Hematoxylin and eosin staining of liver tissues from WT and miR-223 KO mice (0, 4, and 6 hours after Jo2 injection). E: Caspase-3 immunostaining of liver tissues from WT and miR-223 KO mice (0, 4, and 6 hours after Jo2 injection) and the percentages of caspase-3–positive cells. F: Relative fold expression of miR-223 in WT and miR-223 KO livers, as determined by quantitative real-time PCR. Data represent means ± SD of fold changes (C). N = 6 per group (A). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Original magnification, ×100 (D and E).
On gross examination, the livers of miR-223 KO mice showed less injury compared with the livers of WT mice (Figure 1B). At the 4-hour time point after Jo injection, the WT livers became red and dark because of parenchymal hemorrhage/congestion; in contrast, the miR-223 KO livers displayed a normal pink/pale appearance. At 6 hours after Jo2 injection, the WT livers developed massive hepatic hemorrhaging and became extremely dark, whereas the miR-223 KO livers continued to show near normal appearance. The miR-223 KO mice showed significantly lower levels of transaminases (ALT and AST) compared with the WT mice (Figure 1C).
Histological examination of the harvest liver tissues (H&E stain) revealed massive parenchymal hemorrhage and parenchymal damage in WT livers, but no damage in miR-223 KO livers (Figure 1D). Caspase-3 immunostaining of the harvested liver tissues confirmed a significantly lower percentage of apoptotic hepatocytes in miR-223 KO livers compared with WT livers (Figure 1E). Deletion of miR-223 in the miR-223 KO livers was confirmed by quantitative real-time PCR analysis (Figure 1F). Taken together, our findings indicate that miR-223 deletion protects against Fas-induced liver injury.
miR-223 Deletion Protects against Fas-Induced Hepatocyte Apoptosis
Caspase-8 activation is essential for death receptor–mediated apoptosis, and this process leads to subsequent activation of caspase-3/7 and caspase-9. Because these three caspases are known to be implicated in Fas-induced apoptosis, we documented the activation of caspase-3, caspase-8, and caspase-9 in the liver tissue samples from WT and miR-223 KO mice with or without Jo2 treatment. Caspase activity assays revealed lower levels of caspase-3, caspase-8, and caspase-9 activities in miR-223 KO livers compared with WT livers after 4 and 6 hours of Jo2 treatment (Figure 2A). Accordingly, Western blot analysis showed reduced cleavage of caspase-3, caspase-8, and caspase-9 as well as decreased cleavage of poly (ADP-ribose) polymerase in miR-223 KO livers (Figure 2B).
Figure 2.

Deletion of miR-223 protects against Fas-induced caspase activation and poly (ADP-ribose) polymerase (PARP) cleavage. A: Wild-type (WT) and miR-223 KO mice were i.p. injected with 0.5 μg/g body weight Jo2. The liver tissues were harvested 0, 4, and 6 hours after Jo2 injection and processed for caspase (Casp) activity assays. The levels of caspase-3, caspase-8, and caspase-9 in miR-223 knockout (KO) livers are significantly lower compared with WT livers. B: Western blot analysis of liver tissue samples for caspase-3, caspase-8, and caspase-9 and PARP. Jo2 induces less cleavage of caspase-3, caspase-8, caspase-9, and PARP in miR-223 mice compared with WT mice. The result represents three individual experiments. The results represent means ± SD of fold changes (A). N = 6 per group (A). ∗P < 0.05, ∗∗P < 0.01.
Deletion of miR-223 Enhances IGF1R Signaling during Fas-Induced Liver Injury
We performed in silico analyses for target gene prediction by using algorithms from TargetScan and MirnaTarget (http://www.targetscan.org and http://www.microrna.org/microrna/getMirnaForm.do, respectively; last accessed June 6, 2013), which led to the identification of IGF1R as a target of miR-223. Western blot analysis showed elevated IGF1R in miR-223 KO livers compared with WT livers after Jo2 treatment (Figure 3). Enhanced phosphorylation of IGF1R and its downstream molecule, Akt, was also observed in miR-223 KO livers compared with WT livers after Jo2 treatment. The levels of IGF1R, p-IGF1R, and p-Akt did not significantly differ between WT and miR-223 KO livers when the mice were not treated with Jo2.
Figure 3.

Deletion of miR-223 enhances insulin-like growth factor 1 receptor (IGF1R) signaling during Fas-induced liver injury. Wild-type (WT) and miR-223 knockout (KO) mice were i.p. injected with 0.5 μg/g body weight Jo2. In silico analysis for target gene prediction using TargetScan led to identification of Igf1r as a candidate gene of miR-223 target. Western blot analysis was performed to measure the levels of IGF1R and p-IGF1R as well as its downstream target Akt. The miR-223 KO livers show higher levels of IGF1R, p-IGF1R, and p-Akt compared with WT livers after Jo2 treatment. Representative Western blots and the average densitometry data are shown. Data indicate means ± SD. N = 6 per group. ∗P < 0.05.
Inhibition of IGF1R Partially Reverses miR-223 Deletion-Mediated Protection against Fas-Induced Liver Injury
To determine the role of IGF1R in mediating miR-223 effect in the liver, we treated WT and miR-223 KO mice with IGF1R inhibitor (NVP-AEW541) 30 minutes before 0.5 μg/g of body weight Jo2 injection; the animals were sacrificed 4 hours after Jo2 injection. Treatment with the IGF1R inhibitor NVP-AEW541 partially sensitized miR-223 KO mice to Jo2-induced liver injury, as demonstrated by analyses for serum ALT/AST levels (Figure 4A), histological examination of the liver tissues (H&E staining) (Figure 4B), caspase-3 immunostaining of the liver tissues (Figure 4C), and assessment for hepatic caspase-3, caspase-8, and caspase-9 activities (Figure 4D). These findings suggest the involvement of IGF1R in miR-223 deletion-mediated protection against Fas-induced liver injury. In our system, we observed that pretreatment with the vehicle dimethyl sulfoxide attenuated Jo2-induced transaminase increase and caspase activation (compared with the data in Figures 1 and 2).
Figure 4.

The effect of insulin-like growth factor 1 receptor (IGF1R) inhibitor on Fas-induced liver injury. Wild-type (WT) and miR-223 knockout (KO) mice were injected with 25 mg/kg of body weight IGF1R inhibitor (NVP-AEW541) or vehicle [dimethyl sulfoxide (DMSO)] 30 minutes before injection with 0.5 μg/g body weight Jo2 (the animals were sacrificed 4 hours after Jo2 injection. A: The levels of serum transaminases [alanine aminotransferase (ALT) and aspartate aminotransferase (AST)]. B: Representative hematoxylin and eosin staining of the liver tissues. C: Caspase-3 immunostaining of the liver tissues. The percentages of caspase-3–positive cells are shown. D: Caspase-3, caspase-8, and caspase-9 activities in the liver tissues. The data are expressed as means ± SD (A) or means ± SD of fold changes (D). N = 6 per group (A); N = 3 (D). ∗P < 0.05, ∗∗P < 0.01. Original magnification, ×100 (B and C).
miR-223 Directly Targets IGF1R in Hepatocytes
We identified a putative miR-223 binding site in the 3′-UTR of IGF1R (Figure 5A). To determine whether this predictive site is involved in miR-223 targeting of IGF1R in hepatocytes, we constructed a mouse IGF1R 3′-UTR luciferase reporter plasmid (containing the putative miR-223 binding site); the reporter construct was transfected into mouse primary hepatocytes, and the cells were then treated with miR-223 mimic or scramble control miRNA. miR-223 mimic treatment significantly reduced the IGF1R 3′-UTR luciferase reporter activity; this effect was abolished when the miR-223 binding site in the IGF1R 3′-UTR was mutated (Figure 5B). These findings establish that miR-223 directly targets IGF1R in hepatocytes.
Figure 5.

Insulin-like growth factor 1 receptor (IGF1R) is a direct target of miR-223. A: Putative miR-223 binding site in IGF1R–3′-untranslated region (UTR). The IGF1R–3′-UTR sequence that contains seven base nucleotides interacting with miR-223 and the sequence of IGF1R–3′-UTR-mutant generated in this study (with mutation of two nucleotides in the seed sequence) are shown. B: IGF1R 3′-UTR luciferase reporter activity assay. Primary hepatocytes isolated from wild-type (WT) mice were cotransfected with miR-223 mimic plus IGF1R 3′-UTR reporter plasmid (with or without miR-223 binding site mutation). miR-223 mimic decreases the IGF1R 3′-UTR luciferase activity when the cells were transfected with the WT IGF1R 3′-UTR reporter construct; this effect is abolished when the miR-223 binding site at the IGF1R 3′-UTR is mutated. ∗∗P < 0.01.
miR-223 Plays an Important Role in Fas-Induced Primary Hepatocyte Apoptosis
To assess the impact of miR-223 on Fas-induced hepatocyte apoptosis, we isolated hepatocytes from WT mice; the cells were then transfected with miR-223 inhibitor or control miRNA before Jo2 treatment. We observed that miR-223 inhibitor treatment attenuated Jo2-induced activation of caspase-3, caspase-8, and caspase-9 (Figure 6A). As a parallel approach, we further isolated hepatocytes from miR-223 KO mice; the cells were then transfected with miR-223 mimic or control miRNA before Jo2 treatment. In the latter system, we observed that miR-223 mimic potentiated Jo2-induced activation of caspase-3, caspase-8, and caspase-9 (Figure 6A). These findings demonstrate that miR-223 in hepatocytes enhances Fas-induced apoptosis.
Figure 6.

A: The effect of hepatocyte miR-223 on Jo2-induced apoptosis. Left panels: Primary hepatocytes isolated from wild type (WT) were transfected with miR-223 inhibitor or control miRNA inhibitor for 24 hours; the cells were then treated with 0.5 μg/mL Jo2 for 4 hours, and the cell lysates were analyzed for caspase-3, caspase-8, and caspase-9 activities. Right panels: miR-223 isolated from miR-223 knockout (KO) mice were transfected with miR-223 mimic (syn-mmu-miR-223-3p) or control miRNA mimic for 24 hours. The cells were then treated with 0.5 μg/mL Jo2 for 4 hours, and the cell lysates were analyzed for caspase-3, caspase-8, and caspase-9 activities. B: The effect of siRNA knockdown of insulin-like growth factor 1 receptor (IGF1R). Hepatocytes isolated from WT or miR-223 KO mice were transfected with IGF1R siRNA or control siRNA for 24 hours, followed by cells treated with 0.5 μg/mL Jo2 for 4 hours. The cell lysates were then obtained to determine caspase-3, caspase-8, and caspase-9 activities. The efficiency of IGF1R depletion by siRNA is shown verified by Western blot analysis. The data are expressed as means ± SD of fold changes (A and B). N = 3 (A and B). ∗P < 0.05.
IGF1R Is Implicated in Fas-Induced, miR-223–Modulated Hepatocyte Apoptosis
To further investigate the role of IGF1R in Fas-induced hepatocyte apoptosis, we used siRNA to knock down IGF1R in WT and miR-223 KO hepatocytes. Although siRNA knockdown of IGF1R in WT hepatocytes variably influenced Jo2-induced activation of caspases, siRNA knockdown of IGF1R in miR-223 KO hepatocytes consistently enhanced Jo2-induced activation of caspase-3, caspase-8, and caspase-9 (Figure 6B). These findings further support the role of IGF1R in Fas-induced, miR-223–modulated hepatocyte apoptosis.
Restoring miR-223 Expression in miR-223 KO Mice Enhances Fas-Induced Liver Injury
miR-223 KO mice at 8 weeks of age were administered lentiviral particles containing pre–miR-223 (L/miR-223; N = 6) or scrambled control miRNA (L/control; N = 6) in 200 μL of sterile saline via tail vein injections. After 7 days, the mice were treated via i.p. injection with 0.5 μg/g of body weight Jo2 for 4 hours. Overexpression of miR-223 increased Jo2-induced liver injury, as indicated by examination of gross images (Figure 7A), serum transaminase levels (Figure 7C), liver tissue histological evaluation (Figure 7D), caspase-3 immunostaining (Figure 7E), and caspase activity assays (Figure 7F). The efficiency of miR-223 overexpression was confirmed by quantitative real-time PCR analysis (Figure 7B). These findings demonstrate that restoration of miR-223 enhances Fas-induced liver injury in vivo. For mice receiving control lentiviral particles, we observed that Jo2 treatment increased caspase-3 activity, with a slight increase in caspase-9 and caspase-8 activities and ALT/AST levels; this variation likely relates to the use of control lentiviral particles.
Figure 7.

Restoration of miR-223 in miR-223 knockout (KO) mice enhances Fas-induced liver injury. miR-223 KO mice at 8 weeks of age were injected with lentiviral particles expressing pre–miR-223 (L/miR-223) or with lentiviral particles expressing control miRNA (L/control) via tail vein injections. Seven days after tail vein injections, the mice were treated with 0.5 μg/g body weight Jo2 for 4 hours; the animals were sacrificed and the livers were harvested for further analyses [hematoxylin and eosin (H&E) stain, caspase-3 immunostain, caspase activity assay, and Western blot analysis]. A: Gross images depicting greater Jo2-induced liver injury in L/miR-223–injected mice compared with L/control-injected animals. B: The levels of miR-223 in the livers of miR-223 KO mice receiving L/control or L/miR-223, as determined by quantitative real-time PCR analysis. C: The levels of serum transaminases [alanine aminotransferase (ALT) and aspartate aminotransferase (AST)] in the L/control and L/miR-223–injected mice (with or without Jo2 treatment). D: H&E staining of the liver tissues showing more prominent liver injury in L/miR-223–injected compared with L/control-injected mice after Jo2 treatment. E: Caspase-3 immunostaining of the liver tissues showing more prominent hepatocyte apoptosis in L/miR-223–injected compared with L/control-injected mice after Jo2 treatment. The percentages of caspase-3–positive cells are depicted in the bar graph. F: Caspase Glo assay shows increased caspase-3, caspase-8, and caspase-9 activities in L/miR-223–injected compared with L/control-injected mice after Jo2 treatment. The results are presented as means ± SD (C) or means ± SD of fold changes (F). N = 3 for each group (A and C–E). ∗P < 0.05, ∗∗P < 0.01. Original magnification, ×100 (E).
IGF1R Overexpression Prevents Fas-Induced Liver Injury
To further determine the role of IGF1R in Fas-induced liver injury, we performed hydrodynamic tail vein injection of the IGF1R expression plasmid (pBabe-bleo-Igf1r) or the control plasmid in WT mice; 72 hours after the tail vein injection, the mice were i.p. treated with 0.5 μg/mg Jo2 for 4 hours. We observed that forced overexpression of IGF1R prevented a Jo2-induced increase of serum transaminases (Figure 8A). Overexpression of IGF1R also attenuated Jo2-induced increase of caspase-3 and caspase-8 activities (Figure 8B). Successful overexpression of IGF1R protein in the liver tissues injected with the IGF1R expression plasmid was confirmed by Western blot analysis (Figure 8C). These results support the importance of IGF1R for protection against Fas-induced liver injury. We observed that Jo2 treatment induced a relatively low level of caspase 3/7 activation or transaminases increase in mice receiving hydrodynamic tail vein injection of control plasmid; this was likely because of the hydrodynamic tail vein injection procedure or the use of control plasmid.
Figure 8.

Insulin-like growth factor 1 receptor (IGF1R) overexpression protects wild-type (WT) mice against Fas-induced liver injury. WT mice received 20 μg/mL of IGF1R expression plasmid (pBabe-bleo-IGF1R) or control plasmid via hydrodynamic tail vein injection. Forty-eight hours after tail vein injection, the mice were treated with 0.5 μg/g body weight of Jo2 via i.p. injection. A: Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. IGF1R overexpression attenuates Jo2-induced transaminase increase. B: Caspase-3 and caspase-8 activities in the liver tissues. IGF1R overexpression attenuates Jo2-induced activation of caspase-3 and caspase-8. C: Immunostaining for activated caspase-3 in the liver tissues. The bar graph shows percentages of caspase-3–positive cells. ∗P < 0.05, ∗∗P < 0.01. Original magnification, ×100 (C). Vc, vector control plasmid.
The Effect of miR-223 on Fas-Induced Apoptosis in HepG2 Cells
Given the effect of miR-223 on Fas-induced mouse hepatocyte apoptosis in vitro and liver injury in vivo, as documented in the above sections, we sought to further examine whether miR-223 might influence Fas-induced apoptosis in transformed human hepatocytes (HepG2 cells). To this end, the HepG2 cells were transfected with synthetic human miR-223 mimic or control miRNA mimic, followed by treatment with 200 ng/mL FasL or vehicle for 24 hours. We observed that transfection of HepG2 cells with miR-223 mimic did not significantly alter FasL-induced apoptosis. The caspase-3/7 and caspase-8 activities were similar between HepG2 cells transfected with miR-223 mimic and control miRNA mimic (Supplemental Figure S1). Therefore, although miR-223 is able to enhance Jo2-induced mouse hepatocyte apoptosis in vitro and in vivo, it does not significantly alter the sensitivity of HepG2 cells to FasL-induced apoptosis in vitro.
Discussion
miRNAs have been implicated in several liver diseases, including acute liver injury, nonalcoholic fatty liver disease, and nonalcoholic steatohepatitis, in hepatitis B virus/hepatocellular carcinoma infections, and in hepatocarcinogenesis.26, 27, 28, 29 In the present study, we describe a novel role of miR-223 in Fas-induced liver injury in mice. Our data indicate that miR-223 deletion protects against Fas-induced mouse hepatocyte apoptosis and liver injury. After Jo2 treatment, the miR-223 KO mice survived longer with less hepatocyte apoptosis/liver injury compared with the WT mice; such protection was reversed by lentiviral delivery of miR-223. The role of miR-223 in Fas-induced mouse hepatocyte apoptosis was further documented in primary hepatocytes isolated from WT and miR-223 KO mice (miR-223 inhibition attenuated Jo2-induced activation of caspase-3, caspase-8, and caspase-9 in WT hepatocytes; miR-223 mimic enhanced Jo2-induced activation of caspase-3, caspase-8, and caspase-9 in miR-223 KO hepatocytes). Collectively, our findings demonstrate an important role of hepatocyte miR-223 for regulation of Fas-induced mouse hepatocyte apoptosis.
IGF1R is a transmembrane tyrosine kinase receptor implicated in liver injury and hepatic cell growth.30, 31 In our study, we observed that up-regulation of the IGF1R signaling pathway protected miR-223–deficient mice against Fas-induced liver injury. Our data suggest that miR-223 regulates Fas-induced hepatocyte apoptosis and liver injury by directly targeting IGF1R. This assertion is on the basis of the following observations: i) IGF1R protein expression levels were significantly increased in miR-223 KO livers and hepatocytes; ii) luciferase reporter activity assays using IGF1R–3′-UTR reporter constructs, with or without miR-223 binding site mutation, confirmed IGF1R as a direct target of miR-223 in hepatocytes; iii) transfection of hepatocytes with IGF1R siRNA enhanced Fas-induced apoptosis in vitro; iv) IGF1R inhibition restored Fas-induced hepatocyte apoptosis and live injury in vivo; and v) IGF1R overexpression in WT mice prevented Jo2-induced hepatocyte apoptosis and liver injury in vivo.
Fas is a cell surface protein that belongs to the tumor necrosis factor receptors superfamily32; stimulation of Fas by its ligand (FasL) causes rapid assembly of the death-inducing signaling complex leading to apoptosis.33, 34, 35 It is noticeable that Fas-induced hepatocyte apoptosis is implicated in various liver diseases, including viral hepatitis, alcoholic and nonalcoholic steatohepatitis, ischemia/reperfusion injury, and acute hepatic liver failure.13, 14, 15, 16, 17, 18, 19, 20 In this context, data presented in the current study establish an important role of hepatocyte miR-223 for regulation of Fas-induced apoptosis. Our results are further underlined by the increasingly recognized contribution of miR-223 to liver pathobiology and the involvement of Fas-induced apoptosis in liver injury and liver diseases.
In summary, this study describes a novel role of miR-223 for regulation of Fas-induced mouse hepatocyte apoptosis and liver injury by targeting the IGFR1. Further studies are needed to evaluate the implication of the miR-223/IGFR1 signaling pathway in human hepatocytes and to determine whether this signaling cascade can be targeted for the treatment of human liver injury and liver diseases.
Footnotes
Supported by NIH grant R01 CA102325 (T.W.).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.08.020.
Supplemental Data
Supplemental Figure S1.
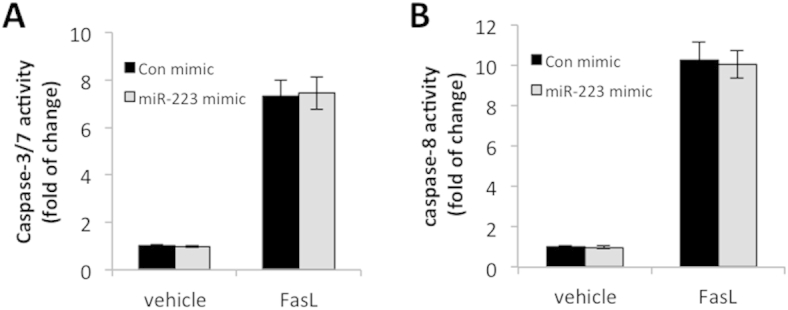
The effect of miR-223 on Fas-induced apoptosis in HepG2 cells. HepG2 cells were transfected with synthetic human miR-223 mimic and control (Con) miRNA mimic and treated with 200 ng/mL recombinant human FasL for 24 hours. The activities of caspase-3/7 (A) and caspase-8 (B) were measured by the Caspase-Glo Assay Kit. The experiments were repeated three times. The data are expressed as means ± SD.
References
- 1.Johnnidis J.B., Harris M.H., Wheeler R.T., Stehling-Sun S., Lam M.H., Kirak O., Brummelkamp T.R., Fleming M.D., Camargo F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 2.Fukao T., Fukuda Y., Kiga K., Sharif J., Hino K., Enomoto Y., Kawamura A., Nakamura K., Takeuchi T., Tanabe M. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell. 2007;129:617–631. doi: 10.1016/j.cell.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 3.Fazi F., Rosa A., Fatica A., Gelmetti V., De Marchis M.L., Nervi C., Bozzoni I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 4.Taibi F., Metzinger-Le Meuth V., Massy Z.A., Metzinger L. miR-223: an inflammatory oncomiR enters the cardiovascular field. Biochim Biophys Acta. 2014;1842:1001–1009. doi: 10.1016/j.bbadis.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Haneklaus M., Gerlic M., O'Neill L.A., Masters S.L. miR-223: infection, inflammation and cancer. J Intern Med. 2013;274:215–226. doi: 10.1111/joim.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vickers K.C., Landstreet S.R., Levin M.G., Shoucri B.M., Toth C.L., Taylor R.C., Palmisano B.T., Tabet F., Cui H.L., Rye K.A., Sethupathy P., Remaley A.T. MicroRNA-223 coordinates cholesterol homeostasis. Proc Natl Acad Sci U S A. 2014;111:14518–14523. doi: 10.1073/pnas.1215767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu C.H., Xu C.F., Li Y.M. Association of microRNA-223 expression with hepatic ischemia/reperfusion injury in mice. Dig Dis Sci. 2009;54:2362–2366. doi: 10.1007/s10620-008-0629-8. [DOI] [PubMed] [Google Scholar]
- 8.Yang F., Lou G., Zhou X., Zheng M., He J., Chen Z. MicroRNA-223 acts as an important regulator to Kupffer cells activation at the early stage of Con A-induced acute liver failure via AIM2 signaling pathway. Cell Physiol Biochem. 2014;34:2137–2152. doi: 10.1159/000369658. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi K., Oda Y., Toyoda Y., Fukami T., Yokoi T., Nakajima M. Regulation of cytochrome b5 expression by miR-223 in human liver: effects on cytochrome P450 activities. Pharm Res. 2014;31:780–794. doi: 10.1007/s11095-013-1200-7. [DOI] [PubMed] [Google Scholar]
- 10.Wong Q.W., Lung R.W., Law P.T., Lai P.B., Chan K.Y., To K.F., Wong N. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology. 2008;135:257–269. doi: 10.1053/j.gastro.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Aucher A., Rudnicka D., Davis D.M. MicroRNAs transfer from human macrophages to hepato-carcinoma cells and inhibit proliferation. J Immunol. 2013;191:6250–6260. doi: 10.4049/jimmunol.1301728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J., Wu C., Che X., Wang L., Yu D., Zhang T., Huang L., Li H., Tan W., Wang C., Lin D. Circulating microRNAs, miR-21, miR-122, and miR-223, in patients with hepatocellular carcinoma or chronic hepatitis. Mol Carcinog. 2011;50:136–142. doi: 10.1002/mc.20712. [DOI] [PubMed] [Google Scholar]
- 13.Galle P.R., Krammer P.H. CD95-induced apoptosis in human liver disease. Semin Liver Dis. 1998;18:141–151. doi: 10.1055/s-2007-1007150. [DOI] [PubMed] [Google Scholar]
- 14.Ryo K., Kamogawa Y., Ikeda I., Yamauchi K., Yonehara S., Nagata S., Hayashi N. Significance of Fas antigen-mediated apoptosis in human fulminant hepatic failure. Am J Gastroenterol. 2000;95:2047–2055. doi: 10.1111/j.1572-0241.2000.02268.x. [DOI] [PubMed] [Google Scholar]
- 15.Okazaki A., Hiraga N., Imamura M., Hayes C.N., Tsuge M., Takahashi S., Aikata H., Abe H., Miki D., Ochi H., Tateno C., Yoshizato K., Ohdan H., Chayama K. Severe necroinflammatory reaction caused by natural killer cell-mediated Fas/Fas ligand interaction and dendritic cells in human hepatocyte chimeric mouse. Hepatology. 2012;56:555–566. doi: 10.1002/hep.25651. [DOI] [PubMed] [Google Scholar]
- 16.Pianko S., Patella S., Ostapowicz G., Desmond P., Sievert W. Fas-mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption, and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J Viral Hepat. 2001;8:406–413. doi: 10.1046/j.1365-2893.2001.00316.x. [DOI] [PubMed] [Google Scholar]
- 17.Gujral J.S., Liu J., Farhood A., Jaeschke H. Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology. 2004;40:998–1007. doi: 10.1002/hep.20380. [DOI] [PubMed] [Google Scholar]
- 18.Minana J.B., Gomez-Cambronero L., Lloret A., Pallardo F.V., Del Olmo J., Escudero A., Rodrigo J.M., Pelliin A., Vina J.R., Vina J., Sastre J. Mitochondrial oxidative stress and CD95 ligand: a dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology. 2002;35:1205–1214. doi: 10.1053/jhep.2002.32969. [DOI] [PubMed] [Google Scholar]
- 19.Feldstein A.E., Canbay A., Angulo P., Taniai M., Burgart L.J., Lindor K.D., Gores G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 20.Hatano E. Tumor necrosis factor signaling in hepatocyte apoptosis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S43–S44. doi: 10.1111/j.1440-1746.2006.04645.x. [DOI] [PubMed] [Google Scholar]
- 21.Li G., Han C., Xu L., Lim K., Isse K., Wu T. Cyclooxygenase-2 prevents fas-induced liver injury through up-regulation of epidermal growth factor receptor. Hepatology. 2009;50:834–843. doi: 10.1002/hep.23052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li G., Chen W., Han C., Wu T. Cytosolic phospholipase A(2)α protects against Fas- but not LPS-induced liver injury. J Hepatol. 2011;55:1281–1290. doi: 10.1016/j.jhep.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parasuraman S., Raveendran R., Kesavan R. Blood sample collection in small laboratory animals. J Pharmacol Pharmacother. 2010;1:87–93. doi: 10.4103/0976-500X.72350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu D., Li C., Chen Y., Burnett C., Liu X.Y., Downs S., Collins R.D., Hawiger J. Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J Biol Chem. 2004;279:48434–48442. doi: 10.1074/jbc.M407190200. [DOI] [PubMed] [Google Scholar]
- 25.Li P., Jayarama S., Ganesh L., Mordi D., Carr R., Kanteti P., Hay N., Prabhakar B.S. Akt-phosphorylated mitogen-activated kinase-activating death domain protein (MADD) inhibits TRAIL-induced apoptosis by blocking Fas-associated death domain (FADD) association with death receptor 4. J Biol Chem. 2010;285:22713–22722. doi: 10.1074/jbc.M110.105692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemoinne S., Thabut D., Housset C., Moreau R., Valla D., Boulanger C.M., Rautou P.E. The emerging roles of microvesicles in liver diseases. Nat Rev Gastroenterol Hepatol. 2014;11:350–361. doi: 10.1038/nrgastro.2014.7. [DOI] [PubMed] [Google Scholar]
- 27.Szabo G., Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:542–552. doi: 10.1038/nrgastro.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giordano S., Columbano A. MicroRNAs: new tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology. 2013;57:840–847. doi: 10.1002/hep.26095. [DOI] [PubMed] [Google Scholar]
- 29.Wang X.W., Heegaard N.H., Orum H. MicroRNAs in liver disease. Gastroenterology. 2012;142:1431–1443. doi: 10.1053/j.gastro.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caro J.F., Poulos J., Ittoop O., Pories W.J., Flickinger E.G., Sinha M.K. Insulin-like growth factor I binding in hepatocytes from human liver, human hepatoma, and normal, regenerating, and fetal rat liver. J Clin Invest. 1988;81:976–981. doi: 10.1172/JCI113451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scharf J.G., Schmidt-Sandte W., Pahernik S.A., Ramadori G., Braulke T., Hartmann H. Characterization of the insulin-like growth factor axis in a human hepatoma cell line (PLC) Carcinogenesis. 1998;19:2121–2128. doi: 10.1093/carcin/19.12.2121. [DOI] [PubMed] [Google Scholar]
- 32.Krammer P.H. CD95's deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 33.Strasser A., Jost P.J., Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–192. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peter M.E., Budd R.C., Desbarats J., Hedrick S.M., Hueber A.O., Newell M.K., Owen L.B., Pope R.M., Tschopp J., Wajant H., Wallach D., Wiltrout R.H., Zornig M., Lynch D.H. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–450. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 35.Lavrik I.N., Krammer P.H. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012;19:36–41. doi: 10.1038/cdd.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]