

Abstract
Enzyme and gene replacement strategies have developed into viable therapeutic approaches for the treatment of Pompe disease (acid α-glucosidase (GAA) deficiency). Unfortunately, the introduction of GAA and viral vectors encoding the enzyme can lead to detrimental immune responses that attenuate treatment benefits and can impact patient safety. Preclinical and clinical experience in addressing humoral responses toward enzyme and gene therapy for Pompe disease have provided greater understanding of the immunological consequences of the provided therapy. B- and T-cell modulation has been shown to be effective in preventing infusion-associated reactions during enzyme replacement therapy in patients and has shown similar success in the context of gene therapy. Additional techniques to induce humoral tolerance for Pompe disease have been the targeted expression or delivery of GAA to discrete cell types or tissues such as the gut-associated lymphoid tissues, red blood cells, hematopoietic stem cells, and the liver. Research into overcoming preexisting immunity through immunomodulation and gene transfer are becoming increasingly important to achieve long-term efficacy. This review highlights the advances in therapies as well as the improved understanding of the molecular mechanisms involved in the humoral immune response with emphasis on methods employed to overcome responses associated with enzyme and gene therapies for Pompe disease.
Introduction
Protein replacement and gene therapy for recessive diseases have been shown to be safe and effective in preclinical and clinical studies. As new treatments begin advancing more toward clinical application, the importance of preventing immune responses has become clear. Experience in the treatment of allergic, autoimmune, lysosomal storage, and blood-clotting disorders has provided an expanded view on immune function. Molecular mechanisms driving humoral immune responses to enzyme replacement therapy (ERT) and gene therapy are now better understood. Although immune responses present with considerable variability across diseases, most show some degree of antibody-mediated neutralization that diminish efficacy. Antibodies directed against the therapeutic protein, viral proteins, or transgene products represent significant hurdles for translation of novel therapies to the clinic.1–3 Innate and cytotoxic responses directed against proteins and nucleic acids as a result of protein or gene therapy play an important role as well. Innate and cytotoxic responses have previously been reviewed.4–7 Over the past 15 years, better understanding and management of B- and T-cell responses to facilitate protein and gene therapies have profoundly increased our knowledge on humoral immune responses. This review provides an overview of current practices in the treatment of Pompe disease, the latest understanding of humoral immune responses and their management, as well as future implications for protein and gene replacement therapies.
Overview of Therapies for Pompe Disease
Protein replacement therapies have been used to treat human diseases for nearly a century. The first treatment described was the use of insulin for the treatment of diabetes mellitus in 1922 (ref. 8). Although individually rare, the lysosomal storage disorders encompass ~70 discrete genetic diseases, several of which are now managed with protein replacement therapies provided by an intravenous infusion of the deficient protein.1,9,10 ERT has developed into an effective treatment for several of these disorders, particularly for the multisystem disorder Pompe disease.11,12 The only lysosomal storage disorder that is also a glycogen storage disorder (glycogen storage disease type II), Pompe disease, results from a deficiency in acid α-glucosidase (GAA), a hydrolase responsible for the breakdown of lysosomal glycogen.13 The result of GAA deficiency is extensive glycogen accumulation in all tissues. Tissues particularly affected are striated muscle, smooth muscle, and neural tissues.14–16 GAA mutation type influences disease severity and the age of onset. Symptoms include profound cardiac hypertrophy, feeding problems, macroglossia, respiratory insufficiency, and skeletal muscle weakness. Early-onset Pompe disease is often fatal before the second year of life if ERT is not initiated.14,15,17,18 Patients have benefited from ERT provided as recombinant human GAA (rhGAA). ERT reduces the cardiomegaly associated with the disorder and prolongs life, although generalized weakness and respiratory insufficiency persists.11,12
GAA is synthesized as a 110 kDa precursor that is proteolytically cleaved into the mature 70 kDa isoform which breaks down the α-1,4- and α-1,6-glycosidic linkages of lysosomal glycogen into glucose.19 After synthesis, GAA traffics from the trans-Golgi network to early endosomes in a mannose-6-phosphate (M6P)-dependent pathway which can also direct the protein for secretion.20 The secretion–reuptake pathway that naturally occurs serves as the mechanism for ERT efficacy. When rhGAA is provided exogenously, the cation-independent-M6P receptors (CI-MPR) on the cell surface internalize circulating rhGAA into clathrin-coated pits which fuse to early endosomes transporting GAA to the lysosome.20
The necessity of the CI-MPR for ERT efficacy has been highlighted in Pompe disease. Conditional CI-MPR knockout mice, under the control of a muscle-specific cre/loxP element, crossed with Pompe disease mice demonstrated persistent GAA deficiency and glycogen accumulation despite treatment with ERT.21,22 Conversely, overexpression of the CI-MPR provided by the β2-agonist clenbuterol promoted increased efficacy with ERT.21,22 This suggests pharmacological compounds that increase CI-MPR expression may be a potent concomitant medication to ERT. β2-Agonists were also reported to decrease glycogen accumulation in CI-MPR-deficient Pompe disease mice treated with ERT.23 These results suggest that β2-agonists act through a CI-MPR-independent mechanism to clear glycogen. Adeno-associated viral (AAV) vector-mediated expression of GAA also results in the secretion of GAA capable of cross-correcting neighboring cells. Clenbuterol treatment improved the efficacy of gene therapy after AAV vector delivery of GAA.22 The synergy of cell autonomous and cross-correction using gene therapy is a distinct advantage over ERT. This suggests that cell-autonomous correction, rather than cross-correction provided by CI-MPR-mediated uptake of exogenous GAA, is the most efficient approach for Pompe disease therapy. Neuronal tissues in particular require cell autonomous correction as ERT cannot cross the blood–brain barrier.10 Although ERT has improved symptoms of Pompe disease, reliance on the secretion/reuptake mechanism to traffic to the lysosome requires very high doses of enzyme. Also, β2-agonists may not be effective in the long term.24,25 Receptor recycling and trafficking is impaired in Pompe disease, adding to the inefficiencies.26 Combinatorial therapies may prove necessary although cell autonomous correction would be ideal.26 ERT is currently the standard of care to treat Pompe disease, and a variety of adjunctive therapies are being produced to enhance the therapeutic effect.
Inherited metabolic diseases such as Pompe disease are model candidates for gene therapy.27–29 Several viral vectors have been utilized but the most success has been with AAV and lentiviral vectors. Many serotypes of AAV have been used to achieve efficacy for Pompe disease: AAV1 (refs. 30–33) AAV2 (ref. 34), AAV5 (refs. 35,36), AAV8 (refs. 36–39), and AAV9 (refs. 15,33,40–44). Initial evidence of efficacy resulting from intramuscular injections of AAV vectors indicated correction of the local pathology and function in Pompe disease mouse models.32–34,40 Systemic delivery of AAV vectors has imparted significant improvement in skeletal muscle force output as well as cardiac and respiratory function most recently in a direct comparison of AAV9-mediated gene therapy and ERT in a mouse model.43 Observations from these and related studies show the ability of AAV to transduce the peripheral and central nervous system by retrograde transduction to a degree that prevents the functional decline of the neuromuscular junction, at least until the degree of pathology is not at a more severe stage of disease.15,40–42 Adequate transduction of the peripheral and central nervous system has been difficult, but the many routes of delivery and serotypes of AAV have provided compelling evidence for successful restoration of physiologic functions superior to ERT.33,35,40,42,43 Evidence from these preclinical studies has led to a successful clinical trial for intradiaphragmatic injections of AAV1-CMV-hGAA in Pompe disease patients.45,46 In this study, both safety and data suggestive of efficacy were observed highlighting the therapeutic advantages of AAV gene therapy for Pompe disease.45–47
Lentiviral vectors also show promise in treating Pompe disease. In vitro,48,49 in vivo,50 and ex vivo51,52 routes of transgene delivery have been investigated in patient cell lines and mouse models. Advances in lentiviral vector design to produce self-inactivating vectors have substantially improved the safety and efficacy of the vectors.53,54 Importantly, a growing body of evidence for in vivo delivery of lentiviral vectors in small and large animal models supports the use of lentiviral vectors for multiple indications.55–58 Glycogen reduction and prevention of Pompe disease-specific cellular changes was evident using in vitro48,49 and in vivo50 approaches for lentiviral vector delivery, but concerns on integration-induced dysregulation of proto-oncogenes exists. The potential for insertional mutagenesis has prompted a shift toward ex vivo gene transfer into hematopoietic stem cells (HSC).29 Insertional mutagenesis cannot be avoided, however, by focusing on ex vivo gene transfer since not every HSC can be analyzed prior to administration and insertional mutagenesis with global transcriptomic changes has been observed in ex vivo transduced HSCs.59 Self-inactivating vectors with a more random integration pattern, improved vector design, and lower vector doses are strategies that have shown success in reducing the risk of insertional mutagenesis while remaining efficacious in models of metabolic disease and immunodeficiencies.51,60–63 Lentiviral transduced HSCs were effective in glycogen reduction in a Pompe disease mouse model, ameliorating the significant cardiac, respiratory, and skeletal muscle pathology.51,52 One hypothesis for the effect is cross-correction of neighboring cells. Transduced HSCs differentiate through erythroid, lymphoid, and myeloid lineages with the integrated transgene cassette passed on after each cell division. The differentiated cells secrete GAA which enters neighboring cells via the CI-MPR. Another possible mechanism, recently demonstrated in a model of cystinosis (another multisystem lysosomal storage disease), is tunneling nanotubes produced by transduced stem cell-derived macrophages that promote vesicular cross-correction.64
Humoral Responses to GAA
ERT and gene therapy are two therapeutic strategies for Pompe disease, yet, deleterious immune responses against GAA are a substantial roadblock for success.1,65–67 The magnitude of the immune response is somewhat related to the severity of the GAA mutation.68,69 The presence of cross-reactive immunological material (CRIM) is determined in part by the expression and functionality of GAA in a Pompe disease patient.70 During the development of the immune system, immune cells are exposed to antigens expressed within the body and autoreactive B- and T-cells are eliminated to prevent autoimmunity. In the periphery, antibody generation results from interactions between professional antigen-presenting cells (APCs) such as dendritic cells, CD4+ T helper cells, and B-cells. Presentation of processed antigens by myeloid APCs and B-cells on major histocompatibility complex II (MHCII), with appropriate cytokine signaling and co-stimulation, results in CD4+ T-cell activation. The cross-talk between B and T-cells ultimately drives B-cell activation and antibody production, thereby establishing a humoral response and antigen-specific plasma cells (Figure 1). MHCI presentation by APCs to CD8+ T-cells can cause a cytotoxic T-cell response.71 Persistent high-titer anti-GAA IgG is the primary immunoglobulin that complicates ERT and gene therapy for Pompe disease. Since CRIM-negative Pompe disease patients have a complete deficiency of GAA, the immune system is naive to the therapeutic protein, perceives GAA as foreign, and develops anti-GAA B- and T-cells.72 Antibodies produced against GAA, via a MyD88-dependent pathway in mice, render the therapy ineffective and potentially dangerous.73,74 Anti-GAA antibodies sequester circulating GAA to a staggering degree. Over 80% of rhGAA can be bound to anti-GAA antibodies.75 Antibody–antigen complexes have the potential to induce immunotoxicities such as anaphylaxis and disseminated intravascular coagulation in mouse models of Pompe disease.75,76 Importantly, neutralization of GAA by antibodies is not as relevant in the context of gene therapy. Exogenous delivery of rhGAA provided as ERT can be neutralized almost completely,75 but cell autonomous correction imparts the natural trafficking of GAA to the lysosome, making the protein less accessible to antibodies from within cells (Figure 2). Gene therapy-derived GAA does initiate an immune response due to secreted GAA trafficking from the trans-Golgi network to the cell surface. Antibodies generated after gene therapy could affect cross-correction and potentially lead to cytotoxic effects. Cytotoxic and antibody responses against the AAV capsid and transgene have occurred in clinical trials for hemophilia B.77 Antibody responses against the capsid and transgene were observed in Pompe disease patients after intradiaphragm injection of AAV1-CMV-hGAA, but no cytotoxic responses were observed.45 Although CRIM-negative patients most readily produce anti-GAA antibodies, CRIM-positive patients can also produce high-titer anti-GAA antibodies leading to difficult decisions in the use of immune suppression strategies.67,78 Early administration of ERT has potential benefits, but determination of CRIM status is an important consideration for successful ERT and gene therapy.67,70,79–81 Due to the multiple components involved in mounting an immune response, many potential targets have been identified for intervention. The following sections will highlight efforts in overcoming humoral responses resulting from Pompe disease therapies.
Figure 1.

Generation of a humoral immune response. CD4+ T-cells engage MHCII loaded with antigen (GAA) on APCs or naive B-cells via TCR. The CD4 coreceptor also binds and facilitates the appropriate signaling through CD3. During the immune response, additional cytokines and costimulatory molecules drive the generation of memory B-cells which undergo Ig class switching from immature IgM to affinity matured IgGs directed against the antigen. Memory B-cells then migrate into the bone marrow and develop into antigen-specific plasma cells which perpetually produce antibodies. APC, antigen-presenting cell; GAA, acid α-glucosidase; Ig, immunoglobulin; MHCII, major histocompatibility complex II; TCR, T-cell receptor.
Figure 2.
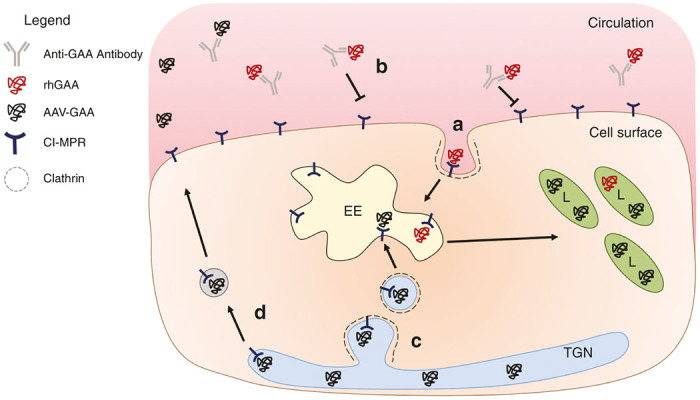
Trafficking and neutralization of GAA. (a) Exogenous rhGAA (red) provided as ERT binds to the CI-MPR which is endocytosed via clathrin-coated pits. GAA is then trafficked to early endosomes which subsequently undergo fusion with lysosomes, delivering the enzyme to the proper intracellular location. (b) ERT initiates an immune response that results in high-titer anti-GAA antibodies that bind and inhibit rhGAA uptake. (c) Cell autonomous correction resulting from gene therapy (AAV-GAA; black) allows for proper production of GAA and trafficking from the trans-Golgi network to lysosomes, representing the more efficient, endogenous pathway, which is protected from antibody-mediated neutralization. (d) GAA trafficking from the trans-Golgi network may escape and exocytose into the circulation (cross-correction), which may be sequestered by anti-GAA antibodies. AAV, adeno-associated virus; CI-MPR, cation-independent mannose-6-phosphate receptor; EE, early endosome; GAA, acid α-glucosidase; L, lysosome; TGN, trans-Golgi network.
Improving the Safety and Efficacy of ERT
A unique feature of the CI-MPR is that it is both a M6P receptor and it binds a separate ligand, insulin-like growth factor II (IGFII).20 Investigators have capitalized on this feature for lysosomal storage diseases and found that inclusion of IGFII signal peptides improved efficacy. LeBowitz et al. first described glycosylation-independent lysosomal targeting (GILT) for treating mouse models of mucopolysaccharidosis type VII (MPS VII; Sly syndrome), Pompe disease, and MPS IIIB (Sanfilippo syndrome).82–84 GILT technology makes use of IGFII-tagged proteins for improved uptake via the CI-MPR without the necessity of M6P residues. The production methods and enzyme activity of GILT-GAA are similar to that of rhGAA from Chinese hamster ovary cell lines which is used for ERT.83,85 Of significance, the trafficking of the therapeutic protein is improved with the GILT-tag.83 With a higher affinity for the CI-MPR compared to rhGAA, the uptake kinetics and glycogen reduction mediated by GILT-GAA not only make it superior to traditional ERT in animal models, but could necessitate decreased doses from 20–40 mg/kg to as low as 5 mg/kg.83 The duration of infusions and burden on the immune system would be significantly reduced if the required doses of enzyme could be lowered to 5 mg/kg or less. Based on preclinical studies, a multicenter phase 1/2 trial using GILT-GAA as a replacement for traditional ERT is underway. However, cross-correction mediated by secreted GAA is a slow and inefficient process requiring very high circulating enzyme concentrations. The IGFII peptide may also trigger hypoglycemia, but initial clinical data suggests that the drug is safe, and changes in diet can control hypoglycemia.86 Although GILT-GAA is more efficient than rhGAA alone, inclusion of other pharmacological interventions may further improve its efficacy. Cell autonomous correction provided via gene therapy delivered GILT-GAA is expected to be the most efficient therapy.
Investigations into improving traditional ERT for Pompe disease are actively underway. One modality is the inclusion of pharmacological chaperones. As reversible allosteric inhibitors, pharmacological chaperones stabilize the conformation of GAA, allowing for proper folding and trafficking, leading to an enhancement in activity and bioavailability in Pompe disease patients and mouse models.87,88 The chaperones are commonly glucose analogues, the imino sugars 1-deoxynojirimycin and N-butyldeoxynojirimycin.88–92 Investigations on the benefit of including chaperones have revealed improved pharmacokinetics and pharmacodynamics of circulating GAA bolstering glycogen substrate reduction.90,91,93 The preclinical evidence has also been predictive of the clinical outcomes. Coadministration of chaperones with ERT have improved circulating GAA levels and GAA activity in Pompe disease patients.92,94 Additional benefit was observed in lowering hypersensitivity reactions and antibody production in patients.94 By ensuring the appropriate folding, increased stability, and uptake of ERT, chaperones may prevent both nonspecific and specific antibody binding. Limiting epitope exposure to APCs, while exposing the glycosylation and phosphorylation moieties in an improved manner, may lead to more efficient CI-MPR-mediated uptake. By diminishing the immunogenicity of native, unfolded GAA, the stimulus to the immune system might be less severe which may be the reason decreased antibody titers were observed. The fact that the patients in the clinical trial were CRIM positive may also be a factor.
Immunotherapies for Pompe Disease
Recent advances preventing IgG-production have proven effective in both preclinical and clinical settings (Figure 3).95 As a means for general immune suppression, the drugs methotrexate, mycophenolate mofetil, and cyclophosphamide have been successfully used to prevent responses to ERT through inhibition of folic acid metabolism (which blocks de novo DNA synthesis) or as DNA alkylating agents (blocking DNA replication). These agents have prevented anti-GAA antibody formation both in mice and Pompe disease patients.96–98 Using low-dose methotrexate, antigen-specific immune tolerance induction is believed to have been achieved through generating the specific induction of IL-10 and TGF-β-producing B-cells, or regulatory B-cells, in a mouse model of Pompe disease.98 Evidence of T-cell elimination by low-dose methotrexate would support that suppressive activity may be an effect of regulatory B-cell responses rather than regulatory T-cell (Treg) responses.99 In a related approach, patients have been placed on a combination of rituximab (a monoclonal anti-CD20 that globally depletes B-cells), methotrexate, and intravenous immunoglobulin.96 This global immune suppression regimen has been effective in the elimination of pathogenic anti-GAA antibodies and has allowed for increased doses of ERT. However, such a protocol would also deplete regulatory B-cells. Some evidence has been provided that it may be possible to discontinue immune suppression with tolerance to GAA maintained.100 Stably induced tolerance after immune suppression in patients awaits verification in more patients under better controlled study conditions.
Figure 3.

Targets for immunotherapy. Multiple targets to prevent humoral immune responses or establish immune tolerance have been pursued for Pompe disease ERT and gene therapy. (a) Targeting of BAFF prevents binding to BAFF-R, which inhibits the maturation of antigen-specific memory B-cells. DNA-damaging agents such as CP, MMF, and MTX result in B-cell depletion. Treatment with anti-CD20 also results in B-cell depletion. Low-dose MTX has recently been shown to induce regulatory B-cells, which allowed for successful immune tolerance induction in a mouse model of Pompe disease. Blocking mTOR signaling through rapamycin treatment inhibits B-cell proliferation. Proteasome inhibitors, such as bortezomib, inhibit the recycling of proteins that would be used to generate antibodies in plasma cells, inducing senescence. (b) T-cell targets, such as CD3 or CD4, induce the depletion of T-cells or prevent the activation of B-cells to produce antibodies, respectively. Treatment with rapamycin induces a signaling cascade that skews T-cells into a regulatory phenotype capable of antigen-specific immune suppression. BAFF, B-cell activating factor; BAFF-R, BAFF receptor; CP, cyclophosphamide; MMF, mycophenolate mofetil; mTOR, mammalian target of rapamycin; MTX, methotrexate.
Immune modulatory regimens that do not include chemotherapeutics have also been investigated. Blocking survival factors that do not ablate the immune system have proven effective in inhibiting antibody formation. The inhibition of B-cell activating factor (BAFF) and mammalian target of rapamycin have been evaluated in Pompe disease mice and patients. BAFF is a pleiotropic cytokine responsible for the maturation of B-cells.101 In a preclinical model of Pompe disease, anti-BAFF treatment (using an antimouse ortholog of the clinical drug, belimumab) promoted an immunologically naive environment such that anti-GAA antibodies were not formed, resulting in improved GAA activity in affected tissues and preventing acute anaphylactic responses.102 To maintain a naive environment, the continuation of belimumab treatment was necessary. Nonetheless, the effect of preventing the formation of GAA-specific plasma cells from maturing and migrating to the bone marrow represents a novel means to address antibody-related immunotoxicities that does not necessitate ablation of the immune system.103
Rapamycin, commonly used as a T-cell modulator, has also been investigated to treat humoral and hypersensitivity reactions in CRIM-negative and CRIM-positive early-onset Pompe disease patients.104 Rapamycin targets mammalian target of rapamycin, which is responsible for regulating cell survival, growth, and proliferation of lymphocytes.105 Despite the well-documented effects on T-cell function, rapamycin is also a potent suppressor of B-cell proliferation.106 Although the mechanism involved in rapamycin-mediated B-cell suppression is not fully understood, the potent effects rapamycin has on lymphocyte survival produces a profound effect in preventing unwanted antibody responses.106 Rapamycin, in combination with rituximab-mediated B-cell depletion, addressed both B- and T-cell responses, was effective in preventing hypersensitivity reactions associated with ERT infusions, and facilitated some functional benefit in clinical outcomes.104 Inclusion of rapamycin increases the capacity to induce immune tolerance as well. Blocking mammalian target of rapamycin signaling with rapamycin in mouse models of hemophilia A and B has shown promise in the induction of tolerance through an expansion of factor VIII- or IX-specific Tregs capable of suppressing inhibitory antibody production.107–111
Targeting of T-cell co-receptors has similarly been effective. The CD3 co-receptor is required for the signal transduction cascade following TCR and CD4 engagement to MHCII.112 Targeting CD3 has been shown to be effective in prevented immune responses in Pompe disease.113,114 Mice treated with anti-CD3 antibody showed marked reduction in CD4+ and CD8+ T-cells yet the ratio between effector and regulatory cell types was skewed toward Tregs. Dependence of tolerance on Tregs was demonstrated by elimination of CD25+ cells, which eliminated tolerance. Anti-CD3 treatment also suppressed anti-GAA antibody formation in the context of preexisting immunity, which would be more indicative of the clinical situation as prophylactic immune suppression is not often in place prior to the initiation of ERT. The mechanism of suppression may then be a direct interaction of Tregs with anti-GAA antibody producing plasma cells. In clinical studies, the inclusion of proteasome inhibitors was necessary to induce senescence of plasma cells.115 Plasma cells are a difficult subset of B-cells to suppress as antibody production is not inhibited with anti-CD20 or anti-BAFF therapy. Once established within the bone marrow, plasma cells no longer express CD20 or require BAFF to the same degree as circulating B-cells and plasmablasts, which are immature plasma cells.103,116,117 Recently, however, it has been shown in mouse models that Tregs can specifically suppress antibody production in refractory plasma cells.118
Although not as advanced as the developments for ERT, some of the immune modulatory strategies are now also being tested to block humoral responses in the context of gene therapy. In a preclinical model of Pompe disease, CD4 co-receptor blockade adequately prevented the formation of anti-GAA antibodies following infusion of AAV9-CBA-hGAA by limiting CD4+ T-cell helper functions.119 Similar to anti-BAFF therapy, which does not globally ablate B-cell immunity,102,103 the anti-CD4 antibody was found to be nondepleting of T-cells while preventing hypersensitivity reactions and mortality associated with ERT. Interestingly, only short-course anti-CD4 treatment was effective as prolonged anti-CD4 treatment after gene transfer had no impact on preventing mortality. Prophylactic treatment with anti-CD4 in mice injected with AAV displayed decreased anti-capsid antibody titers which allowed for greater liver transduction provided by an additional vector that targeted GFP expression to the liver.119 These results led to the conclusion that anti-CD4 treatment similarly prevented the development of neutralizing anticapsid antibodies in addition to blocking anti-GAA antibodies.
Recent gene therapy clinical studies have shown that it is possible to use immune suppression regimens to prevent responses to GAA as well as to the vector. A CRIM-negative child with Pompe disease treated with ERT while on B- and T-cell immunomodulation104 was enrolled in a gene therapy clinical trial for intramuscular AAV1-CMV-hGAA administration.120 Children not receiving immune suppression in the study showed the expected increase in anti-AAV1 capsid antibodies. In sharp contrast, the child under immune suppression remained seronegative for both anticapsid and anti-GAA antibodies through 1 year post-dosing with functional improvements in respiratory outcome measures.120 These results laid the groundwork for management of immune responses in the event of readministration of gene therapy.121 Particularly for pediatric patients, multiple doses of gene therapy may prove necessary as the individual develops. As evidenced by an AAV clinical trial for Leber’s congenital amaurosis (an inherited form of blindness), the 5 years post gene transfer functional decline of vision, indicated by progressive deterioration in the initially improved area of vision, suggests that additional exposure or early delivery of gene therapy may be necessary to achieve a sustained effect.122 As it pertains to Pompe disease, maintaining a stable, high level of GAA systemically as a child matures may be a challenge. Addressing preexisting neutralizing antibodies, which would allow patients to be permissive to multiple administrations of gene therapy throughout the course of their life, will likely be the most effective course of treatment.
Altogether these notions underlie the importance of managing the immune response to gene or ERT for long-term success. Interventions such as pharmacological chaperones and immune suppression allow for immediate application in a clinical situation. Induction of antigen-specific immune tolerance capable of dampening or preventing immune responses stands as the most sought after goal to ultimately achieve in the efforts to overcome humoral immune responses for Pompe disease.
Antigen-Specific Immune Tolerance Induction Strategies
Many approaches to immunomodulation have developed for controlling immune responses toward ERT, and there have been many effective developments in the establishment of immune tolerance toward GAA for Pompe disease therapy (Figure 4). The specifically targeted expression or exposure of GAA to discrete cell types and tissues has allowed for the development of functional Tregs capable of attenuating immune responses. As critical regulators of tolerance, CD4+CD25+FoxP3+ Tregs and type 1 regulatory T-cells (TR1; CD4+CD25-FoxP3-CD49b+LAG-3+) use both cell contact-dependent (via CTLA-4 and PD-1) as well as cytokine-dependent (via IL-10, IL-13, IL-14, IL-35, and TGF-β) mechanisms for immune suppression.81,123–127 Ablation of Tregs results in severe autoimmunity as evidenced by Foxp3 knockout (scurfy) mice which exhibit a lymphoproliferative disease as a result of a lack of T-cell tolerance. Similarly, defects in FOXP3 in humans lead to the autoimmune disease, immunodysregulation polyendocrinopathy enteropathy X-linked syndrome. As critical as Tregs are for central tolerance during development and prevention of autoimmunity, they are similarly instrumental for inducing tolerance toward exogenous antigens, such as therapeutic proteins like GAA. To achieve induced or central tolerance, studies into targeting the mucosal immune system, red blood cells (RBCs), HSCs, and liver-directed gene therapy have all shown promise in preventing the substantial immune responses that occur during treatment for Pompe disease.
Figure 4.

Generation of antigen-specific Tregs. Tregs can be generated from CD4+ T-cells through multiple pathways conducive to gene transfer for immune tolerance induction. Targeting of the GALT through oral tolerance results in antigen uptake by macrophages and dendritic cells that facilitate both CD25+FoxP3+LAP- and CD25-FoxP3-LAP+ Treg induction through an RA-, IDO-, IL-10-, and TGF-β-dependent mechanism. Targeting HSCs allows for induction of central tolerance by exposing developing lymphoid precursors to autoantigens through the coordinated activities of SIRTUIN1 and AIRE. Autoreactive T-cells are eliminated, and the antigen provided by gene transfer is recognized as self, which will not initiate an immune response. RBC delivery of the antigen and hepatic gene transfer result in identical mechanisms as the antigen-loaded RBCs are cleared by the liver. Through interactions of multiple liver-specific cells and high concentrations of immunosuppressive signals, antigen-specific CD25+FoxP3+ Tregs and CD25-FoxP3-CD49b+LAG-3+ TR1 cells are produced. After induction, these cells circulate and inhibit areas undergoing an antigen-specific response through cell contact- and cytokine-dependent mechanisms. AIRE, autoimmune regulator; GALT, gut-associated lymphoid tissue; HSC, hematopoietic stem cell; RBC, red blood cell.
The gut-associated lymphoid tissue (GALT) has developed a sophisticated series of highly specialized cells that, under normal conditions, promote an anti-inflammatory environment capable of inducing oral tolerance.128 In efforts to expose neo-antigens to the GALT and induce oral tolerance, feeding of rhGAA to C57BL/6 and BALB/c mice was performed with immunizations with rhGAA.129 It was observed that the mice that were fed rhGAA every other day for 5 days did not produce GAA-specific IgG or IgE. This seminal study on oral tolerance for Pompe disease has since progressed to using bioencapsulated GAA for oral delivery. Tobacco transplastomic plant lines expressing a cholera-toxin B subunit (CTB) GAA fusion protein produced similar results in preventing anti-GAA IgG1 and IgG2a at a greater than 10-fold lower dose compared to feeding with nonencapsulated rhGAA.130 Inclusion of CTB-GAA within plant cells allows for lower doses due to improved CTB-mediated uptake across the mucosal lining and protection from stomach enzymes and acids when encased in cellulose. This is an important consideration, uptake of CTB-fusion proteins by dendritic and F4/80+ cells within the GALT leads to the induction of antigen-specific regulatory CD4+CD25-LAP+ T-cells and conventional CD4+CD25+FoxP3+ Tregs during plant-based oral tolerance induction.131 The interactions between CD103+ and plasmacytoid dendritic cells with CD4+ T-cells after oral GAA delivery initiates a cascade of secondary signals, namely, retinoic acid, indoleamine-2,3-dioxygenase, TGF-β, and IL-10, which promote GAA-specific oral tolerance.131 Plant-based oral tolerance stands as an attractive option for managing immune responses as transgenic plants can be produced in mass quantities that can be easily administered to patients with relatively little cost.132
A novel approach to induce immune tolerance for Pompe disease, that also employs bioencapsulation, has recently been developed using RBCs.133 Encapsulation of rhGAA into RBCs through dialysis allowed for a very high concentration of rhGAA to be entrapped within the RBCs without affecting the integrity of the cells.133,134 RBCs containing rhGAA transfused into C57BL/6 mice were cleared by the liver in greatest abundance by F4/80+ macrophages and to a lesser extent by splenic CD11b+ macrophages.133 This clearance resulted in antigen uptake and presentation by MHCII that, in the liver, can skew CD4+ T-cells to a regulatory phenotype. The transfusions proved effective in dampening antigen-specific IgG, IgG1, and IgG2c antibody production.133 The inhibition of antibody production was maintained through the study duration of 2 months.133 It will be interesting if these experiments are repeated in a Pompe disease model as these results could have a profound impact in the clinic. RBCs are easily accessed, and if preclinical evidence is indicative of the clinical outcome, Pompe disease patients have an increased hematocrit when ERT is chronically administered, making RBCs an abundant and minimally invasive cell type to modify, especially compared to HSCs.76 Reasonable turnaround time for modified RBCs for a patient would be anticipated as the dialysis procedure to load rhGAA into the RBCs is straightforward, and the use of autologous cells precludes safety concerns related to rejection. Preexisting immunity was not addressed, but, like with immunomodulation, prophylactic intervention was effective in preventing deleterious responses. Efficacy in reversing preexisting immunity will be an important question to address in subsequent studies. Whether through transplastomic plant lines or RBCs, bioencapsulated delivery of GAA represents a promising and expanding area of tolerance induction for Pompe disease.
Lentiviral transduction of HSCs has been shown to be capable of correcting Pompe disease pathology in a mouse model.51 The targeted expression within HSCs was similarly shown to induce GAA-specific tolerance. After sublethal irradiation, transplantation of transduced HSCs was effective in reconstituting the immune system and prevented antibody formation after rhGAA and adjuvant immunizations. The development of immune tolerance in this manner most closely resembles the process of central tolerance as opposed to induced tolerance in the periphery. As thymocytes develop from the lymphoid progenitors derived from transduced HSCs, GAA is expressed within cortical and medullary thymic epithelial cells orchestrated by SIRTUIN 1, a deacetylase, and autoimmune regulator, a master transcriptional regulator, to facilitate central tolerance to autoantigens and negative selection of autoreactive T-cells.135–137 In a situation where GAA, the autoantigen, is never expressed due to deletions or truncations within the gene, SIRTUIN 1, autoimmune regulator and their downstream functions cannot promote the negative selection of GAA-specific T-cells. By facilitating GAA expression in the thymus through gene therapy, central tolerance can be achieved. Of concern, however, is the conditioning regimen that was used in the mouse study.51 A sublethal total radiation dose of 6 Gy was performed on Pompe disease mice to ablate the bone marrow to provide space for the transduced HSCs to engraft. It is not known how well a Pompe disease patient would tolerate total body irradiation. Although it is understood that physical space must exist for HSC engraftment, pursuing less devastating conditioning regimens, such as with busulfan with or without fludarabine, has been shown to be a superior regimen in mice and nonhuman primates compared to irradiation.138,139 In a clinical trial using lentiviral vector transduced HSCs for the treatment of β-thalassemia, a patient was exclusively conditioned with busulfan and has since no longer required RBC transfusions.140 Busulfan conditioning was also used in pediatric indications for metachromatic leukodystrophy and Wiskott–Aldrich syndrome.62,63 Fludarabine was additionally provided in the Wiskott–Aldrich clinical trial.62 In both instances, stable gene transfer occurred with disease-free survival. Although long-term safety data are not currently available, the findings are encouraging and suggest that total body irradiation is not necessary. As lentiviral transduction of HSCs begins to move into clinical trials for Pompe disease and other lysosomal storage disorders,29 the conditioning regimen should be carefully considered with emphasis of using as mild of a regimen as possible considering the fragility of Pompe disease patients.
One of the more often utilized and successful routes for inducing immune tolerance has been liver-directed gene therapy using AAV to generate antigen-specific Tregs.118,126 Although direct intrathymic delivery has been pursued to achieve central tolerance to GAA, the ease of targeting the liver via injection of a peripheral vein represents a more attractive and less invasive route of delivery for inducing tolerance.141 The liver is uniquely equipped to interact with circulating antigens and is naturally immune tolerant expressing high levels of IL-10, TGF-β, and PD-L1.142 The interactions between hepatocytes, endothelial cells, liver sinusoidal endothelial cells, hepatic stellate cells, tissue resident lymphocytes, dendritic cells, and Kupffer cells surveying the antigens within the circulation act discretely to promote a tolerogenic microenvironment similar to the GALT.143 It has been demonstrated that the restriction of transgene expression to hepatocytes, as well as their interactions with APCs, is necessary to prevent deleterious immune responses.125,144 Recent evidence in mouse models has suggested that the immunoregulatory phenotype is initiated via the activation of the Jagged1/Notch signaling pathway.145,146 It was shown that blocking Notch signaling prevented IL-10 secretion by CD4+ T-cells. After gene transfer, continuous T-cell receptor interactions with antigen, provided by liver-restricted gene expression, stabilizes FoxP3 expression and upregulates GITR, PD-1, and CTLA-4 on IL-10 producing CD4+ T-cells, imparting the regulatory phenotype.125,126,147
Liver-directed gene therapy has had benefit for inducing immune tolerance in Pompe disease mouse models. First, successful prevention of antibody production was achieved when employing a liver-specific promoter to express GAA.148 AAV8 was the vector serotype of choice, and it was shown that its natural propensity to target the liver was not as essential as using a liver-specific promoter. Attempts at inducing immune tolerance using an equivalent serotype delivering a constitutive chicken β-actin (CB) promoter-driven construct initiated a deleterious response. As a means to impart increased therapeutic benefit for ERT, liver-directed gene therapy was similarly effective. Immune tolerance allowed for repeated infusions of ERT without anaphylaxis and greater glycogen clearance in affected tissues.149 In a follow-up study, it was shown suppression of antibody production was Treg-dependent. Elimination of CD25+ T-cells through anti-CD25 antibody administration ablated Treg-mediated protection from antibody responses.150 This conclusion is further supported by specific elimination of FoxP3+ cells.118 Lastly, tolerance induction through liver gene transfer was effective, but incomplete, in enhancing gene therapy provided by a separate vector. Although the method of using two vectors in this study was effective in superseding immune responses from a CB-driven construct, challenge with rhGAA initiated a response. The dose of rhGAA, its nonmurine origin, as well as the tendency for CB-driven constructs to initiate substantial immune responses were likely the causes of the immune response.151 The inclusion of multiple vectors to treat Pompe disease will likely be necessary as the immunogenicity of rhGAA, and the systemic nature of the disease, will require a multifaceted approach to treat. Especially considering benefit from liver-derived GAA may not be sufficient for full body correction as secreted GAA behaves similarly to ERT and is too large to cross the blood–brain barrier.10 Therefore, utilizing a serotype that can cross this barrier, such as AAV9, which demonstrates a more global transduction profile, may prove more appropriate.44,152,153 Engineered or de-targeted AAV serotypes may also be considered to specifically transduce tissues of interest.154–156 A dual-vector AAV9 approach that targets gene expression specifically to the liver and neuromuscular tissues has the potential to be superior in inducing immune tolerance and correction of Pompe disease pathology.
Conclusions
In developing therapeutic strategies for Pompe disease, there has been a need to understand and control immune responses against GAA. The same challenges exist for all recessive diseases where limited endogenous protein is produced. Evading immune responses for Pompe disease has been pursued through a variety of approaches. Modifications in the dose and formulation of ERT have shown efficacy when GILT-tags are included, but success with ERT is limited without immunomodulation. Pharmacological interventions provided as small-molecule chaperones or immunosuppressive agents have lessened the frequency of infusion-associated reactions and improved the efficacy of ERT and gene therapy. Long-term immune suppression can have lasting, negative consequences, however,157 and there is a risk of de-sensitization to certain medications. Importantly, all of the interventions discussed are at the experimental stage, and therefore, no universally accepted standard for addressing immune responses has been adopted. Targeted delivery of bioencapsulated GAA has been very promising for inducing tolerance. Oral tolerance induction has proven an effective clinical intervention for severe allergic responses, most recently to peanuts.158 An oral tolerance approach for hemophilia A,159 B,131,160 and Pompe disease130 has been shown to be effective in preclinical models but has yet to be implemented in the clinic. RBCs dialyzed with rhGAA is another approach to induce tolerance that needs further testing in Pompe disease animal models. Ex vivo lentiviral transduction of HSCs holds great promise in achieving central tolerance. As safer vectors and more sensitive assays to ascertain insertional mutagenesis are developed, the likelihood of a lentiviral-based therapy for Pompe disease improves. AAV gene therapy is at the forefront of next-generation therapies for Pompe disease. AAV-mediated hepatic expression has proven effective but limited in corrected the neuromuscular pathology. Developing a clinical product that improves physiologic outcomes and induces tolerance will likely be the most successful intervention.
The most efficacious approaches to induce immune tolerance for Pompe disease are predicated upon the induction of GAA-specific Tregs or elimination of B- and T-cells to prevent antibody production. Preclinical and clinical experience with these methods has warranted enthusiasm for the future. In a naive setting, tolerance can be induced to prevent the development of immune responses to GAA, yet, designing therapies that address preexisting immunity has remained a challenge. The described approaches to overcome immune response related to protein replacement and gene therapy have broad implications for many monogenic, allergic, and autoimmune disorders, translating to safer and more effective therapies for patients.
Acknowledgments
This work was supported in part by the NIH/NCATS Clinical and Translational Science Award to the University of Florida UL1 TR000064 (to B.J.B. and P.A.D.), NIH/NICHD traineeship T32 HD043730 (to P.A.D.), Wenner-Gren Foundation Fellowship-Stockholm, Kronprinsessan Lovisas Förening, and Swedish Society of Medicine grants (to S.N.), NIH/NHLBI research awards R01 HL107904 (to R.W.H.), P01 HL59412 and R01 HD052682 (to B.J.B.). The content is solely the responsibility of the authors, and the National Institutes of Health, National Center for Advancing Translational Sciences, National Institute of Child Health & Human Development, National Heart, Lung and Blood Institute, Wenner-Gren Foundation-Stockholm, Kronprinsessan Lovisas Förening, or Swedish Society of Medicine were not involved in the writing of the review or in the decision to submit the article for publication.
Footnotes
B.J.B. is a founder and owner of founder equity of Applied Genetic Technologies Corporation (AGTC) and an unpaid member of the Scientific Advisory Board of Audentes Therapeutics, Solid Bioventures, LLC, and Bristol-Meyers Squibb. He could be entitled to patent royalties for inventions issued or pending concerning AAV technology. P.A.D. and M.C. could be entitled to patent royalties for pending inventions concerning AAV technology. Johns Hopkins University and University of Florida could be entitled to patent royalties for inventions issued or pending concerning AAV technology. S.N., L.M., and R.W.H. declare that they have no competing interests.
References
- Brooks, DA, Kakavanos, R and Hopwood, JJ (2003). Significance of immune response to enzyme-replacement therapy for patients with a lysosomal storage disorder. Trends Mol Med 9: 450–453. [DOI] [PubMed] [Google Scholar]
- Ponder, KP (2008). Immune response hinders therapy for lysosomal storage diseases. J Clin Invest 118: 2686–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markusic, DM and Herzog, RW (2012). Liver-directed adeno-associated viral gene therapy for hemophilia. J Genet Syndr Gene Ther 1: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mays, LE and Wilson, JM (2011). The complex and evolving story of T cell activation to AAV vector-encoded transgene products. Mol Ther 19: 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL, Martino, AT, Aslanidi, GV, Jayandharan, GR, Srivastava, A and Herzog, RW (2011). Innate immune responses to AAV vectors. Front Microbiol 2: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F and High, KA (2013). Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122: 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basner-Tschakarjan, E, Bijjiga, E and Martino, AT (2014). Pre-clinical assessment of immune responses to adeno-associated virus (AAV) vectors. Front Immunol 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banting, FG, Best, CH, Collip, JB, Campbell, WR and Fletcher, AA (1922). Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J 12: 141–146. [PMC free article] [PubMed] [Google Scholar]
- Meikle, PJ, Hopwood, JJ, Clague, AE and Carey, WF (1999). Prevalence of lysosomal storage disorders. JAMA 281: 249–254. [DOI] [PubMed] [Google Scholar]
- Boustany, RM (2013). Lysosomal storage diseases–the horizon expands. Nat Rev Neurol 9: 583–598. [DOI] [PubMed] [Google Scholar]
- Kishnani, PS, Corzo, D, Nicolino, M, Byrne, B, Mandel, H, Hwu, WL et al. (2007). Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 68: 99–109. [DOI] [PubMed] [Google Scholar]
- Byrne, BJ, Kishnani, PS, Case, LE, Merlini, L, Müller-Felber, W, Prasad, S et al. (2011). Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab 103: 1–11. [DOI] [PubMed] [Google Scholar]
- van der Ploeg, AT and Reuser, AJ (2008). Pompe’s disease. Lancet 372: 1342–1353. [DOI] [PubMed] [Google Scholar]
- DeRuisseau, LR, Fuller, DD, Qiu, K, DeRuisseau, KC, Donnelly, WH Jr, Mah, C et al. (2009). Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci USA 106: 9419–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk, DJ, Todd, AG, Lee, S, Soustek, MS, ElMallah, MK, Fuller, DD et al. (2015). Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum Mol Genet 24: 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi, SP, Phadke, MS and Kerkar, PG (2015). Giant heart of classical infantile-onset Pompe disease with mirror image dextrocardia. Circ Cardiovasc Imaging 8: e003637. [DOI] [PubMed] [Google Scholar]
- Lee, KZ, Qiu, K, Sandhu, MS, Elmallah, MK, Falk, DJ, Lane, MA et al. (2011). Hypoglossal neuropathology and respiratory activity in pompe mice. Front Physiol 2: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani, PS, Hwu, WL, Mandel, H, Nicolino, M, Yong, F and Corzo, D; Infantile-Onset Pompe Disease Natural History Study Group (2006). A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 148: 671–676. [DOI] [PubMed] [Google Scholar]
- Moreland, RJ, Jin, X, Zhang, XK, Decker, RW, Albee, KL, Lee, KL et al. (2005). Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J Biol Chem 280: 6780–6791. [DOI] [PubMed] [Google Scholar]
- Ghosh, P, Dahms, NM and Kornfeld, S (2003). Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4: 202–212. [DOI] [PubMed] [Google Scholar]
- Koeberl, DD, Luo, X, Sun, B, McVie-Wylie, A, Dai, J, Li, S et al. (2011). Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol Genet Metab 103: 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah, BL, Madden, L, Li, S, Nance, S, Bird, A, Bursac, N et al. (2014). Adjunctive β2-agonist treatment reduces glycogen independently of receptor-mediated acid α-glucosidase uptake in the limb muscles of mice with Pompe disease. FASEB J 28: 2272–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberl, DD, Li, S, Dai, J, Thurberg, BL, Bali, D and Kishnani, PS (2012). β2 Agonists enhance the efficacy of simultaneous enzyme replacement therapy in murine Pompe disease. Mol Genet Metab 105: 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberl, DD, Kishnani, PS and Chen, YT (2007). Glycogen storage disease types I and II: treatment updates. J Inherit Metab Dis 30: 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilnicka, E and Nielsen, M (1986). Repeated treatment with clenbuterol produces desensitization of rat brain beta- and alpha 2-adrenoceptors without changes of alpha 1-adrenoceptors. Eur J Pharmacol 121: 107–111. [DOI] [PubMed] [Google Scholar]
- Cardone, M, Porto, C, Tarallo, A, Vicinanza, M, Rossi, B, Polishchuk, E et al. (2008). Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 1: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah, CS, Soustek, MS, Todd, AG, McCall, A, Smith, BK, Corti, M et al. (2013). Adeno-associated virus-mediated gene therapy for metabolic myopathy. Hum Gene Ther 24: 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne, BJ, Falk, DJ, Clément, N and Mah, CS (2012). Gene therapy approaches for lysosomal storage disease: next-generation treatment. Hum Gene Ther 23: 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagemaker, G (2014). Lentiviral hematopoietic stem cell gene therapy in inherited metabolic disorders. Hum Gene Ther 25: 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah, C, Cresawn, KO, Fraites, TJ Jr, Pacak, CA, Lewis, MA, Zolotukhin, I et al. (2005). Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther 12: 1405–1409. [DOI] [PubMed] [Google Scholar]
- Mah, CS, Falk, DJ, Germain, SA, Kelley, JS, Lewis, MA, Cloutier, DA et al. (2010). Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol Ther 18: 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker, M, Fraites, TJ Jr, Porvasnik, SL, Lewis, MA, Zolotukhin, I, Cloutier, DA et al. (2004). Rescue of enzyme deficiency in embryonic diaphragm in a mouse model of metabolic myopathy: Pompe disease. Development 131: 3007–3019. [DOI] [PubMed] [Google Scholar]
- Elmallah, MK, Falk, DJ, Nayak, S, Federico, RA, Sandhu, MS, Poirier, A et al. (2014). Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in pompe mice. Mol Ther 22: 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraites, TJ Jr, Schleissing, MR, Shanely, RA, Walter, GA, Cloutier, DA, Zolotukhin, I et al. (2002). Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol Ther 5: 571–578. [DOI] [PubMed] [Google Scholar]
- Qiu, K, Falk, DJ, Reier, PJ, Byrne, BJ and Fuller, DD (2012). Spinal delivery of AAV vector restores enzyme activity and increases ventilation in Pompe mice. Mol Ther 20: 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresawn, KO, Fraites, TJ, Wasserfall, C, Atkinson, M, Lewis, M, Porvasnik, S et al. (2005). Impact of humoral immune response on distribution and efficacy of recombinant adeno-associated virus-derived acid alpha-glucosidase in a model of glycogen storage disease type II. Hum Gene Ther 16: 68–80. [DOI] [PubMed] [Google Scholar]
- Ziegler, RJ, Bercury, SD, Fidler, J, Zhao, MA, Foley, J, Taksir, TV et al. (2008). Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum Gene Ther 19: 609–621. [DOI] [PubMed] [Google Scholar]
- Sun, B, Zhang, H, Franco, LM, Young, SP, Schneider, A, Bird, A et al. (2005). Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Ther 11: 57–65. [DOI] [PubMed] [Google Scholar]
- Wang, G, Young, SP, Bali, D, Hutt, J, Li, S, Benson, J et al. (2014). Assessment of toxicity and biodistribution of recombinant AAV8 vector-mediated immunomodulatory gene therapy in mice with Pompe disease. Mol Ther Methods Clin Dev 1: 14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, AG, McElroy, JA, Grange, RW, Fuller, DD, Walter, GA, Byrne, BJ et al. (2015). Correcting neuromuscular deficits with gene therapy in Pompe disease. Ann Neurol 78: 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk, DJ, Mah, CS, Soustek, MS, Lee, KZ, Elmallah, MK, Cloutier, DA et al. (2013). Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol Ther 21: 1661–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElMallah, MK, Falk, DJ, Lane, MA, Conlon, TJ, Lee, KZ, Shafi, NI et al. (2012). Retrograde gene delivery to hypoglossal motoneurons using adeno-associated virus serotype 9. Hum Gene Ther Methods 23: 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk, DJ, Soustek, MS, Todd, AG, Mah, CS, Cloutier, DA, Kelley, JS et al. (2015). Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol Ther Methods Clin Dev 2: 15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster, DJ, Dykstra, JA, Riedl, MS, Kitto, KF, Belur, LR, McIvor, RS et al. (2014). Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front Neuroanat 8: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, BK, Collins, SW, Conlon, TJ, Mah, CS, Lawson, LA, Martin, AD et al. (2013). Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther 24: 630–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne, PI, Collins, S, Mah, CC, Smith, B, Conlon, T, Martin, SD et al. (2014). Phase I/II trial of diaphragm delivery of recombinant adeno-associated virus acid alpha-glucosidase (rAAaV1-CMV-GAA) gene vector in patients with Pompe disease. Hum Gene Ther Clin Dev 25: 134–163. [DOI] [PubMed] [Google Scholar]
- Conlon, TJ, Erger, K, Porvasnik, S, Cossette, T, Roberts, C, Combee, L et al. (2013). Preclinical toxicology and biodistribution studies of recombinant adeno-associated virus 1 human acid α-glucosidase. Hum Gene Ther Clin Dev 24: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard, E, Douillard-Guilloux, G, Batista, L and Caillaud, C (2008). Correction of glycogenosis type 2 by muscle-specific lentiviral vector. In Vitro Cell Dev Biol Anim 44: 397–406. [DOI] [PubMed] [Google Scholar]
- Sato, Y, Kobayashi, H, Higuchi, T, Shimada, Y, Era, T, Kimura, S et al. (2015). Disease modeling and lentiviral gene transfer in patient-specific induced pluripotent stem cells from late-onset Pompe disease patient. Mol Ther Methods Clin Dev 2: 15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyosen, SO, Iizuka, S, Kobayashi, H, Kimura, T, Fukuda, T, Shen, J et al. (2010). Neonatal gene transfer using lentiviral vector for murine Pompe disease: long-term expression and glycogen reduction. Gene Ther 17: 521–530. [DOI] [PubMed] [Google Scholar]
- van Til, NP, Stok, M, Aerts Kaya, FS, de Waard, MC, Farahbakhshian, E, Visser, TP et al. (2010). Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 115: 5329–5337. [DOI] [PubMed] [Google Scholar]
- Douillard-Guilloux, G, Richard, E, Batista, L and Caillaud, C (2009). Partial phenotypic correction and immune tolerance induction to enzyme replacement therapy after hematopoietic stem cell gene transfer of alpha-glucosidase in Pompe disease. J Gene Med 11: 279–287. [DOI] [PubMed] [Google Scholar]
- Zufferey, R, Dull, T, Mandel, RJ, Bukovsky, A, Quiroz, D, Naldini, L et al. (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol 72: 9873–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schambach, A, Zychlinski, D, Ehrnstroem, B and Baum, C (2013). Biosafety features of lentiviral vectors. Hum Gene Ther 24: 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsham, DN, Schuesler, T, von Kalle, C and Pan, D (2006). In vivo gene transfer into adult stem cells in unconditioned mice by in situ delivery of a lentiviral vector. Mol Ther 14: 514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy, C, Amirache, F, Costa, C, Frecha, C, Muller, CP, Kweder, H et al. (2012). Lentiviral vectors displaying modified measles virus gp overcome pre-existing immunity in in vivo-like transduction of human T and B cells. Mol Ther 20: 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espana-Agusti, J, Tuveson, DA, Adams, DJ and Matakidou, A (2015). A minimally invasive, lentiviral based method for the rapid and sustained genetic manipulation of renal tubules. Sci Rep 5: 11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Z, Stewart, ZA, Sinn, PL, Olsen, JC, Hu, J, McCray, PB Jr et al. (2015). Ferret and pig models of cystic fibrosis: prospects and promise for gene therapy. Hum Gene Ther Clin Dev 26: 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesana, D, Sgualdino, J, Rudilosso, L, Merella, S, Naldini, L and Montini, E (2012). Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J Clin Invest 122: 1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátrai, J, Chuah, MK and VandenDriessche, T (2010). Recent advances in lentiviral vector development and applications. Mol Ther 18: 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonaro Sarracino, D, Tarantal, AF, Lee, CC, Martinez, M, Jin, X, Wang, X et al. (2014). Effects of vector backbone and pseudotype on lentiviral vector-mediated gene transfer: studies in infant ADA-deficient mice and rhesus monkeys. Mol Ther 22: 1803–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Biasco, L, Scaramuzza, S, Ferrua, F, Cicalese, MP, Baricordi, C et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341: 1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi, A, Montini, E, Lorioli, L, Cesani, M, Fumagalli, F, Plati, T et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158. [DOI] [PubMed] [Google Scholar]
- Naphade, S, Sharma, J, Gaide Chevronnay, HP, Shook, MA, Yeagy, BA, Rocca, CJ et al. (2015). Brief reports: lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 33: 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, DA (1999). Immune response to enzyme replacement therapy in lysosomal storage disorder patients and animal models. Mol Genet Metab 68: 268–275. [DOI] [PubMed] [Google Scholar]
- Bigger, BW, Saif, M and Linthorst, GE (2015). The role of antibodies in enzyme treatments and therapeutic strategies. Best Pract Res Clin Endocrinol Metab 29: 183–194. [DOI] [PubMed] [Google Scholar]
- van Gelder, CM, Hoogeveen-Westerveld, M, Kroos, MA, Plug, I, van der Ploeg, AT and Reuser, AJ (2015). Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inherit Metab Dis 38: 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani, PS, Goldenberg, PC, DeArmey, SL, Heller, J, Benjamin, D, Young, S et al. (2010). Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 99: 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmatz, P (2015). Enzyme replacement therapies and immunogenicity in lysosomal storage diseases: is there a pattern? Clin Ther 37: 2130–2134. [DOI] [PubMed] [Google Scholar]
- Kroos, M, Pomponio, RJ, van Vliet, L, Palmer, RE, Phipps, M, Van der Helm, R et al. GAA Database Consortium. (2008). Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum Mutat 29: E13–E26. [DOI] [PubMed] [Google Scholar]
- Basner-Tschakarjan, E and Mingozzi, F (2014). Cell-mediated immunity to AAV vectors, evolving concepts and potential solutions. Front Immunol 5: 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak, S, Sivakumar, R, Cao, O, Daniell, H, Byrne, BJ and Herzog, RW (2012). Mapping the T helper cell response to acid α-glucosidase in Pompe mice. Mol Genet Metab 106: 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries, JM, van der Beek, NA, Kroos, MA, Ozkan, L, van Doorn, PA, Richards, SM et al. (2010). High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol Genet Metab 101: 338–345. [DOI] [PubMed] [Google Scholar]
- Zhang, P, Luo, X, Bird, A, Li, S and Koeberl, DD (2012). Deficiency in MyD88 signaling results in decreased antibody responses to an adeno-associated virus vector in murine Pompe disease. Biores Open Access 1: 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronsema, KJ, Bischoff, R, Pijnappel, WW, van der Ploeg, AT and van de Merbel, NC (2015). Absolute quantification of the total and antidrug antibody-bound concentrations of recombinant human α-glucosidase in human plasma using protein G extraction and LC-MS/MS. Anal Chem 87: 4394–4401. [DOI] [PubMed] [Google Scholar]
- Nayak, S, Doerfler, PA, Porvasnik, SL, Cloutier, DD, Khanna, R, Valenzano, KJ et al. (2014). Immune responses and hypercoagulation in ERT for Pompe disease are mutation and rhGAA dose dependent. PLoS One 9: e98336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, TT, Banugaria, SG, Case, LE, Wenninger, S, Schoser, B and Kishnani, PS (2012). The impact of antibodies in late-onset Pompe disease: a case series and literature review. Mol Genet Metab 106: 301–309. [DOI] [PubMed] [Google Scholar]
- Bali, DS, Goldstein, JL, Banugaria, S, Dai, J, Mackey, J, Rehder, C et al. (2012). Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet 160C: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaná, E, Yao, LP, Pariser, AR and Rosenberg, AS (2012). The role of immune tolerance induction in restoration of the efficacy of ERT in Pompe disease. Am J Med Genet C Semin Med Genet 160C: 30–39. [DOI] [PubMed] [Google Scholar]
- Sack, BK, Herzog, RW, Terhorst, C and Markusic, DM (2014). Development of gene transfer for induction of antigen-specific tolerance. Mol Ther Methods Clin Dev 1: 14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBowitz, JH, Grubb, JH, Maga, JA, Schmiel, DH, Vogler, C and Sly, WS (2004). Glycosylation-independent targeting enhances enzyme delivery to lysosomes and decreases storage in mucopolysaccharidosis type VII mice. Proc Natl Acad Sci USA 101: 3083–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maga, JA, Zhou, J, Kambampati, R, Peng, S, Wang, X, Bohnsack, RN et al. (2013). Glycosylation-independent lysosomal targeting of acid α-glucosidase enhances muscle glycogen clearance in pompe mice. J Biol Chem 288: 1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan, SH, Aoyagi-Scharber, M, Le, SQ, Vincelette, J, Ohmi, K, Bullens, S et al. (2014). Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc Natl Acad Sci USA 111: 14870–14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani, PS, Nicolino, M, Voit, T, Rogers, RC, Tsai, AC, Waterson, J et al. (2006). Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr 149: 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne, B, Barohn, R, Barshop, B, Bratkovic, D, Desnuelle, C, Geberhiwot, T, et al. (2013). POM-001 phase 1/2 study of BMN 701, GILT-tagged recombinant human (rh) GAA in late-onset Pompe disease: initial experience in 22 patients. Mol Genet Metab 108: S28. [Google Scholar]
- Parenti, G, Zuppaldi, A, Gabriela Pittis, M, Rosaria Tuzzi, M, Annunziata, I, Meroni, G et al. (2007). Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease. Mol Ther 15: 508–514. [DOI] [PubMed] [Google Scholar]
- Flanagan, JJ, Rossi, B, Tang, K, Wu, X, Mascioli, K, Donaudy, F et al. (2009). The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase. Hum Mutat 30: 1683–1692. [DOI] [PubMed] [Google Scholar]
- Porto, C, Cardone, M, Fontana, F, Rossi, B, Tuzzi, MR, Tarallo, A et al. (2009). The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol Ther 17: 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna, R, Flanagan, JJ, Feng, J, Soska, R, Frascella, M, Pellegrino, LJ et al. (2012). The pharmacological chaperone AT2220 increases recombinant human acid α-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PLoS One 7: e40776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna, R, Powe, AC Jr, Lun, Y, Soska, R, Feng, J, Dhulipala, R et al. (2014). The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid alpha-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease. PLoS One 9: e102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti, G, Fecarotta, S, la Marca, G, Rossi, B, Ascione, S, Donati, MA et al. (2014). A chaperone enhances blood α-glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol Ther 22: 2004–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto, C, Ferrara, MC, Meli, M, Acampora, E, Avolio, V, Rosa, M et al. (2012). Pharmacological enhancement of α-glucosidase by the allosteric chaperone N-acetylcysteine. Mol Ther 20: 2201–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfler, PA, Kelley, JS, Nayak, S, Lawson, LA and Byrne, BJ (2014). Pharmacological chaperones prevent the precipitation of rhGAA by anti-GAA antibodies during enzyme replacement therapy. Mol Genet Metab 111: S38. [Google Scholar]
- Wang, J, Lozier, J, Johnson, G, Kirshner, S, Verthelyi, D, Pariser, A et al. (2008). Neutralizing antibodies to therapeutic enzymes: considerations for testing, prevention and treatment. Nat Biotechnol 26: 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn, NJ, Messinger, YH, Rosenberg, AS and Kishnani, PS (2009). Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med 360: 194–195. [DOI] [PubMed] [Google Scholar]
- Banugaria, SG, Patel, TT, Mackey, J, Das, S, Amalfitano, A, Rosenberg, AS et al. (2012). Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab 105: 677–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly, MS, Martin, RP, Mitra-Kaushik, S, Phillips, L, D’Angona, A, Richards, SM et al. (2014). Transient low-dose methotrexate generates B regulatory cells that mediate antigen-specific tolerance to alglucosidase alfa. J Immunol 193: 3947–3958. [DOI] [PubMed] [Google Scholar]
- Genestier, L, Paillot, R, Fournel, S, Ferraro, C, Miossec, P and Revillard, JP (1998). Immunosuppressive properties of methotrexate: apoptosis and clonal deletion of activated peripheral T cells. J Clin Invest 102: 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markic, J, Polic, B, Kuzmanic-Samija, R, Marusic, E, Stricevic, L, Metlicic, V et al. (2012). Immune modulation therapy in a CRIM-positive and IgG antibody-positive infant with Pompe disease treated with alglucosidase alfa: a case report. JIMD Rep 2: 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, FB, Saulep-Easton, D, Figgett, WA, Fairfax, KA and Mackay, F (2013). The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev 24: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfler, PA, Nayak, S, Herzog, RW, Morel, L and Byrne, BJ (2015). BAFF blockade prevents anti-drug antibody formation in a mouse model of Pompe disease. Clin Immunol 158: 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz, JL, Crowley, JE, Tomayko, MM, Steinel, N, O’Neill, PJ, Quinn, WJ 3rd et al. (2008). BLyS inhibition eliminates primary B cells but leaves natural and acquired humoral immunity intact. Proc Natl Acad Sci USA 105: 15517–15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder, ME, Nayak, S, Collins, SW, Lawson, LA, Kelley, JS, Herzog, RW et al. (2013). B-cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr 163: 847–54.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets, VA and Rathmell, JC (2012). Metabolic pathways in T cell fate and function. Trends Immunol 33: 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limon, JJ and Fruman, DA (2012). Akt and mTOR in B cell activation and differentiation. Front Immunol 3: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghimi, B, Sack, BK, Nayak, S, Markusic, DM, Mah, CS and Herzog, RW (2011). Induction of tolerance to factor VIII by transient co-administration with rapamycin. J Thromb Haemost 9: 1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak, S, Cao, O, Hoffman, BE, Cooper, M, Zhou, S, Atkinson, MA et al. (2009). Prophylactic immune tolerance induced by changing the ratio of antigen-specific effector to regulatory T cells. J Thromb Haemost 7: 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar, D, Biswas, M, Liao, G, Seay, HR, Perrin, GQ, Markusic, DM et al. (2014). Ex vivo expanded autologous polyclonal regulatory T cells suppress inhibitor formation in hemophilia. Mol Ther Methods Clin Dev 1: 14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas, M, Sarkar, D, Kumar, SR, Nayak, S, Rogers, GL, Markusic, DM et al. (2015). Synergy between rapamycin and FLT3 ligand enhances plasmacytoid dendritic cell-dependent induction of CD4+CD25+FoxP3+ Treg. Blood 125: 2937–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia, M, Stabilini, A and Roncarolo, MG (2005). Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 105: 4743–4748. [DOI] [PubMed] [Google Scholar]
- Kuhns, MS, Davis, MM and Garcia, KC (2006). Deconstructing the form and function of the TCR/CD3 complex. Immunity 24: 133–139. [DOI] [PubMed] [Google Scholar]
- Ohashi, T, Iizuka, S, Shimada, Y, Higuchi, T, Eto, Y, Ida, H et al. (2012). Administration of anti-CD3 antibodies modulates the immune response to an infusion of α-glucosidase in mice. Mol Ther 20: 1924–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, B, Banugaria, SG, Prater, SN, Patel, TT, Fredrickson, K, Ringler, DJ, et al. (2014). Non-depleting anti-CD4 monoclonal antibody induces immune tolerance to ERT in a murine model of Pompe disease. Mol Genet Metab Reports 1: 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banugaria, SG, Prater, SN, McGann, JK, Feldman, JD, Tannenbaum, JA, Bailey, C et al. (2013). Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: lessons learned from Pompe disease. Genet Med 15: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro-Shelef, M and Calame, K (2005). Regulation of plasma-cell development. Nat Rev Immunol 5: 230–242. [DOI] [PubMed] [Google Scholar]
- Lin, W, Seshasayee, D, Lee, WP, Caplazi, P, McVay, S, Suto, E et al. (2015). Dual B cell immunotherapy is superior to individual anti-CD20 depletion or BAFF blockade in murine models of spontaneous or accelerated lupus. Arthritis Rheumatol 67: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markusic, DM, Hoffman, BE, Perrin, GQ, Nayak, S, Wang, X, LoDuca, PA et al. (2013). Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol Med 5: 1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, SO, Li, S, Brooks, ED, Masat, E, Leborgne, C, Banugaria, S et al. (2015). Enhanced efficacy from gene therapy in Pompe disease using coreceptor blockade. Hum Gene Ther 26: 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti, M, Elder, M, Falk, D, Lawson, L, Smith, B, Nayak, S et al. (2014). B-cell depletion is protective against anti-AAV capsid immune response: a human subject case study. Mol Ther Methods Clin Dev 1: 14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti, M, Cleaver, B, Clément, N, Conlon, TJ, Faris, KJ, Wang, G et al. (2015). Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in Pompe disease: preclinical to clinical planning. Hum Gene Ther Clin Dev 26: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, SG, Cideciyan, AV, Roman, AJ, Sumaroka, A, Schwartz, SB, Heon, E et al. (2015). Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med 372: 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, A, Oberle, N and Krammer, PH (2012). Molecular mechanisms of treg-mediated T cell suppression. Front Immunol 3: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomares, O, Martín-Fontecha, M, Lauener, R, Traidl-Hoffmann, C, Cavkaytar, O, Akdis, M et al. (2014). Regulatory T cells and immune regulation of allergic diseases: roles of IL-10 and TGF-β. Genes Immun 15: 511–520. [DOI] [PubMed] [Google Scholar]
- Liao, G, O’Keeffe, MS, Wang, G, van Driel, B, de Waal Malefyt, R, Reinecker, HC et al. (2014). Glucocorticoid-induced TNF receptor family-related protein ligand is requisite for optimal functioning of regulatory CD4(+) T cells. Front Immunol 5: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, O, Dobrzynski, E, Wang, L, Nayak, S, Mingle, B, Terhorst, C et al. (2007). Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood 110: 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak, S, Sarkar, D, Perrin, GQ, Moghimi, B, Hoffman, BE, Zhou, S et al. (2011). Prevention and reversal of antibody responses against factor IX in gene therapy for hemophilia B. Front Microbiol 2: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Sherman, A, Liao, G, Leong, KW, Daniell, H, Terhorst, C et al. (2013). Mechanism of oral tolerance induction to therapeutic proteins. Adv Drug Deliv Rev 65: 759–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi, T, Iizuka, S, Shimada, Y, Eto, Y, Ida, H, Hachimura, S et al. (2011). Oral administration of recombinant human acid α-glucosidase reduces specific antibody formation against enzyme in mouse. Mol Genet Metab 103: 98–100. [DOI] [PubMed] [Google Scholar]
- Su, J, Sherman, A, Doerfler, PA, Byrne, BJ, Herzog, RW and Daniell, H (2015). Oral delivery of acid alpha glucosidase epitopes expressed in plant chloroplasts suppresses antibody formation in treatment of Pompe mice. Plant Biotechnol J 13: 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Su, J, Sherman, A, Rogers, GL, Liao, G, Hoffman, BE et al. (2015). Plant-based oral tolerance to hemophilia therapy employs a complex immune regulatory response including LAP+CD4+ T cells. Blood 125: 2418–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, KC and Daniell, H (2015). Low-cost oral delivery of protein drugs bioencapsulated in plant cells. Plant Biotechnol J 13: 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremel, M, Guerin, N, Campello, G, Barthe, Q, Berlier, W, Horand, F et al. (2015). Innovative approach in Pompe disease therapy: induction of immune tolerance by antigen-encapsulated red blood cells. Int J Pharm 491: 69–77. [DOI] [PubMed] [Google Scholar]
- Cremel, M, Guérin, N, Horand, F, Banz, A and Godfrin, Y (2013). Red blood cells as innovative antigen carrier to induce specific immune tolerance. Int J Pharm 443: 39–49. [DOI] [PubMed] [Google Scholar]
- St-Pierre, C, Trofimov, A, Brochu, S, Lemieux, S and Perreault, C (2015). Differential features of AIRE-induced and AIRE-independent promiscuous gene expression in thymic epithelial cells. J Immunol 195: 498–506. [DOI] [PubMed] [Google Scholar]
- Kisand, K, Peterson, P and Laan, M (2014). Lymphopenia-induced proliferation in aire-deficient mice helps to explain their autoimmunity and differences from human patients. Front Immunol 5: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuprin, A, Avin, A, Goldfarb, Y, Herzig, Y, Levi, B, Jacob, A et al. (2015). The deacetylase Sirt1 is an essential regulator of Aire-mediated induction of central immunological tolerance. Nat Immunol 16: 737–745. [DOI] [PubMed] [Google Scholar]
- Wilkinson, FL, Sergijenko, A, Langford-Smith, KJ, Malinowska, M, Wynn, RF and Bigger, BW (2013). Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol Ther 21: 868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantal, AF, Giannoni, F, Lee, CC, Wherley, J, Sumiyoshi, T, Martinez, M et al. (2012). Nonmyeloablative conditioning regimen to increase engraftment of gene-modified hematopoietic stem cells in young rhesus monkeys. Mol Ther 20: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Payen, E, Negre, O, Wang, G, Hehir, K, Fusil, F et al. (2010). Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 467: 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, Q, Moreland, RJ, Gao, L, Taylor, KM, Meyers, E, Cheng, SH et al. (2010). Induction of immune tolerance to a therapeutic protein by intrathymic gene delivery. Mol Ther 18: 2146–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispe, IN (2009). The liver as a lymphoid organ. Annu Rev Immunol 27: 147–163. [DOI] [PubMed] [Google Scholar]
- Crispe, IN (2003). Hepatic T cells and liver tolerance. Nat Rev Immunol 3: 51–62. [DOI] [PubMed] [Google Scholar]
- Breous, E, Somanathan, S, Vandenberghe, LH and Wilson, JM (2009). Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 50: 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghardt, S, Erhardt, A, Claass, B, Huber, S, Adler, G, Jacobs, T et al. (2013). Hepatocytes contribute to immune regulation in the liver by activation of the Notch signaling pathway in T cells. J Immunol 191: 5574–5582. [DOI] [PubMed] [Google Scholar]
- Bailis, W, Yashiro-Ohtani, Y, Fang, TC, Hatton, RD, Weaver, CT, Artis, D et al. (2013). Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity 39: 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl, JC, Drees, C, Heger, K, Heink, S, Fischer, JC, Nedjic, J et al. (2014). Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity 41: 722–736. [DOI] [PubMed] [Google Scholar]
- Franco, LM, Sun, B, Yang, X, Bird, A, Zhang, H, Schneider, A et al. (2005). Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther 12: 876–884. [DOI] [PubMed] [Google Scholar]
- Sun, B, Bird, A, Young, SP, Kishnani, PS, Chen, YT and Koeberl, DD (2007). Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet 81: 1042–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, B, Kulis, MD, Young, SP, Hobeika, AC, Li, S, Bird, A et al. (2010). Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol Ther 18: 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P, Sun, B, Osada, T, Rodriguiz, R, Yang, XY, Luo, X et al. (2012). Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther 23: 460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandendriessche, T, Thorrez, L, Acosta-Sanchez, A, Petrus, I, Wang, L, Ma, L et al. (2007). Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost 5: 16–24. [DOI] [PubMed] [Google Scholar]
- Zincarelli, C, Soltys, S, Rengo, G and Rabinowitz, JE (2008). Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16: 1073–1080. [DOI] [PubMed] [Google Scholar]
- Marsic, D, Govindasamy, L, Currlin, S, Markusic, DM, Tseng, YS, Herzog, RW et al. (2014). Vector design Tour de Force: integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol Ther 22: 1900–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, S, Horowitz, ED, Troupes, AN, Brown, SM, Pulicherla, N, Samulski, RJ et al. (2013). Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J Biol Chem 288: 28814–28823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C, Diprimio, N, Bowles, DE, Hirsch, ML, Monahan, PE, Asokan, A et al. (2012). Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol 86: 7752–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson, KR, Focosi, D, Major, EO, Petrini, M, Richey, EA, West, DP et al. (2009). Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol 10: 816–824. [DOI] [PubMed] [Google Scholar]
- Du Toit, G, Roberts, G, Sayre, PH, Bahnson, HT, Radulovic, S, Santos, AF et al. LEAP Study Team. (2015). Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med 372: 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, A, Su, J, Lin, S, Wang, X, Herzog, RW and Daniell, H (2014). Suppression of inhibitor formation against FVIII in a murine model of hemophilia A by oral delivery of antigens bioencapsulated in plant cells. Blood 124: 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, J, Zhu, L, Sherman, A, Wang, X, Lin, S, Kamesh, A et al. (2015). Low cost industrial production of coagulation factor IX bioencapsulated in lettuce cells for oral tolerance induction in hemophilia B. Biomaterials 70: 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]