

Abstract
Interleukin 6 (IL 6) and interleukin 1 (IL-1) regulate the expression of actue phase plasma proteins in rat and human hepatoma cells. Phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA), partially mimics the stimulatory effect of IL-6 but reduces that effect of IL-1. TPA and IL-6 act synergistically. These regulatory properties of TPA are also manifested in HepG2 cells transiently transfected with an indicator gene construct carrying the IL-1/IL-6 regulatory enhancer element of the rat α1-acid glycoprotein gene. IL-6 and IL-1 act independently of TPA-inducible kinase C, and of changes in intracellular Ca2+ concentrations. However, prolonged pretreatment of HepG2 cells with TPA results in a drastically reduced cytokine response that is proportional to the loss of cell surface binding activity for the cytokine. These data suggest that hormones activating protein kinase C probably play a contributing role in stimulating the expression of acute phase plasma protein genes but they may be crucial in controlling the responsiveness of liver cells to inflammatory cytokines during subsequent stages of the hepatic acute phase reaction.
Within hours following a systemic injury, the mammalian liver responds with a coordinate increase in the production of a subset of plasma proteins, namely the acute phase reactants (1–3). Activated monocytes and macrophages have been recognized as a major source of cytokines mediating the hepatic acute phase reaction (4–10). The principle liver-regulating monokines have been identified as interleukin 1β (IL-1β)1 (11), tumor necrosis factor (12, 13), and interleukin 6 (IL-6; identical to monocytic hepatocyte-stimulating factor (8, 14), B-cell stimulatory factor-2 (15), interferon-β2 (16), and 26-kDa protein (17)). Each factor alone is capable of modulating the expression of a subset of acute phase reactants. IL-1 and tumor necrosis factor strongly stimulate the synthesis of α1-acid glycoprotein (AGP), complement component 3, and haptoglobin but inhibit the synthesis of fibrinogen in rat hepatocytes, rat H-35 hepatoma cells, and human hepatoma (HepG2 and Hep3B) cells (14, 18–20). IL-6, although effective on most acute phase protein genes, preferentially stimulates the synthesis of fibrinogen, thiostatin (α1-cysteine protease inhibitor), α2-macroglobulin, and hemopexin in rat liver cells (14, 21, 22), and fibrinogen in human hepatoma cells (14, 20). Glucocorticoids enhance the stimulatory effect of the monokines on the synthesis of most, but not all, acute phase proteins (14, 19–21). A strong synergistic action among IL-1, IL-6, and glucocorticoids in the regulation of AGP and haptoglobin has been reported in both rat and human hepatoma cells (14, 20, 23).
Recently, Evans et al. (24) observed that treatment of FAZA rat hepatoma and HepG2 cells with 12-O-tetradecanoylphorbol-13-acetate (TPA) could mimic the stimulation of fibrinogen synthesis by hepatocyte-stimulating factor of activated monocytes (25). This observation was interpreted by these investigators as indicating that hepatocyte-stimulating factor (IL-6) transduces its signal to the fibrinogen genes by a mechanism involving protein kinase C. Considering that a homogenous preparation of hepatocyte-stimulating factor was not used, the assignment of kinase C as intermediary in IL-6 action appears premature. In this paper, we demonstrate that the action of human recombinant IL-6 is independent of kinase C activity and of intracellular Ca2+ concentration changes, and that its action is only partially reproduced by TPA in rat and human hepatoma cells. TPA, however, can significantly modulate the cell response to IL-6 and IL-1, in part, by reducing available cell surface cytokine binding sites.
MATERIALS AND METHODS
Cells
A newly selected subclone (T-7-18) of Reuber H-35 cells (26), was cultured in Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated fetal calf serum (19). HepG2 cells (a gift of Dr. B. Knowles, Wistar Institute) was cultured in minimal essential medium (MEM) with 10% heat-inactivated fetal calf serum.
Factors
Homogenous preparation of COS cell-derived human recombinant IL-6 (1 × 106 units/mg) (27) was provided by Dr. G. Wong, Genetics Institute, Cambridge, MA, and human recombinant IL-1α (3 × 108 units/mg) (28) by Dr. D. Urdal, Immunex Corp., Seattle, WA. Stock solutions were prepared in MEM at a concentration of 5 units/μl. TPA and ionomycin were obtained from Sigma and dissolved in dimethyl sulfoxide at concentrations of 5 and 3.8 μg/μl, respectively.
Treatment
Confluent cell monolayers in 10-cm dishes were washed twice with serum-free MEM. The cells were incubated with 5 ml of MEM containing optimal concentrations of the indicated factors (20).2 After 8 h, RNA was extracted from the cells as described (29).
RNA Analysis
Aliquots of 15 μg of cellular RNA were separated on 1.5% agarose gels containing formaldehyde (30), transferred to nitrocellulose (31) and hybridized with 32P-labeled cDNA inserts encoding rat AGP, thiostatin, α- and γ-fibrinogen (32), human α1-antichymotrypsin (33), haptoglobin (34), and AGP (35) (the latter two generously provided by Dr. R. Cortese, Heidelberg, Federal Republic of Germany).
Gene Constructs
Three 142-base pair distal regulatory elements (DRE) of the rat AGP gene (responsive to IL-1, IL-6 and keratinocytic hepatocyte-stimulating factors) were inserted in inverted orientation and arranged in tandem into the NdeI site (at position −120 relative to the transcription start site) of plasmid pAGP(140)-CAT (23) yielding plasmid pAGP(3xDRE)-140-CAT. (pAGP(140)-CAT contains the glucocorticoid-responsive enhancer element (GRE) and promoter of the rat AGP gene, from position −120 to +21, linked to the chloramphenicol acetyltransferase gene (CAT gene) in pSVOCAT (36)). To normalize for transfection efficiency, the plasmid pIE-MUP was used (23). (pIE-MUP contains the immediate-early promoter/enhancer region of human cytomegalovirus linked to the entire coding region of the mouse major urinary protein (MUP) gene 25D4). Expression of MUP is not significantly affected by any of the treatments used in here (data not presented).
Transfection
Calcium phosphate precipitates of a mixture of plasmid DNA (18 μg of pAGP(3xDRE)-140-CAT and 1–2 μg of pIE-MUP/ml) were transfected into HepG2 cells in 6-well cluster plates (4 × 105 cells/10 cm2) (37). The cells were treated with 20% glycerol (38) and allowed to recover for 24 h, and then the medium was replaced by 1 ml of serum-free MEM. After 16 h of additional incubation, the medium was removed and 1 ml of fresh MEM containing the indicated factors was added. Following an additional 8 h of incubation, the culture medium was combined with the previously removed medium, dialyzed against 25 mM (NH4)HCO3, and lyophilized. The amount of MUP in the total medium fraction was determined by rocket immunoelectrophoresis. The cells were extracted in 100 μl of 1 M Tris-HCl, pH 7.8. The cell extracts were heat-treated (60 °C for 5 min) and 0.3–30 μl was assayed for chloramphenicol acetyltransferase activity. These amounts were used in order to stay within the linear range of the assay system (39). Specific chloramphenicol acetyltransferase activity was expressed in percent conversion of substrate to product per hour and nanogram of MUP.
Measurements of Cell Surface Receptors
Biologically active 125I-IL-1α (chloramine-T treated; 1 × 103 cpm/fmol) (28) was generously provided by Dr. S. Dower, Immunex Corp., Seattle, WA. IL-6 (0.8 μg) was labeled with 0.5 mCi of Bolton-Hunter reagent (Amersham Corp.) (40) and separated on Sephadex G-25 columns as described (41). Molecular integrity of the 125I-IL-6 was assessed by electrophoresis on 11% sodium dodecyl sulfate polyacrylamide gel. Recovery of protein and biological activity was quantitated by calibrating the system with trace IL-6 standard (42).
Confluent HepG2 cell monolayers in 6-well cluster plates (10 cm2 culture area; 1.2–1.5 × 106 cells) were used for IL-6 binding assay and HepG2 cells in 24-well cluster plates (2 cm2; 0.4 × 106 cells) were used for IL-1 assay. The cells were incubated in 500 μl (or 200 μl) of binding medium (MEM, 1% bovine serum albumin, 20 mM Hepes, pH 7.2) with increasing concentrations of 125I-IL-1 and 125I-IL-6 for 4 h at 11 °C. Under these conditions, binding equilibrium with the cell surface receptors was achieved. After repeated washing of the cells with binding medium, the cell-associated radioactivity and cell numbers in parallel wells were determined.
RESULTS
Response of H-35 and HepG2 Cells to TPA
Treatment of H-35 cells with TPA for 8 h led to an increase in mRNA coding for acute phase proteins that ranged from barely detectable (thiostatin) to 2–3-fold (α- and γ-fibrinogen and AGP) (Fig. 1 and Table I). At optimal concentrations, the stimulatory TPA effect was only a fraction of that of IL-6 which caused a 10–50-fold increase. The first 8 h following addition of the tested factors was found to be best for comparison of TPA and IL-6 action. The response of the cells to TPA peaked between 6 and 12 h but returned to basal level after 24 h (data not shown), whereas that of IL-6 was detectable at 8 h, although its maximal level was achieved after 24 to 48 h, depending upon the gene examined (19, 21). The effect of TPA was greatest at concentrations of 0.1–0.5 μM. At concentrations above 1 μM, a cytostatic reaction, resulting in a reduction in the basal expression of many plasma protein mRNAs was observed. Co-treatment of TPA and IL-6 produced a small additive cellular response (Table I).
Fig. 1. Effect of TPA and IL-6 on H-35.
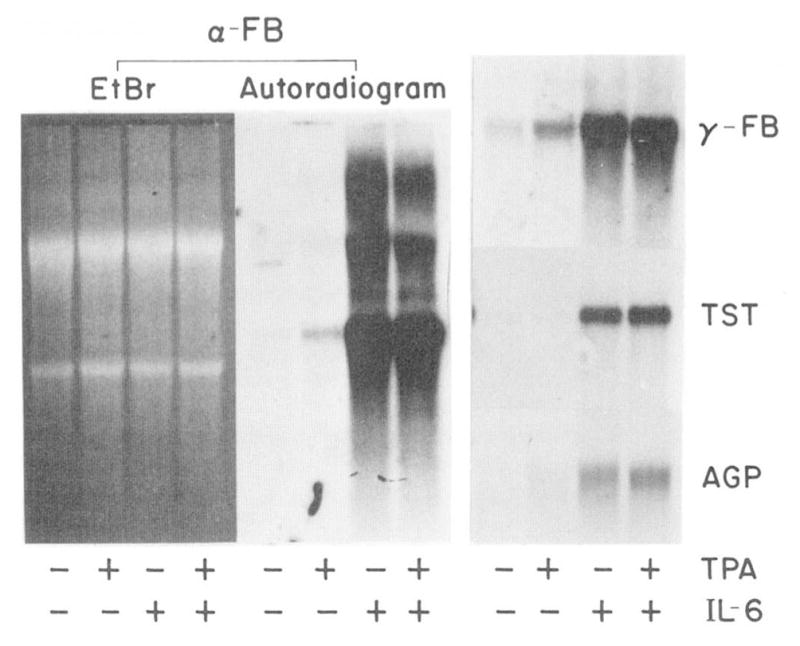
H-35 cells were treated for 8 h with serum-free MEM containing 1 μM dexamethasone alone or with 0.1 μM TPA and/or 250 units/ml IL-6. Aliquots of cellular RNA (15 μg) were analyzed by Northern blot hybridization for the levels of α-fibrinogen (α-FB), γ-fibrinogen (γ-FB), thiostatin (TST), and AGP. The autoradiograms were exposed for 24 h. The ethidium bromide (EtBr)-stained RNA pattern of the α-fibrinogen analysis is pictured to demonstrate equal loading of RNA.
Table I. Relative effect of TPA and IL-6 on expression of acute phase plasma protein mRNAs.
The changes in mRNAs were determined by densitometric quantitation of the hybridization on Northern blot analyses such as shown in Fig. 1. The signal measured for control cells was defined as 1.0.
| Relative mRNA concentration
|
||||
|---|---|---|---|---|
| No additives | TPA | IL-6 | TPA + IL6 | |
| H-35 cells | ||||
| α-Fibrinogen | 1.0 | 2.5 | 55.6 | 58.9 |
| γ-Fibrinogen | 1.0 | 2.1 | 12.3 | 15.1 |
| Thiostatin | 1.0 | 1.2 | 22.2 | 25.3 |
| AGP | 1.0 | 2.4 | 10.1 | 12.4 |
| HepG2 cells | ||||
| Haptoglobin | 1.0 | 2.1 | 4.0 | 11.4 |
| AGP | 1.0 | 2.7 | 5.3 | 18.3 |
| α1-Antichymotrypsin | 1.0 | 3.3 | 9.2 | 16.1 |
The regulatory potential of TPA was not limited to H-35 cells; it was also observed in HepG2 cells (Fig. 2, Table I). In these cells, TPA elicited a low, but significant increase in the level of haptoglobin, AGP, and α1-antichymotrypsin mRNA. Corresponding changes in the synthesis rates of these proteins was observed, including a 2-fold increase in fibrinogen production (data not shown). Although HepG2 cells tolerated TPA concentration up to 2 μM without showing cytotoxic effects, the stimulatory action of TPA was not appreciably elevated above that shown in Fig. 2 by using 0.5 or 1.5 μM TPA (data not shown, see also Table II, below). The response of HepG2 cells to optimal concentration of IL-6 (20) was severalfold that of TPA. However, a strong synergistic action of TPA and IL-6 was noted.
Fig. 2. Effect of TPA and IL-6 on HepG2 cells.
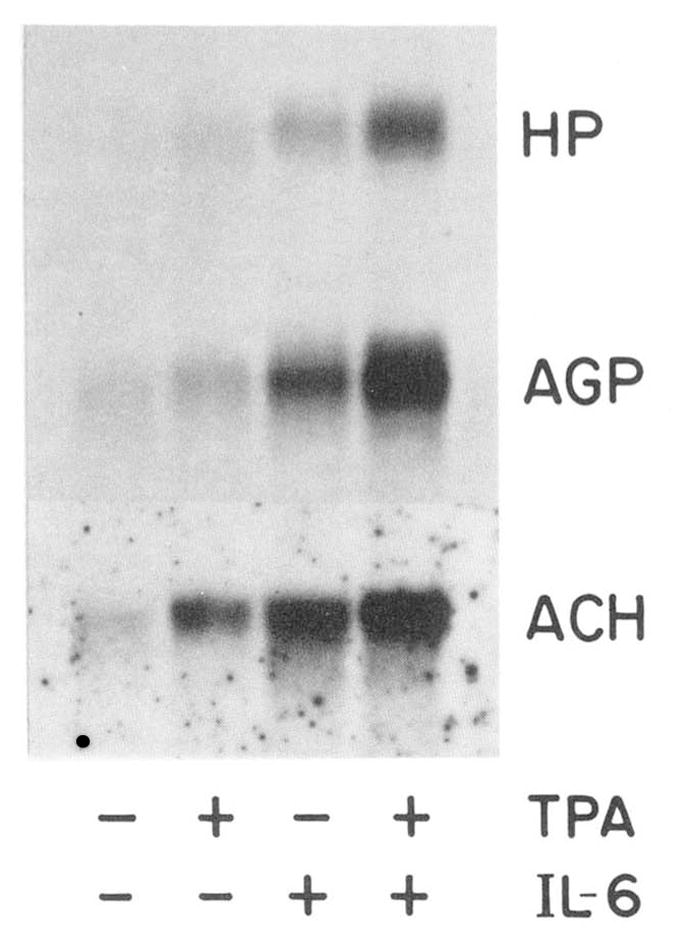
HepG2 cells were treated for 8 h with MEM containing 0.1 μM dexamethasone alone or with 0.15 μM TPA and/or 100 units/ml IL-6. Cellular RNA (15 μg) were analyzed by Northern blot hybridization for haptoglobin (HP), AGP, and α1-antichymotrypsin (ACH) mRNA. Autoradiogram of the mature RNA bands are shown after 3-day (HP, AGP) and 1-day (ACH) exposures.
Table II. Regulatory effect of TPA and ionomycin on the expression of rat AGP-CAT construct in transiently transfected HepG2 cells.
HepG2 cells in 6-well cluster plates were transformed with a mixture of plasmid DNAs (18 μg of pAGP(3×DRE)-140-CAT or pAGP(140)-CAT and 2 μg of pIE-MUP/ml) and subsequently cultured as described in the legend to Fig. 3. The treatments with the indicated components were carried out for 8 h in serum-free MEM containing 0.1 μM dexamethasone. The following concentrations were used where not indicated: ionomycin, 0.35 μM; IL-6, 100 units/ml; IL-1, 250 units/ml. The chloramphenicol acetyltransferase activities in the cell extract were normalized to the amount of MUP secreted into the medium of the same cells during the final 24 h culture period. Mean values and standard deviations of three separate but identically treated wells are shown. Values for cells transfected with pAGP(140)-CAT represent the average of duplicate wells.
| Treatment | Specific chloramphenicol acetyltransferase activity |
|---|---|
| % conversion/h × ng MUP | |
| pAGP(3×DRE)-140-CAT | |
| No addition | 0.10 ± 0.02 |
| TPA | |
| 1.50 μM | 0.44 ± 0.02 |
| 0.50 μM | 0.40 ± 0.01 |
| 0.15 μM | 0.32 ± 0.03 |
| 0.05 μM | 0.21 ± 0.02 |
| Ionomycin | 0.13 ± 0.05 |
| TPA (0.5 μM) + ionomycin | 0.37 ± 0.02 |
| IL-6 | 1.37 ± 0.12 |
| IL-6 + TPA (0.5 μM) | 4.08 ± 0.25 |
| IL-6 + TPA (0.5 μM) + ionomycin | 4.46 ± 0.84 |
| IL-1α | 2.88 ± 0.35 |
| IL-1α + TPA (0.5 μM) | 1.15 ± 0.10 |
| IL-1α + TPA (0.5 μM) + ionomycin | 1.26 ± 0.05 |
| IL-1α + IL-6 | 9.93 ± 1.08 |
| IL-1α + IL-6 + TPA (0.5 μM) | 5.97 ± 0.40 |
| IL-1α + IL-6 + TPA (0.5 μM) + ionomycin | 5.63 ± 0.65 |
| pAGP(140)-CAT | |
| No addition | 0.05 |
| TPA (0.5 μM) | 0.02 |
| IL-6 | 0.09 |
| IL-6 + TPA | 0.07 |
| IL-1α | 0.04 |
| IL-1α + IL-6 | 0.10 |
If IL-6 action is indeed mediated via activation of the phosphatidylinositol phosphate pathway, the partial reproduction of the IL-6 response by phorbol ester-activated kinase C might be explained by the lack of a concomitant increase in cytoplasmic Ca2+ concentration (43). To test this possibility, we modulated intracellular Ca2+ levels by adding ionomycin at concentrations of 0.05–0.5 μM (ionomycin is cytotoxic at concentrations >1 μM) to the culture medium of either H-35 or HepG2. Ionomycin had no detectable consequence on TPA- and IL-6-regulated mRNA accumulation (data not shown).
TPA Acts Via the IL-1/IL-6 Regulatory Region of the Rat α1-Acid Glycoprotein Gene
To assess whether TPA modulates the expression of the acute phase protein genes by interfering with the IL-6 signal transduction system, we measured the effect of TPA on the activity of pAGP(3×DRE)-140-CAT transiently transfected into HepG2 cells. Plasmid pAGP(3×DRE)-140-CAT contains three copies of the 142-base pair IL-1/Il-6 regulatory region of rat AGP gene (i.e. DRE) located 5′ to the GRE/promoter of the AGP gene that controls the transcription activity of the CAT gene (23). We used three DRE regions to enhance responsiveness to the cytokines IL-6 and IL-1. We have verified by testing various rat AGP gene segments (from −5300 to +8000) that the only region responsive to transcriptional enhancement by TPA was confined to the DRE (data not presented). The DRE region in the rat AGP gene does not, however, contain any sequences resembling published TPA-inducible elements in other genes (44–46). The specificity of the TPA response through the DRE-containing construct is indicated by the data in Table II.
The chimeric AGP-CAT gene construct was regulated in HepG2 cells (Fig. 3, Table II) in the same manner as the endogenous AGP gene (Fig. 2, Table I, and Ref. 28). Within 8 h of treatment, TPA enhanced the specific chloramphenicol acetyltransferase activity 4-fold, whereas IL-6 enhanced it 14-fold. The two factors combined yielded a 40-fold stimulation. Although dexamethasone enhances the expression of the plasmid through the GRE element (36), the relative magnitude of TPA- and cytokine-specific stimulation was not significantly altered by dexamethasone (Fig. 3).
Fig. 3. Regulated expression of a rat AGP-CAT gene construct in HepG2 cells.

HepG2 cells in 6-well cluster plates were transfected with plasmid DNA mixture (18 μg of pAGP(3xDRE)-140-CAT and 1 μg of pIE-MUP/ml). After a recovery period of 24 h and culture in serum-free MEM for 16 h, the cells were treated for 8 h with MEM containing the indicated factors (0.15 μM TPA, 100 units/ml IL-6, 250 units/ml IL-1α; 0.1 μM dexamethasone (Dex)). The thin layer chromatography pattern of the chloramphenicol acetyltransferase (CAT) activity in 10 μl of cell extract after 20 h autoradiography is reproduced. The rocket immunoelectrophoresis of the MUP secreted by the same cells during the final 24 h period is shown at the top. The specific chloramphenicol acetyltransferase activity was determined by using 0.3–30 μl of the cell extract and the values were related to the amount of MUP produced.
A potentiation in the effect of IL-6 occurred not only with TPA but also with IL-1. The synergistic action with TPA was, however, substantially less than with IL-1 (Table II). Because TPA quenched the IL-1 response rather than acted additively with it, it was apparent that TPA did not mimic IL-1 action Table II). The specific modulating effects of TPA occurred at maximal and submaximal concentrations of IL-6 and IL-1 (data shown).
Addition of ionomycin to the culture medium did not significantly alter any of the described regulatory properties of TPA, IL-6, and IL-1 (Table II). This suggests that changes in intracellular calcium concentrations are not essential for transmitting the cytokine signals to the regulated AGP-CAT construct.
IL-6 and IL-1 Act Independently of TPA-activated Protein Kinase C, but TPA Modulates the Overall Response
To determine whether the cell response to IL-6 involved kinase C, we subjected pAGP(3xDRE)-140-CAT-transfected HepG2 cells to a 12- or 24-h pretreatment with TPA. It has been demonstrated in several systems that prolonged exposure of cells to TPA results in a marked reduction or even loss of inducible kinase C activity (47–49). Even after a 12-h TPA treatment, a subsequent challenge with a higher concentration of TPA was ineffective (Table III). The 12-h TPA pretreatment, however, did not abolish stimulation by IL-6 and IL-1, although a level of expression was reduced 30 and 70%, respectively. In addition, the synergistic enhancement of IL-6 activity or the reduction of IL-1 action by TPA was virtually eliminated by the 12-h pretreatment with TPA.
Table III. Effect of TPA pretreatment on IL-6 and IL-1 response of HepG2 cells.
HepG2 cells were transfected with pAGP(3×DRE)-140-CAT and pIE-MUP as in Table II. After a 24-h recovery period, the culture medium was replaced by serum-free MEM and to one set of cultures (24 h pretreatment) 0.15 μM TPA was added. All media were replaced again by fresh media after 8 h to collect secreted MUP. After an additional 4 h, TPA treatment of the second set of cultures (12 h pretreatment) began. Twelve h later, all media were collected and the cells were washed three times with MEM. Cells in each group of cultures were incubated for 8 h either with 1 ml MEM and 0.1 μM dexamethasone alone or with 0.5 μM TPA, 100 units/ml IL-6, and 250 units/ml IL-1α. Chloramphenicol acetyltransferase activity and MUP production were determined as indicated in Table II. The amounts of MUP secreted (nanograms per well during the final 24-h culture period) were 62 ± 16, 75 ± 14, and 92 ± 15 for the 0, 12, and 24 h pretreatment group, respectively. The specific chloramphenicol acetyltransferase activities represent mean values of duplicate wells.
| Treatment | Specific chloramphenicol acetyltransferase activity
|
||
|---|---|---|---|
| 0 ha | 12 ha | 24 ha | |
| % conversion/h × ng MUP | |||
| No addition | 0.04 | 0.02 | 0.01 |
| TPA | 0.23 | 0.04 | 0.01 |
| IL-6 | 1.13 | 0.76 | 0.32 |
| IL-6 + TPA | 3.33 | 0.82 | 0.34 |
| IL-1 | 2.83 | 0.85 | 0.15 |
| IL-1 + TPA | 1.12 | 0.78 | 0.22 |
Pretreatment with TPA.
When pretreatment of the cells with TPA was extended to 24 h, the response to cytokine treatment was further reduced, mostly notably that of IL-1. The loss of responsiveness after TPA pretreatment was not simply attributable to general reduction of protein synthesis (e.g. chloramphenicol acetyltransferase enzyme), since the expression of the cotransfected control plasmid, pIE-MUP, as determined by the amount of major urinary protein secreted into the medium, was not impaired (Table III). Moreover, TPA did not appear to increase the degradation of mRNA encoding acute phase proteins previously synthesized as a result of IL-6 stimulation (data not shown).
The inhibiting effect of TPA pretreatment on the expression of endogenous acute phase protein genes was determined by Northern blot analysis (data not shown). Although the IL-6 of AGP, haptoglobin, and α-antichymotrypsin mRNA accumulation was essentially uneffected by 24-h TPA pretreatment (similar levels as shown in Fig. 2), the prominent synergistic action of IL-1 and IL-6 on AGP and haptoglobin mRNA (20) was completely abolished. A similar result was also obtained with H-35 cells, in which pretreatment with 0.15 μM TPA for 24 h led to an elimination of the IL-1 specific regulation of AGP, haptoglobin, and complement C3 mRNA (19).
Prolonged TPA Treatment Reduces Cell Surface Receptor Activity
TPA could conceivably lower the response of HepG2 cells to IL-1 and IL-6 by interfering with any of the many steps between cell surface receptor, second messenger pathways, and trans-acting regulatory elements. Because several studies have documented a phorbol ester-induced reduction of cell surface hormone receptor activity (e.g. for epidermal growth factor (50, 51), insulin (52), tumor necrosis factor (53), and acetylcholine (54)), we examined the influence of TPA on the IL-1 and IL-6 binding activity of HepG2 cells.
In these experiments, IL-6 was labeled with Bolton-Hunter reagent to a specific radioactivity of 2200 cpm/fmol. The labeled IL-6 remained structurally intact (Fig. 4A) and retained essentially full biological activity when tested on HepG2 cells for increased production of acute phase proteins (Fig. 4B). The number of IL-6 binding sites on the HepG2 cell surface was determined using the method of Scatchard (55) (Fig. 5). In four independent experiments, a mean ± S.D. of 450 ± 100 high affinity binding sites of IL-6 per HepG2 cell was calculated with an equilibrium dissociation constant of 1.5 × 10−11 M. However, a substantial portion of IL-6 was bound to about 5000 sites per cell with a KD of 5 × 10−10 M.
Fig. 4. Gel electrophoretic pattern and activity of 125I-IL-6.
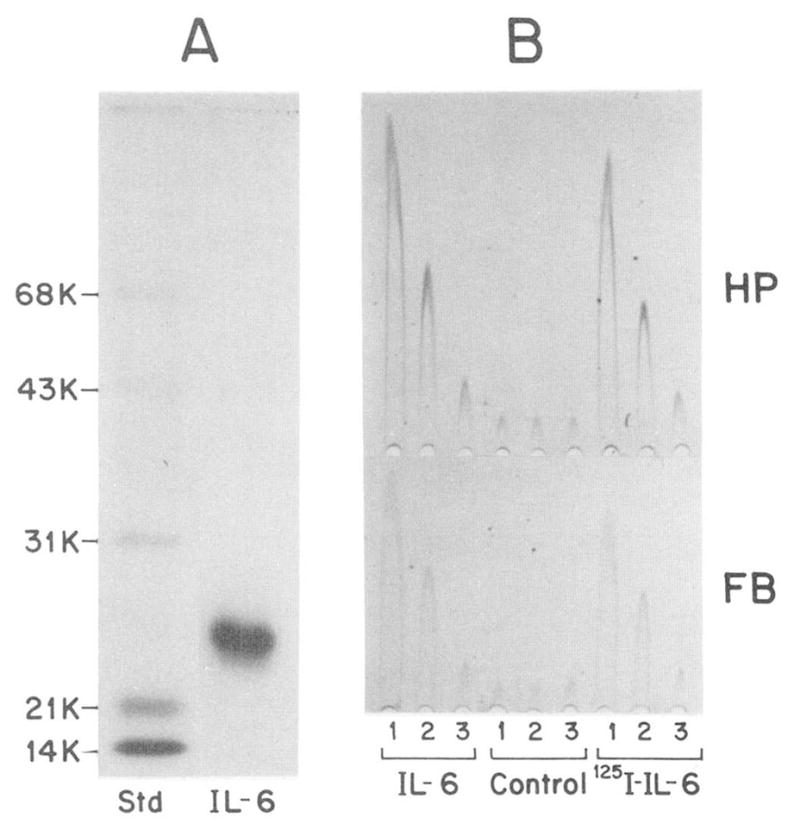
In A, an aliquot of IL-6 labeled with the 125I-Bolton Hunger reagent (65,000 cpm) was separated together with molecular weight standard (Std) on an 11% sodium dodecyl sulfate polyacrylamide gel. The fluorogram shown was exposed for 3 h. (The 125I-IL-6 protein band at Mr 24,000 yielded 59,800 cpm.) In B, medium (MEM, 1% fetal calf serum, 1 μM dexamethasone) containing 10, 1, or 0.1 ng/ml of unlabeled IL-6 or 125I-IL-6 (lanes 1, 2, and 3, respectively), was tested on HepG2 cells. The stimulated production of haptoglobin (HP) and fibrinogen (FB) was measured by rocket Immunoelectrophoresis (20). Control represents HepG2 cells treated with medium containing the buffer used for chromatography of 125I-IL-6 (50 mM sodium phosphate, pH 7.5, 0.25% gelatin; Ref. 41). The amount of buffer added was the same as used in the experimental assays.
Fig. 5. Binding of IL-1 and IL-6 to HepG2 cells.

Confluent monolayers of HepG2 cells were pretreated for 24 h with serum-free MEM alone (○), or with MEM containing either 0.1 μM dexamethasone (△), 0.15 μM TPA (●), or dexamethasone and TPA (▲). The cells were washed once with binding buffer and then incubated for 4 h at 4 °C with increasing concentrations of 125I IL-1 or 125I-IL-6. The binding of label to the 4 cm2 (IL-1) or 10 cm2 (IL-6) monolayer cultures was expressed according to Scatchard (55). The mean values of duplicate cultures are shown.
Titration of HepG2 cells with biologically active 125I-IL-1α yielded a single class binding activity (Fig. 5). Four independent experiments indicated a mean ± S.D. of 13,400 ± 2,000 IL-1 binding sites per HepG2 cell with a KD of 1.9 × 10−10 M.
Treatment of HepG2 cells with 0.15 μM TPA for 24 h resulted in the virtual removal of IL-1 binding activity (Fig. 5). Total IL-6 binding was also reduced, most notably the high affinity binding component. The effect of TPA on cytokine binding activity TPA was not modified by dexamethasone. Dexamethasone treatment alone caused a roughly 2-fold reduction of IL-1 binding sites but did not significantly influence the IL-6 binding activity (Fig. 5).
Taken together, TPA appears to regulate acute phase protein production by two means. It promotes a transient stimulation of acute phase protein gene expression and synergistic enhancement of the IL-6 signals and it causes a reduction in cell surface receptor activity, preferentially that for IL-1.
DISCUSSION
The relative stimulation of acute phase plasma protein expression by TPA in rat and human hepatoma cells (Table I) is comparable to the values reported by Evans et al. (24). Moreover, both studies show that the response to TPA is only a fraction of that attained by IL-6 (or HSF), as indicated in the examples of the acute phase protein mRNAs in H-35 cells (Fig. 1, Table I), α2-macroglobin mRNA in FAZA cells (Fig. 4A in Ref. 24) or haptoglobin mRNA in HepG2 cells (Fig. 2, Table I; Fig. 4B in Ref. 24). The only major discrepancy between our data and that of Evans et al. (24) is that they did not observe a significant HSF-stimulation of fibrinogen mRNA levels above that achieved by TPA. It may be that the HSF preparation used by Evans et al. (24) was not optimally active and/or it contained inhibitory activities, such as trace amounts of IL-1β, which is known to lower basal or stimulated levels of fibrinogen expression in rat and human hepatoma cells (14, 18, 20). Failure to detect human IL-1 binding activity or IL-1 mediated increase of fibrinogen expression in FAZA cells (24) is not necessarily an indication for nonresponsiveness of the cells to the cytokine (56). The specific stimulation of complement C3, haptoglobin, and AGP synthesis seems to be the diagnostic indicator of the IL-1 response in rat hepatic cells (19).2
The ability of TPA to induce an IL-6-like response in liver hepatoma cells led to the suggestion that activation of the diacylglyceride-dependent protein kinase C is a intermediary step in the IL-6 signal transduction system (24). Two findings, however, indicate that IL-6 probably does not exercise its action via the conventional phosphatidylinositol phosphate pathway, i.e. resulting in a coupled increase of free cytosolic Ca2+ concentration and of protein kinase C activity (24). We found that treatment of the cells with combinations of TPA and ionomycin were not effective in substituting for IL-6 (Table II); in addition, prolonged pretreatments with TPA did abolish TPA response but not the stimulation by IL-6 (Table III). We conclude from this that TPA and IL-6 communicate their signals via separate pathways to regulatory elements of the acute phase protein genes. It is conceivable that the TPA-dependent signals converge with those generated by IL-6 (e.g. activation of common trans-acting factor(s)). This would be a logical explanation for the synergistic enhancement of the IL-6 effect and the ability of TPA to regulate the rat AGP gene through the IL-1/IL-6-responsive element. We cannot rule out, however, that noninteracting signal pathways with effector-specific enhancer elements within the DRE exist, although a sequence similar to the known cis-acting elements responsive to phorbol esters has not been found. Functional dissection of the DRE region is currently in progress, and the presence of sequences specific for TPA and/or IL-6 will indicate the most likely mode of regulation.
The TPA-induced down-regulation of the cell responsiveness to IL-1 and IL-6 (Table III) is one of the most interesting findings of this study. It appears that prolonged exposure to TPA not only leads to loss of the TPA effect on acute phase gene regulation—probably due to down-regulation of protein kinase C activity (47)—but also to a substantial reduction of cell surface receptor activity for IL-1 and, to a lesser degree, for IL-6. This reduction is reminiscent of the TPA-induced loss of epidermal growth factor receptor activity on epithelial cells (reviewed in 57). However, the cellular mechanism underlying the receptor loss in hepatoma cells (e.g. inactivation of the receptor at cell surface, or internalization followed by sequestration or degradation) must first be determined. Once the functional elements of the IL-1 and IL-6 receptors are known, the exact molecular modification (e.g. phosphorylation) leading to the down-regulation can be assessed. Only then will a meaningful comparison with modulation of the epidermal growth factor, or other hormone receptors, be possible.
The ability to down-regulate the responsiveness of liver cells to the inflammatory cytokines may prove to be an important element in controlling liver activity following the acute phase. Within 5–10 days after onset of an inflammatory reaction, the plasma protein production of rodent liver returns to control levels (58–60). This reversal is maintained even when the inflammatory insults are repeated during the “recovery” period (60). We have speculated that the degree of plasma protein gene expression during acute and chronic inflammations is primarily controlled by the humoral concentration of the liver-regulating hormones IL-1, IL-6 (HSF), and glucocorticoids (60). The ineffectiveness of reoccurring tissue injuries to support peak acute phase level of plasma protein production was thought to be either due to an attenuated output of the cytokines by the inflammatory cells, due to the presence of a factor (or factors) quenching the action of the cytokines, or a combination of the two. A reduction of cytokine effect appears to be a real possibility in view of the TPA-dependent modification of the liver cell response. It remains to be determined whether hormones that primarily stimulate protein kinase C in liver cells remain elevated after the acute phase of the inflammatory reaction and thereby act as modulators of the hepatic acute phase response by down-regulating the responsiveness to the stimulating cytokines. These hormones might play a crucial role in determining the liver phenotype in chronic inflammatory diseases.
Acknowledgments
We are greatly indebted to Drs. G. Wong, D. Urdal, and S. Dower for providing recombinant cytokines, Dr. R. Cortese for providing cDNA probes, Karen R. Prowse for help in plasmid construction, and Marcia Held for secretarial work.
Footnotes
This work was supported by National Institutes of Health Grants CA26122 and DK33886.
The abbreviations used are: IL-1, interleukin 1; AGP, α1-acid glycoprotein; CAT gene, chloramphenicol acetyltransferase gene; IL-6, interleukin 6; HSF, hepatocyte stimulating factor; MEM, minimal essential medium; MUP, major urinary protein; TPA, 12-O-tetradecanylphorbol-13-acetate; DRE, distal regulatory elements; GRE, glucorticoid responsive element; Hepes, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid.
S. Marinkovic, G. P. Jahreis, G. G. Wong, and H. Baumann, manuscript submitted for publication.
References
- 1.Koj A. In: Structure and Function of Plasma Proteins. Allison AC, editor. Vol. 1. Plenum Publishing Corp; New York: 1974. pp. 73–125. [Google Scholar]
- 2.Kushner I. Ann N Y Acad Sci. 1982;389:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x. [DOI] [PubMed] [Google Scholar]
- 3.Koj A. In: The Acute-phase Response to Injury and Infection. Gordon AH, Koj A, editors. Elsevier Scientific Publishing Co; Amsterdam: 1985. pp. 145–151. [Google Scholar]
- 4.Kampschmidt RF. J Reticuloendothel Soc. 1978;23:287–297. [PubMed] [Google Scholar]
- 5.Selinger MJ, McAdam KPWJ, Kaplan MM, Sipe JD, Vogel SN, Rosenstreich DL. Nature. 1980;285:498–500. doi: 10.1038/285498a0. [DOI] [PubMed] [Google Scholar]
- 6.Rupp RG, Fuller GM. Exp Cell Res. 1979;118:23–30. doi: 10.1016/0014-4827(79)90579-2. [DOI] [PubMed] [Google Scholar]
- 7.Saunders PK, Fuller GM. Thromb Res. 1983;32:133–145. doi: 10.1016/0049-3848(83)90025-7. [DOI] [PubMed] [Google Scholar]
- 8.Ritchie DG, Fuller GM. Ann N Y Acad Sci. 1983;408:490–502. doi: 10.1111/j.1749-6632.1983.tb23268.x. [DOI] [PubMed] [Google Scholar]
- 9.Le PT, Mortensen RF. J Leukocyte Biol. 1984;35:587–603. doi: 10.1002/jlb.35.6.587. [DOI] [PubMed] [Google Scholar]
- 10.Baumann H, Jahreis GP, Sauder DN, Koj A. J Biol Chem. 1984;259:7331–7342. [PubMed] [Google Scholar]
- 11.Dinarello CA. Rev Infect Dis. 1984;6:51–95. doi: 10.1093/clinids/6.1.51. [DOI] [PubMed] [Google Scholar]
- 12.Beutler B, Cerami A. Nature. 1986;320:584–588. doi: 10.1038/320584a0. [DOI] [PubMed] [Google Scholar]
- 13.Perlmutter DH, Dinarello CA, Punsal PJ, Cotton HR. J Clin Invest. 1986;78:1349–1354. doi: 10.1172/JCI112721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gauldie J, Richards C, Harnish D, Landsorp P, Baumann H. Proc Natl Acad Sci U S A. 1987;84:7251–7255. doi: 10.1073/pnas.84.20.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Kayama K, Iwamatsu A, Tsunasawa S, Sakiyama F, Matsui H, Takahara Y, Taniguichi T, Kishimito T. Nature. 1986;324:73–76. doi: 10.1038/324073a0. [DOI] [PubMed] [Google Scholar]
- 16.Zilberstein A, Ruggieri R, Korn JH, Revel M. EMBO J. 1986;5:2529–2537. doi: 10.1002/j.1460-2075.1986.tb04531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haegeman G, Content J, Volckaert G, Derynck R, Favernier J, Fiers W. Eur J Biochem. 1986;159:625–632. doi: 10.1111/j.1432-1033.1986.tb09931.x. [DOI] [PubMed] [Google Scholar]
- 18.Darlington GJ, Wilson DR, Lachman LB. J Cell Biol. 1986;103:787–793. doi: 10.1083/jcb.103.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumann H, Onorato V, Gauldie J, Jahreis GP. J Biol Chem. 1987;262:9756–9768. [PubMed] [Google Scholar]
- 20.Baumann H, Richards C, Gauldie J. J Immunol. 1987;139:4122–4128. [PubMed] [Google Scholar]
- 21.Baumann H, Mueller-Eberhard U. Biochem Biophys Res Commun. 1987;146:1218–1226. doi: 10.1016/0006-291x(87)90778-9. [DOI] [PubMed] [Google Scholar]
- 22.Andus T, Geiger T, Hirano T, Northoff H, Gauter U, Bauer J, Kishimoto T, Heinrich PC. FEBS Lett. 1987;221:18–22. doi: 10.1016/0014-5793(87)80344-7. [DOI] [PubMed] [Google Scholar]
- 23.Prowse KR, Baumann H. Mol Cell Biol. 1988;8:42–51. doi: 10.1128/mcb.8.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans G, Courtois GM, Kilian PL, Fuller GM, Crabtree GR. J Biol Chem. 1987;262:10850–10854. [PubMed] [Google Scholar]
- 25.Woloski BMRNJ, Fuller GM. Proc Natl Acad Sci U S A. 1985;82:1443–1447. doi: 10.1073/pnas.82.5.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reuber MD. J Natl Cancer Inst. 1961;26:891–899. [PubMed] [Google Scholar]
- 27.Wong GG, Witek-Gianotti GC, Temple PA, Krisz R, Ferenc C, Hewick RM, Clark SC, Ikebuchi K, Ogawa M. J Immunol. 1988 in press. [PubMed] [Google Scholar]
- 28.Dower SK, Kronheim SR, Hopp TP, Cantrell M, Deeley M, Gillis S, Henney CS, Urdal DL. Nature. 1986;324:266–268. doi: 10.1038/324266a0. [DOI] [PubMed] [Google Scholar]
- 29.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 30.Rave N, Crkvenjakov R, Boedtker H. Nucleic Acids Res. 1979;6:3559–3567. doi: 10.1093/nar/6.11.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas PS. Proc Natl Acad Sci U S A. 1980;77:5201–5205. doi: 10.1073/pnas.77.9.5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baumann H, Hill RE, Sauder DN, Jahreis GP. J Cell Biol. 1986;102:370–383. doi: 10.1083/jcb.102.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill RE, Shaw PH, Boyd PA, Baumann H, Hastie ND. Nature. 1984;311:175–177. doi: 10.1038/311175a0. [DOI] [PubMed] [Google Scholar]
- 34.Raugei G, Bensi G, Colantuoni V, Romano V, Santoro C, Costanzo F, Cortese R. Nucleic Acids Res. 1983;11:5811–5819. doi: 10.1093/nar/11.17.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dente L, Cilberto G, Cortese R. Nucleic Acids Res. 1985;13:3941–3952. doi: 10.1093/nar/13.11.3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baumann H, Maquat LE. Mol Cell Biol. 1986;6:2551–2561. doi: 10.1128/mcb.6.7.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham FL, Van der Eb AJ. Virology. 1973;52:456–461. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 38.Lopata MA, Cleveland DW, Sollner-Webb B. Nucleic Acids Res. 1984;12:5707–5717. doi: 10.1093/nar/12.14.5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorman CM, Moffat LF, Howard BH. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolton AE, Hunter WM. Biochem J. 1973;133:529–539. doi: 10.1042/bj1330529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bird TA, Saklatvala J. Nature. 1986;324:263–266. doi: 10.1038/324263a0. [DOI] [PubMed] [Google Scholar]
- 42.Dower SK, Kronheim SR, March CJ, Conlon PJ, Hopp TP, Gillis S, Urdal DL. J Exp Med. 1985;162:501–515. doi: 10.1084/jem.162.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berridge MJ. Annu Rev Biochem. 1987;56:159–193. doi: 10.1146/annurev.bi.56.070187.001111. [DOI] [PubMed] [Google Scholar]
- 44.Angel P, Imazawa M, Chiv R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herlich P, Karin M. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- 45.Kaufman JD, Valandra G, Roderiguez G, Bushar G, Giri C, Norcross MA. Mol Cell Biol. 1987;7:3759–3766. doi: 10.1128/mcb.7.10.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imber JL, Schatz C, Wasylyk C, Chatton B, Wasylyk B. Nature. 1988;332:275–278. doi: 10.1038/332275a0. [DOI] [PubMed] [Google Scholar]
- 47.Kaibuchi K, Tsuda T, Kikuchi A, Tanimoto T, Yamashita T, Takai Y. J Biol Chem. 1986;261:1187–1192. [PubMed] [Google Scholar]
- 48.Frick KK, Womer RB, Scher CD. J Biol Chem. 1988;263:2948–2952. [PubMed] [Google Scholar]
- 49.Martinez-Valdez H, Thompson E, Cohen A. J Biol Chem. 1988;263:4043–4046. [PubMed] [Google Scholar]
- 50.Downward J, Waterfield MD, Parker PJ. J Biol Chem. 1985;260:14538–14546. [PubMed] [Google Scholar]
- 51.Lin CR, Chen WS, Lazar CS, Carpenter CD, Gill GN, Evans RM, Rosenfeld MG. Cell. 1986;44:839–848. doi: 10.1016/0092-8674(86)90006-1. [DOI] [PubMed] [Google Scholar]
- 52.Hachiya HL, Takayama S, White MF, King GL. J Biol Chem. 1987;262:6417–6424. [PubMed] [Google Scholar]
- 53.Holtmann H, Wallach D. J Immunol. 1987;139:1161–1167. [PubMed] [Google Scholar]
- 54.Liles WC, Hunter DD, Meier KE, Nathanson NM. J Biol Chem. 1986;261:5307–5313. [PubMed] [Google Scholar]
- 55.Scatchard G. Ann N Y Acad Sci. 1949;51:660–672. [Google Scholar]
- 56.Rosoff PM, Savage N, Dinarello CA. Cell. 1988;54:73–81. doi: 10.1016/0092-8674(88)90181-x. [DOI] [PubMed] [Google Scholar]
- 57.Carpenter G. Annu Rev Biochem. 1987;56:881–914. doi: 10.1146/annurev.bi.56.070187.004313. [DOI] [PubMed] [Google Scholar]
- 58.Weimer HE, Humelbaugh Can J Physiol Pharmacol. 1967;45:241–247. doi: 10.1139/y67-027. [DOI] [PubMed] [Google Scholar]
- 59.Baltz ML, Gomer K, Davies AJS, Evans PJ, Klaus GGB, Pepys MO. Clin Exp Immunol. 1980;39:355–360. [PMC free article] [PubMed] [Google Scholar]
- 60.Glibetic MD, Baumann H. J Immunol. 1986;137:1616–1622. [PubMed] [Google Scholar]