

Abstract
Amyloid (senile) plaques, one of the two pathologic hallmarks of Alzheimer disease (AD), are associated with dystrophic neurites and glial responses, both astrocytic and microglial. Although plaque burden remains relatively stable through the clinical course of AD, whether these features of local plaque toxicity continue to worsen over the course of the disease is unclear. We performed an unbiased plaque-centered quantification of SMI312+ dystrophic neurites, GFAP+ reactive astrocytes, and IBA1+ and CD68+ activated microglia in randomly selected dense-core (Thioflavin-S+) plaques from the temporal neocortex of 40 AD subjects with a symptom duration ranging from 4 to 20 years, and nine nondemented control subjects with dense-core plaques. Dystrophic neurites (Kendall τ = 0.34, P = 0.001), reactive astrocytes (Kendall τ = 0.30, P = 0.003), and CD68+ (Kendall τ = 0.48, P < 0.0001), but not IBA1 microglia (Kendall τ = 0.045, P = 0.655), exhibited a significant positive correlation with symptom duration. When excluding control subjects, only the positive association between CD68+ microglia and symptom duration remained significant (Kendall τ = 0.39, P = 0.0003). The presence of the APOEε4 allele did not affect these results. We conclude that plaques exert an increasing toxicity in the surrounding neuropil over the clinical course of AD, thereby potentially contributing to cognitive decline.
Amyloid plaques and neurofibrillary tangles are the core features of Alzheimer disease (AD). Among amyloid plaques, the subset of dense-core plaques defined by positive staining with dyes selective for β-pleated sheet structure such as Congo red and Thioflavin-S are considered more toxic and correlate more specifically with the presence of dementia. Dense-core plaques have a number of associated features that include dystrophic neurites, reactive astrocytes, and activated microglial cells. Dystrophic neurites are described as spheroids, swellings, and distorted neurites (dendrites and axons) that are embedded within dense-core plaques or in their close vicinity and exhibit immunoreactivity for neurofilament proteins and hyperphosphorylated tau.1 Reactive astrocytes and activated microglial cells also cluster within and around dense-core plaques. Therefore, the microenvironment of dense-core neuritic plaques is thought to recapitulate all of the steps of the amyloid cascade hypothesis.2, 3
With the use of quantitative postmortem measures, we previously showed that amyloid both plaque burden and plaque size remain relatively constant throughout the clinical course of AD.4, 5, 6 Thanks to the body of evidence accumulated with the use of fibrillar amyloid positron emission tomography in human subjects, there is now consensus that amyloid plaque deposition occurs for the most part before onset of cognitive deficits and plateaus soon after.7, 8, 9 However, whether plaque-associated toxicity, represented by surrounding dystrophic neurites, reactive astrocytes, and activated microglial cells, is stable or worsens during disease progression remains unclear. Two models could be proposed. At one extreme, plaques could cause local damage to the neuropil as they deposit, but then remain relatively static lesions10 as the disease pathologic process becomes increasingly dominated by nonplaque pathologies, including tau-associated lesions such as neuropil threads and tangles, neuronal and synaptic loss, and non–plaque-associated glial reactions.5 At the other extreme, plaques could increasingly contribute to local neural system destruction over the entire course of the disease. Therefore, we tested the hypothesis that the microenvironment in the vicinity of plaques becomes more and more toxic as the disease advances. Specifically, we investigated whether the plaque-associated features of neuritic changes and reactive glia have already reached a plateau at dementia onset or parallel the progression of dementia and continue to accrue over time. Although cross-sectional postmortem studies are not able to unambiguously distinguish the prior history of the brain during life, we reasoned that examining specimens from individuals who had known durations of illness would provide insight into these questions. With the use of quantitative neuropathologic measures in postmortem specimens and robust statistical methods, we demonstrate that these markers of plaque toxicity change throughout the clinical course of AD, leading to an overall increase in local plaque-associated damage as time goes on. The temporal accrual of features of plaque-associated injury is independent of the APOEε4 allele.
Materials and Methods
Subjects
Paraffin-embedded sections from the temporal neocortex (BA 38) of 40 AD subjects and nine nondemented control (CTRL) subjects were obtained from the Massachusetts General Hospital Alzheimer Disease Research Center. Next of kin for study subjects provided informed consent to donate their brain, and the study was approved by the Institutional Review Board at Massachusetts General Hospital. All AD subjects met the clinical11, 12 and neuropathologic diagnostic13, 14, 15 criteria of AD. CTRL subjects had no clinical or neuropathologic evidence of any neurodegenerative disease. AD subjects were selected on the basis of disease duration from symptom onset as assessed by their neurologist at the patient's first clinical encounter with Massachusetts General Hospital Alzheimer Disease Research Center Neurology team (≤5 years, n = 10; 6 to 10 years, n = 10; 11 to 15 years, n = 10, and >15 years, n = 10). The demographic and clinical characteristics of AD and CTRL subjects and of AD subgroups by APOEε4 status are depicted in Table 1. The temporal neocortex was chosen because it is a region with abundant and early amyloid deposition and because our prior quantitative neuropathologic studies in this area have revealed marked glial responses.5, 16, 17
Table 1.
Demographic and Clinical Characteristics of the Study Subjects
| Characteristic | CTRL subjects | AD subjects | P | AD APOEε4 carriers | AD APOEε4 noncarriers | P |
|---|---|---|---|---|---|---|
| Subjects, n | 9 | 40 | 21 | 19 | ||
| Sex, female, n (%) | 4 (44.4) | 26 (65.0) | 0.28 | 16 (76.2) | 10 (52.6) | 0.18 |
| Age at death, mean ± SD (years) | 80.3 ± 14.4 | 77.6 ± 8.6 | 0.12 | 75.8 ± 8.7 | 79.6 ± 8.2 | 0.16 |
| Age at onset, mean ± SD (years) | 66.9 ± 10.2 | 64.4 ± 10.6 | 69.6 ± 9.3 | 0.07 | ||
| Symptom duration, mean ± SD (years) | 10.7 ± 5.0 | 11.3 ± 4.9 | 10.0 ± 5.1 | 0.39 | ||
| APOEε4 carriers, n (%) | 3 (33.3) | 21 (52.5) | 0.46 | |||
| APOEε4 alleles, n (%) | 3 (16.7) | 25 (31.2) | 0.75 | |||
| Postmortem interval, mean ± SD (hours) | 17.4 ± 11.4 | 14.1 ± 6.2 | 0.53 | 14.5 ± 5.8 | 13.7 ± 6.7 | 0.68 |
Postmortem interval was not available for two CTRL subjects. Comparisons were performed with U-test, unpaired t-test, or Fisher's exact test as appropriate.
AD, Alzheimer disease; CTRL, control.
Immunohistochemistry
Eight-micron–thick paraffin sections were cleared with xylenes, rehydrated with decreasing concentrations of ethanol, and subjected to a standard antigen retrieval procedure (microwave for 20 minutes at 95°C in boiling citrate buffer 0.01 mol/L with 0.05% Tween 20, pH 6.0) before immunohistochemistry. Primary antibodies and concentrations used were mouse anti-SMI312 (dilution 1:1000; Covance, Princeton, NJ; catalog no. SMI312R), rabbit anti-GFAP (dilution 1:1000; Sigma-Aldrich, St. Louis, MO; catalog no. G6296), rabbit anti-IBA1 (dilution 1:250; Wako, Osaka, Japan; catalog no. 019-19741), and mouse anti-CD68 (dilution 1:100; Dako, Glostrup, Denmark; catalog no. M0814). Cyanine 3-conjugated anti-rabbit or anti-mouse secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA) and used at a 1:200 concentration. Sections were counterstained with Thioflavin S (Sigma-Aldrich) 0.05% in 50% ethanol for 8 minutes and differentiated in 80% ethanol for 30 seconds before being coverslipped with Vectashield mounting media with DAPI (Vector Labs, Burlingame, CA; catalog no. H-1200).
Quantitative Neuropathologic Studies
Single paraffin-embedded temporal neocortex sections were randomly sampled with the use of a stereology microscope-computer system, and 100 dense-core (Thioflavin-S+) amyloid plaques per subject were selected from randomly sampled microscopic fields by a trained observer (A.S.-P.). Briefly, sections were placed on a motorized stage of an upright Olympus BX51 epifluorescence microscope controlled by a computer with the computer-assisted stereological toolbox software version 2.3.1.5 (Olympus, Tokyo, Japan). Reactive (GFAP+) astrocytes and activated (IBA1+ or CD68+) microglia within 50 μm from the edge of the dense-core plaques were manually counted as previously described.16, 18 This boundary was selected on the basis of our previous animal and human postmortem quantitative studies.5, 16, 18, 19, 20, 21, 22 Only glial cells with a DAPI+ visible nucleus were counted. To prevent double counting of cells in areas rich in plaques, individual astrocytes and microglia that were close to two or more plaques were split among those plaques (ie, 0.5 cells if close to two plaques, 0.33 if close to three plaques, 0.25 if close to four plaques, etc.). The number of SMI312+ axonal swellings/spheroids and distorted neurites >2.5 μm embedded within the Thioflavin-S+ area or in contact with its edges were manually counted as described before.18 Figure 1 shows examples of these quantitative methods.
Figure 1.

Quantitation of plaque-associated features in the temporal neocortex. A and B: Arrows show how GFAP+ astrocytes (A) and CD68+ microglia (B) with a DAPI+ visible nucleus within the 50-μm halo (outlines) of the nearest plaque were counted. One hundred dense-core (Thioflavin-S+) plaques were randomly selected on single paraffin sections with the use of a stereology system.
Statistical Analysis
Analyses were conducted blinded to clinical diagnosis, symptom duration, and APOEε4 status. After completing the above quantifications, each of the 40 AD subjects yielded a histogram distribution with 100 values for each of the neuropathologic quantitative measures. For some CTRL subjects, the number of dense-core plaques was lower than the goal of 100 plaques, despite sampling 100% of the cortex included in the section. CTRL subjects were excluded from analyses of measures for which they had <100 plaques. Data for SMI312+ dystrophic neurites were not available for one CTRL subject.
We used symptom duration as a surrogate of progression because i) duration of illness and clinical severity are generally correlated; ii) duration of illness overcomes biases inherent to formal neuropsychological testing in advanced dementia patients, including floor effect of psychometric measures, loss to follow-up, impact of intercurrent illnesses (eg, cataracts, medications) on test performance, and variable period of time between last follow-up and death; and iii) in prior studies we have found correlation between duration from symptom onset and variables of progression such as number of neurons and tau-related pathology,23 cortical atrophy,5 and glial responses.4, 5
Because histograms are multidimensional, to investigate the correlation between each of the plaque-associated features and symptom duration, we used the signed Kolmogorov-Smirnov (K-S) measure of distance between two histograms and estimated its association with symptom duration with the use of Kendall τ (nonparametric rank) correlation. The K-S distance is the maximum absolute distance between subjects' empirical distribution functions, across all values of the variable. The signed version of K-S distance inherits the sign (positive or negative) of the distance that yields the K-S distance; therefore, a larger positive distance between subject i's histogram and subject j's histogram suggests that subject j has larger values than subject i.
To investigate whether the strength of the association between each plaque-associated feature and symptom duration remains constant throughout the clinical course of the disease or changes over time, we plotted Kendall τ correlation coefficients that were calculated for subsets of subjects defined by moving windows of symptom duration. Cross-correlations between plaque-associated features (ie, CD68+ microglia versus SMI312+ dystrophic neurites) were estimated with Kendall τ test with the use of the medians of the distributions. Finally, the numbers of SMI312+ dystrophic neurites per plaque were compared between APOEε4 carriers and noncarriers with the use of a two-sided Wilcoxon rank-sum test that accommodates clustered data.24
All analyses were conducted including and excluding CTRL subjects. Statistical significance was set at a level of P < 0.05. Analysis and graphs were performed with the statistical software package R version 3.2 (http://www.r-project.org/about.html), except for the two-sided clustered Wilcoxon rank-sum test, which was run in SAS version 9.3 (SAS Institute, Cary, NC).
Results
Plaque-Associated Features Accumulate over the Clinical Course of AD
We have shown before that amyloid plaque burden and plaque size remain relatively constant throughout the clinical course of AD. Here, we investigated whether plaque-associated features also remain constant or, by contrast, continue to accrue as the disease advances. To this goal, we counted the number of SMI312+ axonal swellings and spheroids, GFAP+ astrocytes, IBA1+ microglial cells, and CD68+ microglial cells per plaque in 100 randomly selected dense-core Thioflavin-S+ amyloid plaques from the temporal cortex of 40 AD subjects with a symptom duration that ranged between 4 and 20 years and up to nine CTRL subjects. Counts were performed without knowledge of clinical history or APOE genotype. These analyses yielded up to 49 histograms with 100 values per subject.
Histograms can be summarized with the use of central measures (ie, mean for normal distributions and median for nonnormal distributions) and dispersion measures (ie, interquartile range), but these descriptive measures are limited because they can still be influenced by outliers, and they reduce the multidimensional information to a single number. To avoid these limitations, we compared the histograms among pairs of subjects with the use of the K-S test and correlated their signed K-S difference (or distance) with their difference in symptom duration. A positive correlation between the K-S distance from distribution histograms of pairs of subjects for a certain plaque-associated feature and their difference in symptom duration indicates an accrual of that plaque-associated feature over time.
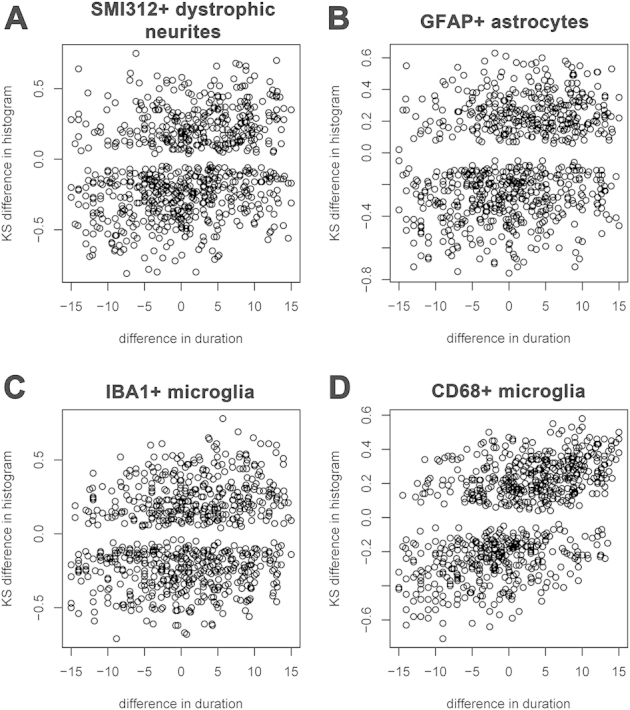
Figure 2 contains plots of the histogram distance versus the difference in disease duration for each pair of subjects. It is seen that the number of SMI312+ dystrophic neurites (Figure 2A), GFAP+ astrocytes (Figure 2B), and CD68+ microglia (Figure 2D) but not IBA1+ microglia (Figure 2C) per plaque increase with increasing duration of illness. As expected from these graphs, a positive correlation was found between symptom duration and number of SMI312+ dystrophic neurites (Kendall τ = 0.34, P = 0.001), GFAP+ astrocytes (Kendall τ = 0.30, P = 0.003), and CD68+ (Kendall τ = 0.48, P < 0.0001) but not IBA1+ microglia (Kendall τ = 0.045, P = 0.655). When CTRL subjects were removed from the analyses, only the association between CD68+ microglia and symptom duration remained significant (Kendall τ = 0.39, P = 0.0003) (Supplemental Figure S1 and Supplemental Table S1).
Figure 2.

Plaque-associated features accumulate over the clinical course of AD. A–D: Plots represent the KS difference between the distribution histogram from pairs of subjects against their difference in symptom duration. Note that the larger the difference in symptom duration between two subjects, the larger the difference in their distribution histograms for SMI312+ dystrophic neurites (A), GFAP+ astrocytes (B), and CD68+ microglia (D) per plaque, indicating accrual over the clinical course of the disease. This association was, however, not significant for IBA1+ microglia per plaque (C). n = 40 AD subjects and 9 CTRL subjects (8 CTRL for SMI312+ dystrophic neurites). AD, Alzheimer disease; CTRL, control; KS, Kolmogorov-Smirnov.
No significant correlation was found between any pair of plaque-associated features, either in AD subjects or in all (AD and CTRL) subjects (Table 2).
Table 2.
Summary of Study Results for All Subjects
| All subjects | Symptom duration |
SMI312+ dystrophic neurites |
GFAP+ astrocytes |
IBA1+ microglia |
CD68+ microglia |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Kendall τ | P | Kendall τ | P | Kendall τ | P | Kendall τ | P | Kendall τ | P | |
| Symptom duration | 0.34∗ | 0.001∗ | 0.30∗ | 0.003∗ | 0.045 | 0.655 | 0.48∗ | <0.0001∗ | ||
| SMI312+ dystrophic neurites | 0.34∗ | 0.001∗ | −0.028 | 0.735 | 0.006 | 0.943 | 0.008 | 0.929 | ||
| GFAP+ astrocytes | 0.30∗ | 0.003∗ | −0.028 | 0.735 | −0.137 | 0.064 | −0.014 | 0.849 | ||
| IBA1+ microglia | 0.045 | 0.655 | 0.006 | 0.849 | −0.137 | 0.064 | 0.109 | 0.151 | ||
| CD68+ microglia | 0.48∗ | <0.0001∗ | 0.008 | 0.929 | −0.014 | 0.849 | 0.109 | 0.151 | ||
Depicted are correlations between the Kolmogorov-Smirnov distances of pairs of distribution histograms of a given plaque-associated feature and the difference in symptom duration of that pair of subjects, and the correlations between the median values of the distribution histograms for a given pair of plaque-associated features (ie, GFAP+ astrocytes versus CD68+ microglia). Data refer to all subjects, that is, 40 AD and 9 CTRL subjects (8 for SMI312+ dystrophic neurites).
AD, Alzheimer disease; CTRL, control.
Statistically significant results.
Lack of Effect of APOEε4 Allele in the Accrual of Plaque-Associated Features over the Clinical Course of AD
The APOEε4 allele is the strongest known genetic risk factor for the development of sporadic AD, and it does so by promoting the accumulation of amyloid β (Aβ) peptide and its deposition in plaques (reviewed in Holtzman et al25), but whether plaques from APOEε4 carriers are more toxic to the surrounding neuropil than plaques from APOEε4 noncarriers remains unclear. There were 21 APOEε4 carriers and 19 APOEε4 noncarriers among the 40 AD subjects and 3 APOEε4 carriers and 6 APOEε4 noncarriers among the nine CTRL subjects in this sample. With the use of this same data set, we have previously reported that dense-core plaques from AD APOEε4 carriers and noncarriers do not differ in the magnitude of glial responses.16 Here, we investigated whether they differ in the magnitude of neuritic change, specifically if dense-core plaques from APOEε4 carriers contain more neuritic dystrophies than those from noncarriers. The number of SMI312+ dystrophic neurites per dense-core plaque was not significantly different between APOEε4 carriers and noncarriers, either for AD subjects (P = 0.77), CTRL subjects (P = 0.11), or all subjects combined (P = 0.96).
We next asked whether the temporal accrual of plaque-associated features described in the previous section is driven by the presence of the APOEε4 allele. Figure 3 plots show that essentially no difference was found in the temporal accrual of SMI312+ dystrophic neurites (Figure 3, A and B), GFAP+ astrocytes (Figure 3, C and D), and IBA1+ microglia (Figure 3, E and F) per plaque between APOEε4 carriers and noncarriers. However, CD68+ microglia exhibited a stronger association with symptom duration in APOEε4 noncarriers than in APOEε4 carriers (Figure 3, G and H).
Figure 3.

Lack of effect of APOEε4 allele on the temporal accrual of plaque-associated features over the clinical course of AD. A–H: Plots represent the KS difference between the distribution histogram from pairs of subjects against their difference in symptom duration for SMI312+ dystrophic neurites (A and B), GFAP+ astrocytes (C and D), IBA1+ microglia (E and F), and CD68+ microglia (G and H) per plaque for APOEε4 noncarriers (A, C, E, and G) and carriers (B, D, F, and H). Note that all plaque-associated features exhibit a similar behavior over time regardless of the presence of the APOEε4 allele except for CD68+ microglia that exhibits a stronger association with symptom duration in APOEε4 noncarriers. n = 40 AD subjects (21 APOEε4 carriers and 19 noncarriers) and 9 CTRL subjects (3 APOEε4 carriers and 6 noncarriers). AD, Alzheimer disease; CTRL, control; KS, Kolmogorov-Smirnov.
Association between Plaque-Associated Features and Symptom Duration Is Linked to the Clinical Course of AD
Next, we investigated whether the strength of the above associations is constant or changing throughout the clinical course of the disease. To this goal, we estimated and plotted the Kendall τ correlation coefficients for subsets of subjects defined by moving windows of symptom duration. The left-hand plots in Figure 4, Figure 5, Figure 6 and 7 show the Kendall τ correlation coefficients for symptom durations < x years; therefore, their leftmost values are the most relevant. The right-hand plots depict the Kendall τ correlation coefficients for symptom durations > x years; therefore, their rightmost values are the most relevant.
Figure 4.

The association between SMI312+ dystrophic neurites per plaque and symptom duration is stronger at initial stages. A and B: Association between Kendall τ correlation coefficients and symptom duration for a length shorter (A) or longer (B) than that indicated in the x axis. Therefore, the most relevant results in A are the leftmost values and in B the rightmost values. n = 40 AD subjects and 8 CTRL subjects. AD, Alzheimer disease; CTRL, control.
Figure 5.

The association between the number of GFAP+ astrocytes per plaque and symptom duration is stronger at initial stages. A and B: Association between Kendall τ correlation coefficients and symptom duration for a length shorter (A) or longer (B) than that indicated in the x axis. Therefore, the most relevant results in A are the leftmost values and in B, the rightmost values. n = 40 AD subjects and 9 CTRL subjects. AD, Alzheimer disease; CTRL, control.
Figure 6.

The association between number of IBA1+ microglia per plaque and symptom duration is overall weak, but stronger at initial and late stages. A and B: Association between Kendall τ correlation coefficients and symptom duration for a length shorter (A) or longer (B) than that indicated in the x axis. Therefore, the most relevant results in A are the leftmost values and in B the rightmost values. n = 40 AD subjects and 9 CTRL subjects. AD, Alzheimer disease; CTRL, control.
Figure 7.

The association between the number of CD68+ microglia per plaque and symptom duration is stronger at initial and late stages. A and B: Association between Kendall τ correlation coefficients and symptom duration for a length shorter (A) or longer (B) than that indicated in the x axis. Therefore, the most relevant results in A are the leftmost values and in B the rightmost values. n = 40 AD subjects and 9 CTRL subjects. AD, Alzheimer disease; CTRL, control.
For SMI312+ dystrophic neurites per plaque, the strength of the association was positive when restricted to short durations and negative when restricted to long durations of >10 years of symptoms. In other words, a longer symptom duration was associated with a reduced number of dystrophic neurites per plaque beyond a decade of symptoms (Figure 4).
The strength of the association between GFAP+ astrocytes per plaque and symptom duration was also largest in magnitude and positive when restricted to short durations and was close to zero or slightly negative when restricted to long durations (Figure 5).
For IBA1+ microglia per plaque, the correlations between Kendall τ and symptom duration (Figure 6) showed a weak association throughout the clinical course of the disease, slightly stronger in the earliest and latest years. This is consistent with the lack of significant association between plaque-associated IBA1+ microglia and symptom duration in this plaque-centered quantitative study. By contrast, the strength of the association between CD68+ microglia per plaque and symptom duration was strong and positive through out the clinical course of the disease, with the strongest associations at the initial and final stages (Figure 7).
Similar results were obtained when CTRL subjects were excluded from these analyses (data not shown).
Discussion
To test the hypothesis that plaque-related local toxicity accrues with increasing duration of the disease, we examined neuronal, astrocytic, and microglial markers of local plaque-associated damage in a large series of autopsied subjects who had symptomatic AD for various durations. Overall, we found an accumulation of markers of local damage around plaques as the disease advanced, arguing that plaques are not static lesions that deposit and then no longer further contribute to disease, but instead that they remain important contributors to neural system dysfunction by increasing disruption of the neuropil.
Plaque-associated dystrophic neurites are thought to be the result of the direct neurotoxic effect of Aβ oligomers,26 reactive oxygen species,27 and the inflammatory milieu28, 29 that exist around dense-core plaques. There are many markers for plaque-associated dystrophic neurites, including amyloid precursor protein, phosphorylated neurofilament, ubiquitin, and phospho-tau. We selected a phosphorylated neurofilament because it was reported to be an early marker, labeling neuritic dystrophies in plaques from nondemented healthy subjects, whereas phospho-tau is considered a later marker.30 Although there is no question that tau-related accumulation of neuropil threads, the accumulation and spread of neurofibrillary tangles, and synapse and neuronal loss all parallel and provide a major contribution to the progression of AD dementia,23, 31, 32, 33, 34, 35, 36 the present data emphasize that plaques also contribute. In agreement with this conclusion, in a large autopsy data set derived from the National Alzheimer's Coordinating Center, we reported that the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) semiquantitative score of neuritic plaques was correlated with the extent of cognitive impairment, even after taking into account the extent of tangle distribution as measured by the Braak stage.37
Aβ species, particularly oligomers, can activate both astrocytes and microglia.38, 39 Recently, genetic data40, 41, 42, 43, 44 corroborated by experimental studies45, 46, 47, 48, 49 have directly implicated microglia in the pathogenesis of AD through mechanisms that involve either Aβ phagocytosis or the secretion of proinflammatory cytokines. Similarly, reactive astrocytes were shown to promote either neuronal hyperexcitability50 or inhibition,51, 52 thereby causing neural network dysfunction. Thus, the observation of increasing numbers of activated microglial cells and reactive astrocytes around plaques the longer the symptom duration supports a critical role of these nonneuronal cells in the progression of the disease. The activated microglia markers IBA1 and CD68 exhibited a similar pattern of association with symptom duration, but CD68 association was stronger. IBA1 is a calcium-binding protein constitutively expressed in microglia and other macrophages and up-regulated in activated microglia,53 whereas CD68 is a lysosomal marker specifically expressed by activated phagocytic cells.54 Although the IBA1 protein levels are increased in the AD brain, we have previously shown that the number of IBA1+ microglia detected by fluorescent immunohistochemistry does not substantially differ between AD and healthy control subjects.17 By contrast, CD68 is a marker of activated microglia much more specific for AD.5 Our present data suggest that increasing duration of AD is specifically associated with a change of microglia toward a phagocytic phenotype.
It is well established that the presence of the APOEε4 allele is associated with a higher amyloid plaque burden.55, 56, 57, 58, 59, 60 An association of the apolipoprotein E4 with enhanced glial responses compared with the E3 isoform was proposed as well.61, 62, 63, 64 However, we have previously reported that plaque-associated glial responses in APOEε4 carriers and noncarriers are similar in magnitude.16 Here, we report that plaque-associated glial responses and neuritic changes (assessed per plaque) accrue throughout the clinical course of AD with essentially a similar rate in the presence and absence of the APOEε4 allele (only CD68+ microglia exhibited a stronger association with symptom duration in APOEε4 noncarriers). This observation is in agreement with the idea that APOEε4 affects primarily the levels of Aβ peptide (thereby leading to a higher plaque burden, a higher risk of AD, and an earlier age of symptom onset), but it has no or little effect on pathologic processes downstream of Aβ.25
Although they should be viewed as preliminary, given the modest sample size and that complex interactions among the variables may not have been adequately modeled, some interesting observations can be extracted from the secondary analyses of rate of change of markers over time. The fact that the increase in number of dystrophic neurites seems to slow down in the longest duration cases might suggest that the neurites, or the neurons that give rise to them, were lost in the long-duration cases. The pattern of association with symptom progression exhibited by reactive astrocytes was remarkably similar to that of dystrophic neurites, strongest at initial stages and weakest or even negative at late stages, suggesting that the astrocytic response is directed toward (or against) plaque-associated neuronal damage rather than plaques themselves. By contrast, activated phagocytic (CD68+) microglia exhibited a distinct pattern, with a stronger association during initial and late stages and a weaker association during intermediate stages. Whether this observation is explained by the occurrence of two waves of microglial activation and/or migration toward plaques, the first aiming at engulfing amyloid deposits and their halo of bioactive soluble Aβ species, and the second aiming at engulfing neurite debris, cannot be answered in human postmortem specimens but warrants further in vivo studies in AD mouse models. We have previously described that the number of IBA1+ and CD68+ microglia close to dense-core plaques correlates positively with plaque size, whereas the number of GFAP+ astrocytes does not, supporting the idea that both glial cell types have different preferential targets within the senile plaque: Aβ versus dystrophic neurites.16 This interpretation is in agreement with recent animal data indicating that depopulation of reactive astrocytes from plaques is associated with an increase in the number of dystrophic neurites per plaque,65 whereas depopulation of microglia has no effect on dystrophic neurites.66
Anti-Aβ immunization was successful at reducing plaque deposition in AD mouse models and in the handful of human patients who have come to autopsy, but substantial clinical benefit or at least stabilization of cognitive decline has not been observed.67, 68, 69 This has led to the postulate that antibody-mediated plaque removal in patients with established disease (ie, mild-to-moderate dementia) might be too late, because plaques may not contribute substantially to cognitive impairment during the symptomatic stages of dementia. The observation that both neuronal and glial markers of damage accumulate near plaques with increasing duration of illness suggests, by contrast, that plaques increasingly disrupt their local microenvironment during the clinical course of AD. Importantly, anti-Aβ immunization is effective at correcting plaque-associated neuritic changes in both AD mouse models70, 71 and human AD brain,18, 72 and it was also shown to ameliorate microglial activation,73, 74 further reinforcing the idea that the presence of plaques is a key factor that contributes to neuritic and glial alterations and neuropil disruption even after decades of illness.
Although limitations of cross-sectional analyses of postmortem specimens must be acknowledged, the strength of our study stems from the following: i) a well-characterized series of subjects with no other concurrent central nervous system diseases; ii) a range of clinical illness duration wide enough (4 to 20 years), so that clinician-derived variability in assessment of symptom onset would be expected to have a minimal impact on the analyses; iii) the stereology-based unbiased selection of the plaques analyzed and the blind quantification of four separate neuronal and glial markers of toxicity in their vicinity; and iv) the use of robust statistics that encompass all the information collected. Taken together, these data support the idea that amyloid plaques are not merely static extracellular deposits of Aβ but dynamic lesions characterized by an increasingly neurotoxic microenvironment, likely contributing to AD-related cognitive decline. The presence of the APOEε4 allele does not seem to have a main impact on the magnitude of plaque-associated neuritic changes and glial responses or their accrual. Although these observations support the continuing development of anti–Aβ-directed therapeutics in AD, they also argue that understanding the biology of plaque-associated dystrophic neurites and glial responses could lead to the development of novel non–Aβ-directed disease-modifying therapies.
Acknowledgments
We thank Alona Muzikansky for conducting the clustered Wilcoxon tests and the patients and relatives involved in research studies at Massachusetts Alzheimer Disease Research Center.
Footnotes
The Massachusetts Alzheimer Disease Research Center is supported by NIH grant P50AG05134 (R.A.B., M.P.F., and B.T.H.). A.S.-P. was supported by nonoverlapping fellowships from the Fondo de Investigaciones Sanitarias-Instituto de Salud Carlos III of the Spanish former Ministerio de Ciencia e Innovación grant CM06/00161 and the Fundación Alfonso Martín-Escudero (Madrid, Spain).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.10.010.
Supplemental Data
Supplemental Figure S1.
Plaque-associated features accumulate over the clinical course of Alzheimer disease. A–D: Plots represent the K-S difference between the distribution histogram from pairs of subjects against their difference in symptom duration for SMI312+ dystrophic neurites (A), GFAP+ astrocytes (B), IBA1+ microglia (C), and CD68+ microglia (D) per plaque. Note the general trend toward a larger difference in distribution histograms with a larger difference in symptom duration between pairs of subjects, although this association is only statistically significant for CD68+ microglia per plaque (D). n = 40 AD subjects only (CTRL subjects were excluded). AD, Alzheimer disease; CTRL, control.
References
- 1.Serrano-Pozo A., Frosch M.P., Masliah E., Hyman B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe D.J. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Ingelsson M., Fukumoto H., Newell K.L., Growdon J.H., Hedley-Whyte E.T., Frosch M.P., Albert M.S., Hyman B.T., Irizarry M.C. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 5.Serrano-Pozo A., Mielke M.L., Gómez-Isla T., Betensky R.A., Growdon J.H., Frosch M.P., Hyman B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol. 2011;179:1373–1384. doi: 10.1016/j.ajpath.2011.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serrano-Pozo A., Mielke M.L., Muzitansky A., Gómez-Isla T., Growdon J.H., Bacskai B.J., Betensky R.A., Frosch M.P., Hyman B.T. Stable size distribution of amyloid plaques over the course of Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:694–701. doi: 10.1097/NEN.0b013e31825e77de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack C.R., Jr., Knopman D.S., Jagust W.J., Shaw L.M., Aisen P.S., Weiner M.W., Petersen R.C., Trojanowski J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jack C.R., Jr., Knopman D.S., Jagust W.J., Petersen R.C., Weiner M.W., Aisen P.S., Shaw L.M., Vemuri P., Wiste H.J., Weigand S.D., Lesnick T.G., Pankratz V.S., Donohue M.C., Trojanowski J.Q. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bateman R.J., Xiong C., Benzinger T.L., Fagan A.M., Goate A., Fox N.C., Marcus D.S., Cairns N.J., Xie X., Blazey T.M., Holtzman D.M., Santacruz A., Buckles V., Oliver A., Moulder K., Aisen P.S., Ghetti B., Klunk W.E., McDade E., Martins R.N., Masters C.L., Mayeux R., Ringman J.M., Rossor M.N., Schofield P.R., Sperling R.A., Salloway S., Morris J.C., Dominantly Inherited Alzheimer Network Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer-Luehmann M., Spires-Jones T.L., Prada C., Garcia-Alloza M., de Calignon A., Rozkalne A., Koenigsknecht-Talboo J., Holtzman D.M., Bacskai B.J., Hyman B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R., Jr., Kawas C.H., Klunk W.E., Koroshetz W.J., Manly J.J., Mayeux R., Mohs R.C., Morris J.C., Rossor M.N., Scheltens P., Carrillo M.C., Thies B., Weintraub S., Phelps C.H. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 13.Hyman B.T., Phelps C.H., Beach T.G., Bigio E.H., Cairns N.J., Carrillo M.C., Dickson D.W., Duyckaerts C., Frosch M.P., Masliah E., Mirra S.S., Nelson P.T., Schneider J.A., Thal D.R., Thies B., Trojanowski J.Q., Vinters H.V., Montine T.J. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 15.Montine T.J., Phelps C.H., Beach T.G., Bigio E.H., Cairns N.J., Dickson D.W., Duyckaerts C., Frosch M.P., Masliah E., Mirra S.S., Nelson P.T., Schneider J.A., Thal D.R., Trojanowski J.Q., Vinters H.V., Hyman B.T., National Institute on Aging, Alzheimer's Association National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serrano-Pozo A., Muzikansky A., Gómez-Isla T., Growdon J.H., Betensky R.A., Frosch M.P., Hyman B.T. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol. 2013;72:462–471. doi: 10.1097/NEN.0b013e3182933788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano-Pozo A., Gómez-Isla T., Growdon J.H., Frosch M.P., Hyman B.T. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol. 2013;182:2332–2344. doi: 10.1016/j.ajpath.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serrano-Pozo A., William C.M., Ferrer I., Uro-Coste E., Delisle M.-B., Maurage C.-A., Hock C., Nitsch R.M., Masliah E., Growdon J.H., Frosch M.P., Hyman B.T. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010;133:1312–1327. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knowles R.B., Wyart C., Buldyrev S.V., Cruz L., Urbanc B., Hasselmo M.E., Stanley H.E., Hyman B.T. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer's disease. Proc Natl Acad Sci U S A. 1999;96:5274–5279. doi: 10.1073/pnas.96.9.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le R., Cruz L., Urbanc B., Knowles R.B., Hsiao-Ashe K., Duff K., Irizarry M.C., Stanley H.E., Hyman B.T. Plaque-induced abnormalities in neurite geometry in transgenic models of Alzheimer disease: implications for neural system disruption. J Neuropathol Exp Neurol. 2001;60:753–758. doi: 10.1093/jnen/60.8.753. [DOI] [PubMed] [Google Scholar]
- 21.D'Amore J.D., Kajdasz S.T., McLellan M.E., Bacskai B.J., Stern E.A., Hyman B.T. In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories. J Neuropathol Exp Neurol. 2003;62:137–145. doi: 10.1093/jnen/62.2.137. [DOI] [PubMed] [Google Scholar]
- 22.Spires T.L., Meyer-Luehmann M., Stern E.A., McLean P.J., Skoch J., Nguyen P.T., Bacskai B.J., Hyman B.T. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gómez-Isla T., Hollister R., West H., Mui S., Growdon J.H., Petersen R.C., Parisi J.E., Hyman B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 24.Datta S., Satten G.A. Rank-sum tests for clustered data. J Am Stat Assoc. 2005;100:908–915. [Google Scholar]
- 25.Holtzman D.M., Herz J., Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H.-Y., Hudry E., Hashimoto T., Kuchibhotla K., Rozkalne A., Fan Z., Spires-Jones T., Xie H., Arbel-Ornath M., Grosskreutz C.L., Bacskai B.J., Hyman B.T. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Alloza M., Dodwell S.A., Meyer-Luehmann M., Hyman B.T., Bacskai B.J. Plaque-derived oxidative stress mediates distorted neurite trajectories in the Alzheimer mouse model. J Neuropathol Exp Neurol. 2006;65:1082–1089. doi: 10.1097/01.jnen.0000240468.12543.af. [DOI] [PubMed] [Google Scholar]
- 28.Griffin W.S., Sheng J.G., Roberts G.W., Mrak R.E. Interleukin-1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54:276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 29.Stoltzner S.E., Grenfell T.J., Mori C., Wisniewski K.E., Wisniewski T.M., Selkoe D.J., Lemere C.A. Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol. 2000;156:489–499. doi: 10.1016/S0002-9440(10)64753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su J.H., Cummings B.J., Cotman C.W. Plaque biogenesis in brain aging and Alzheimer's disease. II. Progressive transformation and developmental sequence of dystrophic neurites. Acta Neuropathol. 1998;96:463–471. doi: 10.1007/s004010050920. [DOI] [PubMed] [Google Scholar]
- 31.DeKosky S.T., Scheff S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 32.Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 33.Bierer L.M., Hof P.R., Purohit D.P., Carlin L., Schmeidler J., Davis K.L., Perl D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 34.Gómez-Isla T., Price J.L., McKeel D.W., Jr., Morris J.C., Growdon J.H., Hyman B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giannakopoulos P., Herrmann F.R., Bussière T., Bouras C., Kövari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 36.Scheff S.W., Price D.A., Schmitt F.A., DeKosky S.T., Mufson E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 37.Serrano-Pozo A., Qian J., Monsell S.E., Frosch M.P., Betensky R.A., Hyman B.T. Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. J Neuropathol Exp Neurol. 2013;72:1182–1192. doi: 10.1097/NEN.0000000000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norris C.M., Kadish I., Blalock E.M., Chen K.-C., Thibault V., Porter N.M., Landfield P.W., Kraner S.D. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho H., Hashimoto T., Wong E., Hori Y., Wood L.B., Zhao L., Haigis K.M., Hyman B.T., Irimia D. Microfluidic chemotaxis platform for differentiating the roles of soluble and bound amyloid-β on microglial accumulation. Sci Rep. 2013;3:1823. doi: 10.1038/srep01823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J.-C., Carrasquillo M.M. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naj A.C., Jun G., Beecham G.W., Wang L.-S., Vardarajan B.N., Buros J. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J.S., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J.-C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., Hardy J., Alzheimer Genetic Analysis Group TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P.V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A.I., Lah J.J., Rujescu D., Hampel H., Giegling I., Andreassen O.A., Engedal K., Ulstein I., Djurovic S., Ibrahim-Verbaas C., Hofman A., Ikram M.A., van Duijn C.M., Thorsteinsdottir U., Kong A., Stefansson K. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambert J.-C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 45.Bradshaw E.M., Chibnik L.B., Keenan B.T., Ottoboni L., Raj T., Tang A., Rosenkrantz L.L., Imboywa S., Lee M., Von Korff A., Alzheimer Disease Neuroimaging Initiative. Morris M.C., Evans D.A., Johnson K., Sperling R.A., Schneider J.A., Bennett D.A., De Jager P.L. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griciuc A., Serrano-Pozo A., Parrado A.R., Lesinski A.N., Asselin C.N., Mullin K., Hooli B., Choi S.H., Hyman B.T., Tanzi R.E. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ulrich J.D., Finn M.B., Wang Y., Shen A., Mahan T.E., Jiang H., Stewart F.R., Piccio L., Colonna M., Holtzman D.M. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20. doi: 10.1186/1750-1326-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y., Cella M., Mallinson K., Ulrich J.D., Young K.L., Robinette M.L., Gilfillan S., Krishnan G.M., Sudhakar S., Zinselmeyer B.H., Holtzman D.M., Cirrito J.R., Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crehan H., Hardy J., Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013;54:139–149. doi: 10.1016/j.nbd.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Ortinski P.I., Dong J., Mungenast A., Yue C., Takano H., Watson D.J., Haydon P.G., Coulter D.A. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Z., Guo Z., Gearing M., Chen G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer's disease model. Nat Commun. 2014;5:4159. doi: 10.1038/ncomms5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jo S., Yarishkin O., Hwang Y.J., Chun Y.E., Park M., Woo D.H., Bae J.Y., Kim T., Lee J., Chun H., Park H.J., Lee da Y., Hong J., Kim H.Y., Oh S.-J., Park S.J., Lee H., Yoon B.-E., Kim Y., Jeong Y., Shim I., Bae Y.C., Cho J., Kowall N.W., Ryu H., Hwang E., Kim D., Lee C.J. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat Med. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ito D., Imai Y., Ohsawa K., Nakajima K., Fukuuchi Y., Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi K., Muramori F., Aoki T., Hayashi M., Miyazu K., Fukutani Y., Mukai M., Koshino F. KP-1 is a marker for extraneuronal neurofibrillary tangles and senile plaques in Alzheimer diseased brains. Dement Geriatr Cogn Disord. 1998;9:13–19. doi: 10.1159/000017015. [DOI] [PubMed] [Google Scholar]
- 55.Rebeck G.W., Reiter J.S., Strickland D.K., Hyman B.T. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 56.Schmechel D.E., Saunders A.M., Strittmatter W.J., Crain B.J., Hulette C.M., Joo S.H., Pericak-Vance M.A., Goldgaber D., Roses A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polvikoski T., Sulkava R., Haltia M., Kainulainen K., Vuorio A., Verkkoniemi A., Niinistö L., Halonen P., Kontula K. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333:1242–1247. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 58.Gomez-Isla T., West H.L., Rebeck G.W., Harr S.D., Growdon J.H., Locascio J.J., Perls T.T., Lipsitz L.A., Hyman B.T. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer's disease. Ann Neurol. 1996;39:62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- 59.Tiraboschi P., Hansen L.A., Masliah E., Alford M., Thal L.J., Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62:1977–1983. doi: 10.1212/01.wnl.0000128091.92139.0f. [DOI] [PubMed] [Google Scholar]
- 60.Serrano-Pozo A., Qian J., Monsell S.E., Betensky R.A., Hyman B.T. APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol. 2015;77:917–929. doi: 10.1002/ana.24369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cudaback E., Li X., Montine K.S., Montine T.J., Keene C.D. Apolipoprotein E isoform-dependent microglia migration. FASEB J. 2011;25:2082–2091. doi: 10.1096/fj.10-176891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu Y., Nwabuisi-Heath E., Dumanis S.B., Tai L.M., Yu C., Rebeck G.W., LaDu M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60:559–569. doi: 10.1002/glia.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rodriguez G.A., Tai L.M., LaDu M.J., Rebeck G.W. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J Neuroinflammation. 2014;11:111. doi: 10.1186/1742-2094-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cudaback E., Yang Y., Montine T.J., Keene C.D. APOE genotype-dependent modulation of astrocyte chemokine CCL3 production. Glia. 2015;63:51–65. doi: 10.1002/glia.22732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kraft A.W., Hu X., Yoon H., Yan P., Xiao Q., Wang Y., Gil S.C., Brown J., Wilhelmsson U., Restivo J.L., Cirrito J.R., Holtzman D.M., Kim J., Pekny M., Lee J.-M. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013;27:187–198. doi: 10.1096/fj.12-208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grathwohl S.A., Kälin R.E., Bolmont T., Prokop S., Winkelmann G., Kaeser S.A., Odenthal J., Radde R., Eldh T., Gandy S., Aguzzi A., Staufenbiel M., Mathews P.M., Wolburg H., Heppner F.L., Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gilman S., Koller M., Black R.S., Jenkins L., Griffith S.G., Fox N.C., Eisner L., Kirby L., Rovira M.B., Forette F., Orgogozo J.-M., AN1792(QS-21)-201 Study Team Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 68.Salloway S., Sperling R., Fox N.C., Blennow K., Klunk W., Raskind M., Sabbagh M., Honig L.S., Porsteinsson A.P., Ferris S., Reichert M., Ketter N., Nejadnik B., Guenzler V., Miloslavsky M., Wang D., Lu Y., Lull J., Tudor I.C., Liu E., Grundman M., Yuen E., Black R., Brashear H.R., Bapineuzumab 301 and 302 Clinical Trial Investigators Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doody R.S., Thomas R.G., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., Raman R., Sun X., Aisen P.S., Siemers E., Liu-Seifert H., Mohs R., Alzheimer's Disease Cooperative Study Steering Committee. Solanezumab Study Group Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 70.Brendza R.P., Bacskai B.J., Cirrito J.R., Simmons K.A., Skoch J.M., Klunk W.E., Mathis C.A., Bales K.R., Paul S.M., Hyman B.T., Holtzman D.M. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115:428–433. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rozkalne A., Spires-Jones T.L., Stern E.A., Hyman B.T. A single dose of passive immunotherapy has extended benefits on synapses and neurites in an Alzheimer's disease mouse model. Brain Res. 2009;1280:178–185. doi: 10.1016/j.brainres.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boche D., Donald J., Love S., Harris S., Neal J.W., Holmes C., Nicoll J.A.R. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer's disease. Acta Neuropathol. 2010;120:13–20. doi: 10.1007/s00401-010-0705-y. [DOI] [PubMed] [Google Scholar]
- 73.Zotova E., Holmes C., Johnston D., Neal J.W., Nicoll J.A., Boche D. Microglial alterations in human Alzheimer's disease following Aβ42 immunization. Neuropathol Appl Neurobiol. 2011;37:513–524. doi: 10.1111/j.1365-2990.2010.01156.x. [DOI] [PubMed] [Google Scholar]
- 74.Zotova E., Bharambe V., Cheaveau M., Morgan W., Holmes C., Harris S., Neal J.W., Love S., Nicoll J.A., Boche D. Inflammatory components in human Alzheimer's disease and after active amyloid-β42 immunization. Brain. 2013;136:2677–2696. doi: 10.1093/brain/awt210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.