

Abstract
DNA repair is essential for the maintenance of genomic integrity and stability. Nucleotide excision repair (NER) is a major pathway responsible for remediation of damage caused by UV light, bulky adducts, and cross-linking agents. We now show that NER capacity is differentially expressed in human tissues. We established primary cultures of peripheral blood lymphocytes (PBLs: N = 33) and foreskin fibroblasts (FF: N = 6), as well as adult breast tissue (N = 22) using a unique culture system, and measured their NER capacity using the unscheduled DNA synthesis (UDS) functional assay. Relative to FF, primary cultures of breast cells exhibited only 24.6 ± 2.1% of NER capacity and PBLs only 8.9 ± 1.2%. Cells from the breast therefore have a unique and distinctive DNA repair capacity. The NER capacities of all three cell types had similar coefficients of variation in the range of 10%–15%, which should be taken into account when running controls for this contextual assay. Unlike previous studies and speculation in the field, we found that NER was not affected by cell morphology, donor age, or proliferation as measured by the S phase index. While the NER capacity of the transformed lymphoblastoid cell line TK6 was within the range of our PBL samples, the breast tumor-derived MDA MB-231 cell line was four-fold higher than normal breast tissue. These studies show that analysis of baseline DNA repair in normal human cell types is critical as a basis for evaluation of the effects of “mutator” genes as etiological factors in the development of cancer.
Keywords: Human mammary epithelial cells, HMECs, Nucleotide excision repair, NER, DNA repair, Tissue specificity, Unscheduled DNA synthesis assay, UDS
Introduction
Living organisms are subjected to a constant barrage of DNA damage caused by exposure to a wide range of exogenous chemical and physical agents, as well as endogenous metabolic processes. Unrepaired DNA damage may lead to the accumulation of somatic mutation and the eventual development of neoplastic transformation.
Three types of genes are known to be involved in the etiology of most cancers: oncogenes [1–3], tumor suppressor genes [4,5], and the subset of tumor suppressor genes known as “mutator” genes, usually involved in DNA repair [6–8]. Mutations in DNA repair genes compromise the long-term ability of a cell to correct genotoxic damage. DNA repair deficiencies can result in an accelerated rate of cellular mutation, potentially serving as the step that confers genomic instability. There are multiple pathways of DNA repair in man, including double-strand break repair, base excision repair, base mismatch repair, and nucleotide excision repair [9].
Long patch, or nucleotide excision repair (NER), is the primary process by which cyclobutane pyrimidine dimers, 6-4 photoproducts, and DNA cross-links are removed from the DNA [10,11]. UV254 nm light, as well as so-called UV mimetic drugs, induce DNA lesions that are corrected by this pathway. Damage lesions caused by genotoxic chemotherapy agents that act as inter- and intrastrand cross-linkers, such as cisplatin [12], covalently bind to DNA creating “bulky” adducts. Other bulky adducts are caused by agents such as N-acetoxyaminoacetylfluorene (AAAF) and melphalan [13,14] and alkylating agents, such as cyclophosphamide [15,16] are also presumably remediated by this pathway. NER is a complicated process requiring the protein products of 20–30 genes [17]. NER involves the recognition of a damage lesion causing distortion of the DNA helix, incisions flanking the lesion on the damaged strand, excision of 27–29 bases including the damage lesion, and replication and ligation to replace the excised information and seal the strand breaks at each end of the newly synthesized region [18–21]. This pathway can also be called into play for other types of DNA damage lesions that have not been corrected by base excision and other single-stranded DNA repair mechanisms [22,23]. In effect, the NER pathway provides redundancy for these other repair systems should they be overwhelmed by a genotoxic exposure [10].
NER of the overall genome can be measured quantitatively using the unscheduled DNA synthesis assay (UDS). The UDS assay involves the measurement of labeled base incorporation into the DNA after in vitro exposure to UV light or certain chemicals. The UDS assay is a cell autonomous, functional assay, in that it allows one to look at the complex process of NER as a whole, at least as it is expressed in a particular cell type [24–26]. As applied in our laboratory, this assay predominantly quantifies the repair of UV-induced DNA 6-4 photoproducts, and elements of both the “global genomic” as well as the “transcription coupled” components of NER contribute to the results [10,26].
The autoradiographic UDS assay requires the analysis of living cells. It has previously been applied primarily to skin fibroblasts and peripheral blood lymphocytes (PBLs) for diagnosis of xeroderma pigmentosum (XP) and other DNA repair diseases impacting specifically on the NER pathway. Classical NER deficiency disorders are characterized by UV sensitivity manifesting mainly in the skin and cornea. In diagnostic studies, a single “normal” sample of the same tissue type is used as a control. Alternatively, foreskin fibroblasts (FF) have historically been used as positive standards in UDS studies, because these cells manifest a consistently high level of repair. Mixtures of FF from several babies have sometimes been used to attempt to account for possible interindividual differences. PBLs are among the most accessible nucleated cell types in the body, and have therefore been used in studies to determine the DNA repair capacity of cancer patients and their relatives. UDS analysis of PBLs and skin fibroblasts have seldom been reported concurrently, however, and never in a form that would allow the two to be placed into the same context.
Studies involving functional assays in general, and specifically functional assays of DNA repair capacity, have been hampered by a technical lack of ability to perform primary explant culture on all cell types. The one notable exception is that of rat hepatocyte primary cultures, which have been used extensively in UDS assays for evaluation of the carcinogenic potential of chemicals [27,28]. Although repair assays can be performed on established, transformed cell lines, the generation of cell lines from normal adult tissue has proven to be a technical challenge. In addition, during the process of passaging, established cell lines undergo clonal evolution that may alter or extinguish many of the original characteristics of the cells, including their intrinsic repair capacity [29,30].
Until recently, cell culture techniques have not existed to support primary culture explants of most human tissues. These tissues require attachment to a substratum of some sort of extracellular matrix (ECM). In particular, lineages that involve epithelial cells have proven to be extremely difficult to culture, because these cells normally rest on a complex, biologically reactive basement membrane produced in vivo by the cell type residing beneath the basal surface of the epithelial cells, the myoepithelial cells [31,32]. We have developed a novel system for establishing primary cultures of breast epithelial cells and stromal fibroblasts [33]. Because most breast tumors arise from epithelial cells, the evaluation of their baseline mutator gene function, i.e., DNA repair capacity, is extremely important, and is now possible in primary epithelial cultures.
It has long been presumed that DNA repair is so essential to the maintenance of genomic integrity that it is constitutively expressed in all cells within an individual. A natural extension of this assumption is that DNA repair capacity is equivalent in all cell-types within an individual. However, the possibility that NER can be regulated is illustrated by the findings that NER is present in different levels in various tissues and cell types in mammalian development. In a previous study of the excision repair capacity of the four distinct extraembryonic lineages that comprise the extraembryonic yolk sac, as well as five cell types derived from the fetus, we have shown that NER in the mouse is lineage-specific during embryogenesis [34]. In the present study, we expand this observation from mouse embryonic tissues to a small sampling of human tissues, including, for the first time, breast epithelial cells.
Materials and methods
Tissue procurement and establishment of cultures
Breast reduction mammoplasty tissues were obtained from patients at Magee-Womens Hospital under Magee-Womens Hospital/University of Pittsburgh IRB # MWH-94-108. A neighboring piece of mammoplasty tissue (from the same 0.25 cm2 sample) to that placed into primary culture was fixed and embedded in paraffin. Sections were examined by a pathologist to verify the histological normality of the tissue.
Peripheral blood lymphocytes (PBLs) were obtained with consent from normal healthy control subjects working at Magee-Womens Hospital, Magee-Womens Research Institute or students at the University of Pittsburgh. Foreskin fibroblast (FF) tissue was obtained as discarded tissue from newborn infants after circumcision.
FF were converted into primary explants as described in Latimer et al. [34]. Briefly, cells were grown in MEM containing 10% fetal calf serum on uncoated chamber slides (Nalge Nunc International, Naperville, IL). These cells were passaged to promote homogeneity and grown continuously in culture for up to 12 passages. These cultures show constant levels of DNA repair until passage 13 and senesce after approximately 20 [35]. For UDS experiments, FF were utilized between passages 7 and 10. Four different preparations of FF were used in this study: one consisted of a pool of three perinatal circumcisions and the remaining three were made from individual circumcisions.
PBLs were obtained from normal healthy male and female controls ages 20–50. Lymphocytes were purified using the ficoll gradient method [36] and placed onto a diluted form of Matrigel (BD Biosciences, Bedford, MA) in RPMI medium supplemented with 15% fetal calf serum. It was discovered that PBLs adhered to Matrigel-coated chamber slides, so the autoradiographic UDS assay could be performed on these adherent cells [37]. Lymphocytes were cultured 5–7 days before performance of the UDS assay. Nineteen out of the 33 samples presented here have been previously published as controls in a study on the effects of stress on NER capacity [37].
Breast reduction mammoplasty tissue was rinsed, processed, and placed into primary cultures within 5 h of surgery. Tissue was mechanically disaggregated and placed on diluted (1:1) Matrigel in a novel tissue culture medium called MWRIα according to a method developed in our laboratory [33]. It has been well established that Matrigel provides an optimal commercially available surface for attachment of epithelial cells [38]. The extracellular matrix (ECM) components of matrigel are apparently close enough to the natural basement membrane for mammary luminal epithelial to adhere and retain a rounded morphology [39]. We therefore used matrigel for both establishment of breast cell cultures and to allow the normally nonadherent PBLs to attach to the solid-surface of the glass slides used for the UDS assay. Primary breast cultures were grown for 7–10 days, imaged using a digital Hammamatsu video camera, and then analyzed for NER capacity using the UDS assay. UDS experiments were performed when the epithelial cells were present as mammospheres (clusters of epithelial cells numbering from 40 to 150 cells [40]). Stromal fibroblasts were also readily distinguishable in these cultures.
TK6 lymphoblastoid cells and MDA MB-231 stage IV breast tumor cells were purchased from the American Type Culture Collection (Rockville, MD).
Unscheduled DNA synthesis assay
NER was measured using autoradiography of unscheduled DNA synthesis (UDS) [25]. After a total of 7 days in culture, without passaging, cultures were irradiated with UV light at 254 nm at a mean fluence of 1.2 J/m2 for 12 s in the absence of culture medium, for a total dose of 14 J/m2. Primary cultures had not reached confluence and were still actively growing at the time the UDS assay was performed. Control established cell lines were plated subconfluently 1–2 days before the UDS assay to ensure that they also were not in a quiescent state brought on by confluence. Careful UV dosimetry was performed using a UV delivery system specifically designed for this assay [41]. This machine contains three UV germicidal bulbs placed at a distance of 3 feet (91.4 cm) from an electric turntable where the chamber slides are placed. A 6-in diameter photographic shutter opens under electronic control to deliver a precisely timed dose of UV light. UV bulbs were warmed up at least 1 h before UDS and UV output was checked before each experiment using a Spectroline DM-254X UV meter.
Each sample was represented by at least two chamber slides. One chamber of each two-chamber slide was shielded from the UV dose to be used as an unirradiated control sample. After UV exposure, all cultures were incubated in medium supplemented with 10 μCi ml [3H]methylthymidine (~80 Ci mmol−1) (PerkinElmer Life Sciences, Boston, MA) for 2 h at 37 °C. Labeling medium was then replaced with unlabeled chasing medium containing 10−3 M nonradioactive thymidine (Sigma, St. Louis, MO) and incubated for a further 2 h to clear radioactive label from the intracellular nucleotide pools. After incubation in the post-labeling medium, cells were fixed in 1X SSC, 33% acetic acid in ethanol, followed by 70% ethanol and finally rinsed in 4% perchloric acid overnight at 4°C. All slides were dried and subsequently dipped in photographic emulsion (Kodak type NTB2) and exposed for 10 to 14 days in complete darkness at 4 °C.
The length of exposure of emulsion was determined in each experiment by preparing “tester” slides. These are extra slides of the positive controls, for these experiments consisting of two slides each of FF and of the established breast cancer cell line MDA MB-231 [42]. These slides were dipped in photographic emulsion, dried, and packaged separately from the rest of the experiment in a sealed slide box. After 10 days these tester slides were developed and grain counting was performed. If the nuclei over the foreskin fibroblasts averaged 50 or more grains per nucleus, then the rest of the experimental slides were developed (including additional FF and MDA MB-231 slides). If the grain count was below this level, the remaining slides were left to expose 1–3 days longer before being developed. The tester slides were only used to determine when the exposure time was optimal, because exposure time can vary depending primarily on the age of the radiolabel and the emulsion.
Grain counting analysis
After photographic development of the emulsion on the slides, the nuclei were stained with Giemsa, then examined at 1000X magnification on a Zeiss Axioskop under oil emersion for grains located immediately over the nuclei of non-S phase cells (S phase cells were distinguished by their high grain counts, at least 10-fold higher than non-S phase, and by a clustered pattern of grains). Local background grain counts were determined in each microscopic field, over an area the same size as a representative nucleus, and this total was subtracted from the grain count of each nucleus in that field. The average number of grains per nucleus were quantified for each side of the chamber slide, both unirradiated and irradiated. The final NER value for each slide was calculated by subtracting the unirradiated mean grains per nucleus from the irradiated mean grains per nucleus, after the initial subtraction of local background in each field. NER was ultimately expressed as a percentage of the activity of concurrently analyzed FF. An average of 4.6 FF slides were scored per experiment, with an average of 145 nuclei per slide, for a total of almost 700 nuclei for each FF sample, with an average of 42.6 grains/nucleus. An average of 2.9 slides were scored for each PBL sample, with an average of 205 nuclei/slide. Thus, almost 600 nuclei were counted for each PBL sample, with an average of 6.6 grains/nucleus. Finally, an average of 4.9 slides were evaluated for each breast reduction sample, with an average of 144 nuclei per slide. An average of 700 nuclei were therefore scored per breast reduction sample, with an average of 14 grains/nucleus.
Statistical analysis
To ensure accuracy and guard against transcription errors, raw grain counts from the UDS assay were processed independently in duplicate, once using StatView (version 5.0.1, SAS Institute, Inc., Cary, NC), and once using the Data Analysis Toolpack of the Excel 2001 spreadsheet program (Microsoft Corp., Redmond, WA). The final count from slides of the same cell type within the same experiment and developed the same day were averaged together and expressed as a percentage of concurrently analyzed FF, or, in the case of FF, a percentage of concurrently analyzed MDA MB-231. NER values for cell lines and FF controls were averaged over all experiments. Comparisons between different cell lines and cell types were performed using both the parametric two-tailed t test and the nonparametric Mann-Whitney U test, with significance determined at alpha < 0.05. In all cases, the P value reported is the higher of the two, which was always generated by the nonparametric test. Comparison of UDS values for cells with epithelial and fibroblastic morphologies from the same samples was performed using a paired t test at alpha < 0.05. The possible effects of donor age, S phase index, and cell type on NER capacity were evaluated using linear regression at the same level of significance.
Results
Tissue specificity of NER
Because UDS is a relative measure of DNA repair capacity, primary cultures of human FF were included in each experiment to serve as a standard of comparison. Subjects used for generation of FF cultures were all within the first week of life. PBL donors were recruited from laboratory and hospital workers and ranged from 20 to 50 years of age (N = 33). Breast reduction patients who provided normal tissue for culture ranged in age from 20 to 70 years of age (N = 22). The established breast cancer pleural effusion-derived cell line MDA MB-231 [42] was also included as a second, supplementary control in all experiments.
Histological evaluations were performed on breast reduction tissue adjacent to that placed into culture, on a block provided from the same 0.25 cm2 sample that was fixed and processed in paraffin. Normal breast tissue can display an array of nonmalignant histologies that include fibrocystic changes, hyperplasia (overgrowth), and calcification. These changes are typical of normal breast tissue [43], and none of the samples selected for UDS analysis contained changes that were outside of this range of normal.
As shown in Fig. 1, our results with the UDS assay revealed distinct DNA repair capacities for all three types of cultures analyzed. Although defined as 100% repair capacity in each experiment, comparison of the FF with MDA MB-231 allowed us to calculate the variability exhibited by our four lots of FF; one of which was pooled from tissue from three infants and three of which were derived from single individuals. Thus, our FF samples showed a coefficient of variation (COV) of 15%. Previous studies, because they have each used a single unique culture of FF as their normal “standard,” have essentially assumed that there was no variability in FF NER capacity. PBLs, another cell type that has been often analyzed with the UDS assay, exhibited only 8.9 ± 1.2% (mean ± standard error) of FF NER capacity, demonstrating that there is a greater than 10-fold range in NER capacities among normal human cell types. This level of DNA repair was significantly lower than that of concurrently analyzed FF (P = 0.001), with a similar interindividual variability (a COV of 14%). Because we did not mitogen stimulate our PBL cultures, these results are consistent with previous studies suggesting unstimulated PBLs are “deficient” in NER [44,45].
Fig. 1.
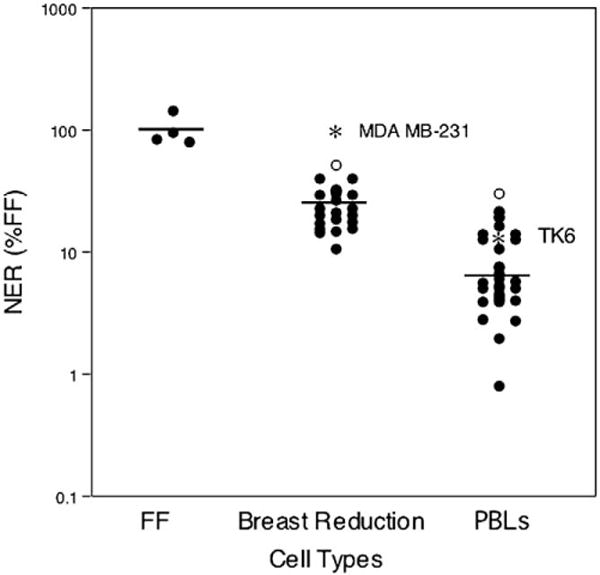
NER capacity of different cell types. NER capacity of FF (arbitrarily set at 100%, N = 6), primary cultures of breast cells (24.6 ± 2.1%, N = 22), and PBLs (8.9 ± 1.2%, N = 33) from normal healthy newborns, women, and adults, respectively. Outliers are represented by open circles (○). The NER capacity of two transformed cell lines derived from the same tissue of origin as the primary cultures are indicated by asterisks in the breast reduction and PBL columns. The mean of each data set is shown by the horizontal line. NER capacity is expressed using a log scale.
Primary breast cell cultures exhibited an intermediate NER capacity of 24.6 ± 2.1% of FF levels, distinct and significantly different from both FF (P = 0.002) and from PBLs (P < 0.001). The interindividual variability of the breast reduction samples was again similar to the other two cell types (COV = 9%). Interestingly, both the PBL and breast reduction results contained one point that was significantly higher than the rest of the population (30% of FF for the PBL sample, 50% of FF for the breast reduction sample). Both of these values are greater than three standard deviations higher than the mean of the rest of their populations, suggesting that they might be considered as “outliers.” Removal of these outliers did not affect the level of significance of the difference in NER capacity between these tissue types, nor did the outliers seem to affect the other statistical comparisons reported in the following sections.
Of the three types of cell samples tested, only the PBLs were available from donors of both sexes. Twenty-four of our PBL samples came from women, with an average NER capacity of 9.1 ± 1.6% of FF (8.2 ± 1.4% without the outlier). The eight male PBL samples were not significantly different in NER capacity from the female, averaging 8.7 ± 1.3% of FF (P = 0.86). Again, sex has not always been expressly matched in UDS studies that used adult controls rather than foreskin fibroblasts; this small comparison suggests that such matching might be unnecessary, at least for comparison of lymphocyte samples.
Analysis of breast cells by morphology
Because there was no attempt at cell-type selection or enrichment other than culture conditions, the primary breast tissue cultures contained cells of two distinct morphologies: epithelial cells which clustered together in three-dimensional organotypic structures called “mammospheres” [40], and more fibroblastic cells that grew as an underlying monolayer, and which might represent the stromal component of the breast (Fig. 2). Because we used the autoradiographic version of the UDS assay, we were able to analyze the NER capacity of both types of cells in our experiments. Epithelial cells were easily identified morphologically as rounded cells that stain with epithelial specific antigen (ESA), whereas the fibroblastic cells do not (data not shown). Tissue lymphocytes are not generally present in our normal breast tissue cultures unless some type of biopsy has been performed on the breast previous to surgery. This was not true of any of the breast reduction cases analyzed in this study. In any case, PBLs have a round cell morphology that is almost entirely nuclear, are present as singleton cells, and are much smaller than breast cells (Fig. 2).
Fig. 2.

Normal breast cells from reduction mammoplasty (A, B) and PBLs (C) after the UDS assay (1000X photomicrographs, bar = 100 microns). The field in (A) contains a cluster of mammary epithelial cells (mammosphere), while the field in (B) contains fibroblastic cells. Cells with intensely dark nuclei, such as those indicated by arrows, are in S-phase.
Three breast cell samples yielded assayable cells that were entirely epithelial in morphology and one sample was entirely fibroblastic in morphology, but the average fraction of epithelial-like cells for all samples was 54%. Interestingly, separate quantification of NER in the epithelial and fibroblastic cells from the normal breast revealed no statistically significant difference between the two cell types, either on a population basis (P = 0.53) or within individuals (P = 0.09; 0.17 without the outlier) (Fig. 3). The NER level exhibited by the epithelial cells from these samples averaged 22.5 ± 1.9% of FF, whereas the fibroblastic cells from the same cultures exhibited an average NER capacity of 25.7 ± 2.7%. Both cell-type subsets from the pooled outlier sample had high NER capacities (35% of FF for the epithelial cells), but the effect was greatest in the fibroblastic cells, 53.4% of FF, which was an outlying value itself. Removal of the outlier reduced the NER capacity of the breast fibroblastic cells to 24.1 ± 2.3% of FF. The NER capacities of the epithelial and fibroblastic cells from the same individuals were highly correlated (P = 0.003), suggesting that genetic factors modify DNA repair activity similarly in both cell types.
Fig. 3.
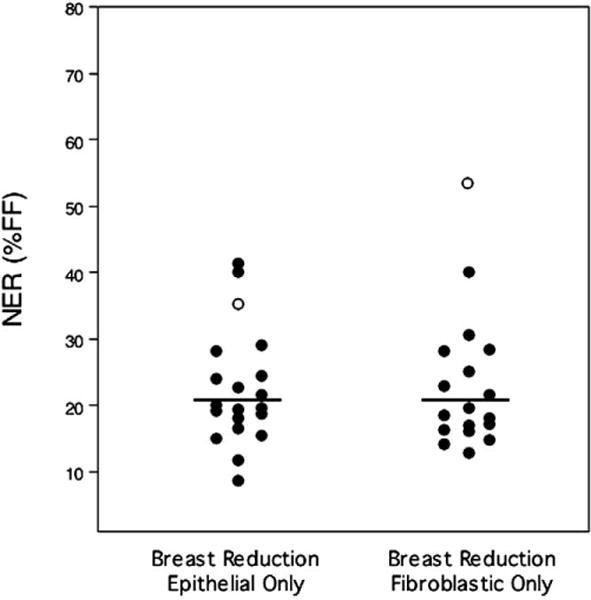
Stratification of breast cell UDS results by cellular phenotype. No significant difference was observed in the NER capacities of cells exhibiting an epithelial vs a fibroblastic morphology. The outlier identified in Fig. 1 is represented by open circles (○). The mean of each data set is shown by the horizontal line.
Effects of age and proliferative index
Two factors have previously been proposed to affect DNA repair capacity: age of donor [46–48], and the mitotic activity of the cell sample [20,49]. It has been observed that uninduced human somatic mutation increases with age when measured in a number of different ways [50,51]. It has been suggested that this is consistent with a generalized loss of DNA repair capacity with age [52], and some data on NER in fibroblasts and PBLs seem to support this hypothesis [53,54]. Analyses of our NER data for a possible effect of age does not support such a hypothesis, however (Fig. 4). Indeed, a significant positive association between NER capacity and age was observed for the PBL data (P = 0.01). This result should be viewed with caution for two reasons: the relative paucity of samples from older individuals (only four would be considered postmenopausal), and the potential effect of the “outlier” mentioned earlier, who was one of these older donors. In fact, if the “outlier” is removed, the association is no longer significant (P = 0.06), although it remains an interesting trend. There is no evidence of a relationship between age and NER capacity in the breast samples, either when considered as combined epithelial and fibroblastic cells (P = 0.75), or for the epithelial (P = 0.58) or fibroblastic cells (P = 0.63) considered separately (data not shown). Removal of the breast reduction “outlier” (a different donor from that of the PBL sample mentioned above), allowed the regression to become negative, but the association remained well below the level of statistical significance (P = 0.50). Analysis of a possible age association was not possible for FF because all samples were harvested from infant circumcisions within the first week of neonatal life.
Fig. 4.
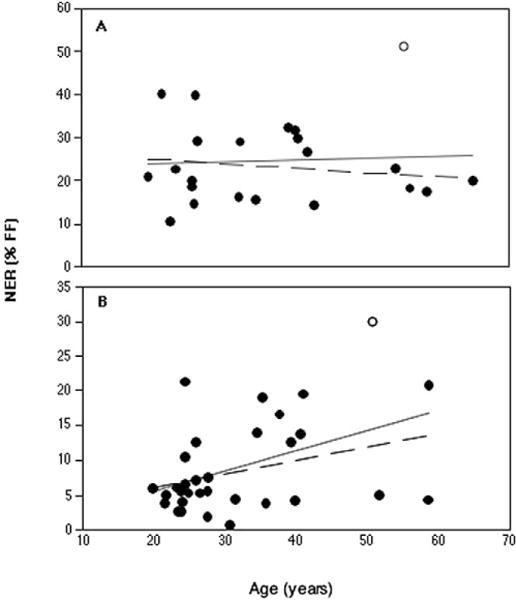
Age regression graph of NER capacity for (A) breast cells, and (B) PBLs. The aberrantly high “outliers” described in the text are represented by open circles. No relationship between NER capacity and age of donor of the human breast cells is indicated, either including all samples (unbroken line) or deleting the outlier (unbroken line). NER capacity of the PBLs significantly increases with age if all samples are included in the analysis (unbroken line), but the relationship falls just below the threshold of significance if the outlier is omitted (broken line).
The S phase index, i.e., percentage of cells in the DNA synthesizing phase of the cell cycle, a measure of mitotic activity, has long been assumed to be positively correlated with DNA repair capacity [15,49]. This assumption is based on the belief that in more rapidly dividing cells there is a greater requirement for DNA repair. The S phase index can easily be derived from the autoradiographic UDS assay. As shown in Fig. 2, cells in S phase incorporate much more label than non-S phase cells, regardless of whether they have been exposed to DNA damaging agents such as UV light. Indeed, the S phase of the cell cycle is the “scheduled” DNA synthesis alluded to by the “unscheduled” DNA synthesis, or UDS used to measure NER. We have previously found that NER capacity is not correlated with the S phase index among mammalian embryonic and extraembryonic lineages [34]. In the present study, FF, breast cells and PBLs exhibited S phase indices of 25%, 20%, and 5%, respectively. We compared NER capacity to S phase index in two ways: between samples from different tissues of origin (Fig. 5A), and within the populations of PBL and breast cell samples (Fig. 5B and C). Between tissues, there is a positive correlation between the UDS results and cell proliferation, but it does not reach significance when each tissue type is represented by a single average value (P = 0.36). If all individual measurements are included, however, the relationship between S phase index and NER capacity is highly significant (P < 0.001), even when an independent cell type indicator variable is added to distinguish the three cell types (which was also significant at P < 0.001). This two-variable model accounted for about 40% of the variability in the NER data, suggesting that factors other than cell type and proliferative capacity influence NER capacity.
Fig. 5.
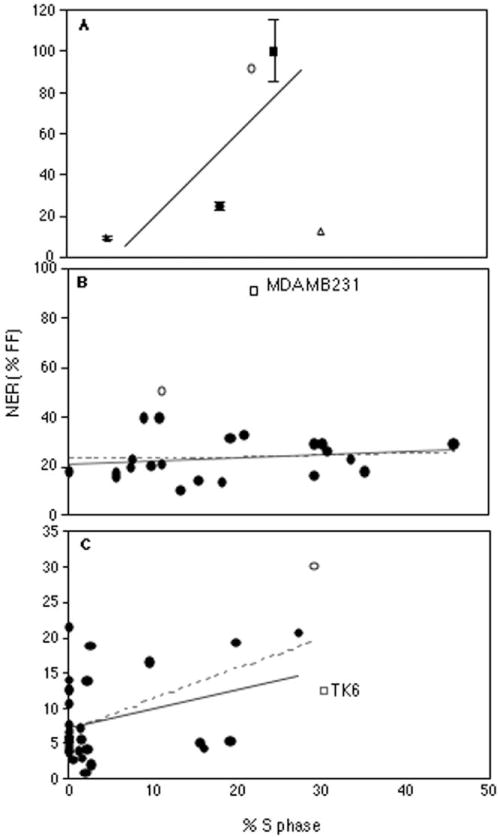
S-phase index regression graphs of NER capacity for (A) the three cell types analyzed in this report, FF (filled square, ■), breast primary cultures (filled circle, ●), and PBLs (filled triangles). Error bars represent standard deviations. The relationship indicated by the line is only significant when individual points are used in the regression rather than the average results for each cell type. Also represented are data from the established breast tumor (MDA MB-231) (open circle, ○) and lymphoblastoid cell lines (TK6) (open triangle, Δ). No relationship between NER capacity and the S phase-index of human breast cells is indicated in (B), either including all samples (broken line) or deleting the outlier (unbroken line). By contrast, there is a statistically significant association between NER capacity and S phase-index in the PBL data (C), but only when the outlier sample is included (broken line). In panels B and C, the outliers are represented by open circles (○), and, for comparison, data from the appropriate established cell line from each cell type is presented (open squares, □).
S phase indices among the population of breast reduction samples had a COV of 8% (ranging from 0.1% to 45%), and there was no evidence of a correlation with NER capacity, either including the NER outlier (P = 0.79), or without it (P = 0.45). The COV of the S phase index measurements among the population of PBL samples was much higher at 45% (range 0 to 27%), and a significant positive correlation was observed between individual S phase indices and NER capacity (P = 0.002). However, once again, when the outlier is removed, the association drops below the level of statistical significance (P = 0.07), although a trend is still evident. Age was not associated with S phase index among the breast cell samples (P = 0.75), but a significant association was observed among the PBL samples (P < 0.001).
Concordance with transformed cell lines
Because the UDS assay measures NER capacity relative to a control, the results are dependent on how accurately both the experimental sample and the control are quantitated. Ideally, slides are developed when the “tester” FF slides show an average of 40–50 grains per nucleus. However, with the much lower NER capacities we observed in PBLs and breast cells it was difficult to count both samples and controls with similar degrees of accuracy. With an NER capacity five to 10-fold higher than those of these samples. FF are no longer an appropriate control. An attempt to address this problem was made by measuring the NER capacities of two established cell lines with appropriate cell-type derivations, the breast tumor-derived cell line MDA MB-231 [42], and the in vitro transformed lymphoblastoid cell line TK6 [55]. MDA MB-231 was included as a second control in all experiments, and exhibited variability between experiments about on the order of the interindividual variabilities observed in the FF, breast cell, and PBL studies, with a COV of 15%. The MDA MB-231 cells exhibited an NER capacity of 91% of FF, while the TK6 line exhibited an NER capacity of 13% of FF (Fig. 1). While the NER capacity of TK6 is well within the range observed in normal lymphocytes, the NER capacity of MDA MB-231 is significantly higher than that of any of the primary breast samples, including the outlier. The unexpectedly high NER capacity of MDA-MB231 limits its usefulness as a control for breast tissue samples, while the TK6 line should be appropriate as a control for PBL studies. Our results are consistent with reports that lymphoblastoid cells have NER capacities similar to those of PHA-stimulated lymphocytes. Both of these transformed cell lines had relatively high S phase indices of 22% (MDA MB-231) and 30% (TK6). Their addition to the data examining the possible effect of S phase index on NER capacity was mixed; the MDA MB-231 data supported the previously observed trend, while the TK6 data did not (Fig. 5A). Overall, there was still no significant correlation between NER capacity and S phase index (P = 0.58).
Discussion
These data provide further evidence for tissue specific differences in NER capacity, suggesting that the human body is heterogeneous in its ability to deal with certain types of genotoxic insult. Because we have performed our studies on primary cultures, the results are as representative as possible of the normal physiological state of the human body. We have also characterized the variability, or “range of normal,” in NER capacity for PBLs and breast cells, and to a lesser degree FF. Only one previous study has provided NER data on a human population survey, and that was performed with a transfection-based assay of transcription-coupled repair [56]. These distributions suggest that a single control “normal” sample is insufficient to provide context for experimental results. Age did not appear to play a major role in this variability. The lack of a difference in NER capacity between cell types contributing to the breast samples demonstrates that adjoining cells may exhibit similar NER activity despite their different lineage, and also suggests that cell morphology does not significantly affect the UDS assay. While NER capacity did seem to correlate with cell proliferation when different cell types were considered, there was no evidence of such an effect within either the PBL or breast cell samples. Finally, our results with the transformed lines TK6 and MDA MB-231 show that while NER capacity may be unaffected by transformation and that an established cell line may remain representative of its original tissue for this trait (TK6), other cell lines may not (MDA MB-231). These studies were possible due to our ability to grow breast cells in primary culture and due to the robust nature of the autoradiographic UDS assay.
Putative factors affecting DNA repair
In this study we saw no consistent effect of aging, cell proliferation, or cell morphology on NER capacity as measured by the UDS assay. In many types of cancer, including breast cancer, advanced age is the greatest risk factor for development of the disease. This is consistent with a proposal that DNA repair capacities decline with age, allowing greater mutagenesis and therefore carcinogenesis. Previous studies using a transfection-based assay have shown a decline in the transcription-coupled component of NER with donor age in human dermal fibroblasts, lymphocytes, and transformed lymphoblastoid cell lines [46–48]. We saw no effect of donor age among our breast cell cultures, and a significant increase in NER capacity with age of donor in our PBL samples. Rather than a loss of activity with age, as would be expected if the integrity of the NER pathway was compromised over time, this result may be associated with the known reduction in hematopoietic progenitor cells that occurs with age, increasingly compromising the diversity of the immune response [57,58]. It may be that those cells with intact NER, and by extension competence in other aspects of DNA metabolism (transcription, replication) preferentially persist into advanced age. On the other hand, our assays were performed on an uncharacterized subset of PBLs that preferentially adhered to our Matrigel-coated slides and continued to metabolize under these conditions, and we cannot know how representative these cells are of the full lymphocyte repertoire available in vivo. A recent study by Goukassian et al. [59] that examined the rate of removal of thymine dimers and 6-4 photoproducts in human dermal fibroblasts using adduct-specific antibodies reported data consistent with our study in similar age groups for breast and PBLs. It is possible that the NER associated “age effect” actually consists of a dramatic postnatal loss of activity that plateaus early in adulthood and is then maintained, or even selectively improved upon, in later years.
Comparing between tissue types, there is a suggestion of a correlation of high cell proliferation with high DNA repair. More cell types will have to be evaluated to determine whether this is a generalizable trend. Addition of the cell line data did not clarify this issue, because MDA MB-231 supported the trend while TK6 did not. We found no evidence of an effect of cell proliferation on NER capacity within samples from the same tissue (PBLs and breast). These results are similar to those of a molecular analysis of expression levels of five NER genes published by Cheng et al. [49]. They found similar levels of gene expression in stimulated and unstimulated lymphocytes, but were able to distinguish the expression levels of rapidly proliferating tissues from those of slowly or nonproliferating tissues. Attempts to rationalize the endocrine theory of cancer with the somatic mutational theory have hypothesized that highly proliferating cells should have lower DNA repair capacity, due to a shortened G1 cell cycle period [60,61], and similar arguments have been made as explanations for the observed relationship between mutation frequency and cell viability [62,63]. Our data suggest that it is doubtful that physiologically relevant changes in proliferation would have much overall impact on NER capacity.
Given our results with FF and PBLs, it was somewhat surprising that there was no observable difference in NER capacity between the two cell types (epithelial and fibroblastic) we analyzed from our breast tissue samples. This observation suggests that it is their degree of sequestration from an environmental insult, such as UV light, that might determine a cell’s baseline expression of NER genes, and therefore repair activity. Alternatively, the epithelial and fibroblastic cells in our cultures may simply be different morphological manifestations of related cell types, if reversible epithelial to mesenchymal conversion takes place normally in vivo, as it does in vitro and in transformed cells [64,65]. In either case, these results suggest that differential cell morphology does not affect NER measurement by the UDS assay, either by modifying the delivery of the UV dose or the exposure of the emulsion. It is far more likely that these cells actually exhibit similar levels of NER activity than the alternative, that they manifest significantly different levels of NER that are exactly compensated by morphological effects.
Finally, a caveat: because we have demonstrated tissue specificity for NER activity, it is possible that other levels of regulation also apply to these pathway. Of concern would be cell cycle-specific regulation resulting in significant changes in NER capacity during S phase.
Tissue specificity of NER capacity and cancer risk
The importance of tissue-specificity studies can be seen in the fact that although individuals with XP have NER defects in PBLs and skin fibroblasts [66,67], they do not show heightened somatic mutation using the allele loss glycophorin A allele loss assay in their hematopoietic bone marrow cells [68]. This brings into question studies in which PBLs have been used as surrogates for many other types of tissues. Tissue specificity studies need to be performed on the normal counterparts of tissues that develop cancer. Because PBLs are relatively easily obtained, the literature is full of epidemiological studies on PBLs of people with various types of cancer, comparing the repair capacity of their PBLs with those of normal controls. It is now possible, as we have shown, to perform this work directly on the tissue(s) and cell type(s) of interest. A similar approach was taken by Monnat and co-workers [69] who performed the HPRT somatic mutation assay on human kidney epithelial cells, and found a significantly higher baseline mutation frequency than had been established in PBLs. The very low NER activity that we have observed in PBLs probably reflects their terminal differentiation and low potential for transformation.
Two previously reported epidemiological studies performed the UDS assay on PBLs from breast cancer patients [70] and from healthy women with first degree relatives with breast cancer [71]. In both of these studies, the UDS assay was performed in the presence of hydroxyurea to inhibit spontaneous DNA synthesis (S-phase cells). These studies concluded that NER capacity was reduced in the PBLs from a significantly greater number of patients and subjects with affected first degree relatives (P < 0.01) than in the lymphocytes of the control populations. It was suggested that subtle inherited deficiencies in NER may be a factor in breast cancer etiology [70]. In contrast, our data suggest that breast cells themselves are particularly susceptible to damage from UV-mimetic chemicals due to an intrinsically low NER activity.
The current trend in the literature appears to be away from functional assays of DNA repair and toward PCR-based molecular analyses of gene expression and of DNA repair gene polymorphism. Without knowing which of the genes are most critical to the pathway, are subject to regulation, or are functionally redundant, it is difficult to know whether molecular analysis of one or a subset of these genes will be representative of the entire NER pathway. Similarly, analyses of repair gene polymorphisms should be based on a demonstration of functional deficiency at the polymorphism itself or an associated haplotype. Serendipitous correlation with a complex biological endpoint such as cancer, may or may not be relevant.
In addition to our results demonstrating low levels of NER DNA repair in breast tissue, it has previously been shown that expression of the BER (base excision repair) glycosylases is lowest in the breast [72,73]. Intrinsically low activity of these DNA repair mechanisms in the breast might suggest an explanation for the tissue-specific cancer predisposition associated with loss of the constitutively expressed BRCA1 and BRCA2 genes, which have also been implicated in DNA repair [74]. The loss of BRCA1- or BRCA2-associated DNA repair would be expected to have the greatest impact in those cells without extensive backup capacity in this regard, such as the breast epithelium. This explanation is similar to the “redundancy” hypothesis suggested by a number of authors [75–77], but is less specific. Whereas these authors have proposed that breast, ovarian, and prostate epithelium uniquely lack redundant systems of double-strand break repair that are present in other tissues (such that loss of BRCA1/2-associated repair is less important or compensated for in most tissues but not those where cancer eventually manifests), we simply suggest that cells already at increased susceptibility to point mutations and DNA crosslinks are likely to be more affected by the increased susceptibility to double strand breaks conferred by loss of BRCA1/2 activity. This susceptibility is in contrast to cells in which compromised double strand break repair is their only vulnerability.
Acknowledgments
This study was supported in part by NIH grant CA 71894-01A1, and by grants from the Ruth Estrin Goldberg Foundation, the Pennsylvania Department of Health, and the Magee-Womens Health Foundation. We greatly appreciate the technical support of Lori Thomas, Linda D. Piersall, Elena Kisin, Karen Hanley-Yanez, Janiene A. Patterson, Ayodola Anise, and Lynn R. Jancieukewic in completing this study. We thank Jennifer M. Johnson for helpful discussion of the manuscript.
References
- 1.Coussens L, Yang-Feng TL, Lioa YC, Chen E, Gray A, McGrath J, Seeburg PH, Liberman TA, Schlessinger J, Francke V. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. 1985;230:1132–1139. doi: 10.1126/science.2999974. [DOI] [PubMed] [Google Scholar]
- 2.Hall JM, Zuppa PJ, Anderson LA, Huey B, Carter C, King MC. Oncogenes and human breast cancer. Am J Hum Genet. 1989;44:577–584. [PMC free article] [PubMed] [Google Scholar]
- 3.Ernberg IT. Oncogenes and tumor growth factors in breast cancer. Acta Oncol. 1990;29:331–334. doi: 10.3109/02841869009090009. [DOI] [PubMed] [Google Scholar]
- 4.Sidransky D, Tokino T, Helzlsouer K, Zehnbauer B, Rausch G, Shelton B, Prestigiacomo L, Vogelstein B, Davidson N. Inherited p53 gene mutations in breast cancer. Cancer Res. 1992;52:2984–2986. [PubMed] [Google Scholar]
- 5.Tripathy D, Benz CC. Activated oncogenes and putative tumor suppressor genes involved in human breast cancers. Breast Cancer Res Treat. 1992;63:15–60. doi: 10.1007/978-1-4615-3088-6_2. [DOI] [PubMed] [Google Scholar]
- 6.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 7.Ionov YM, Pelnado A, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 8.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 9.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. American Society of Microbiology; Washington, D.C: 1995. [Google Scholar]
- 10.Thompson LH. Nucleotide excision repair: its relation to human disease. In: Nickoloff JA, Hoekstra MF, editors. DNA Damage and Repair: DNA Repair in Higher Eukaryotes. Vol. 2. Humana Press; Totowa, NJ: 1998. pp. 335–393. [Google Scholar]
- 11.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 12.Reed E. Platinum–DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat Rev. 1998;24:331–344. doi: 10.1016/s0305-7372(98)90056-1. [DOI] [PubMed] [Google Scholar]
- 13.Kaneko M, Cerutti PA. Excision of N-acetoxy-2-acetylaminofluorene-induced DNA adducts from chromatin fractions of human fibroblasts. Cancer Res. 1980;40:4313–4319. [PubMed] [Google Scholar]
- 14.Grant DF, Bessho T, Reardon JT. Nucleotide excision repair of melphalan monoadducts. Cancer Res. 1998;58:5196–5200. [PubMed] [Google Scholar]
- 15.Andersson BS, Sadeghi T, Siciliano MJ, Legerski R, Murray D. Nucleotide excision repair genes as determinants of cellular sensitivity to cyclophosphamide analogs. Cancer Chemother Pharmacol. 1996;38:406–416. doi: 10.1007/s002800050504. [DOI] [PubMed] [Google Scholar]
- 16.Gamesik MP, Dolan ME, Andersson BS, Murray D. Mechanisms of resistance to the toxicity of cyclophosphamide. Curr Pharmaceut Des. 1999;5:587–605. [PubMed] [Google Scholar]
- 17.Mullenders LHF, Berneberg M. Photoimmunology and nucleotide excision repair: impact of transcription coupled and global genome excision repair. J Photochem Photobiol B. 2001;56:97–100. doi: 10.1016/s1011-1344(01)00244-5. [DOI] [PubMed] [Google Scholar]
- 18.Covertey D, Kenney MK, Rupp WD, Lane DP, Wood RD. Requirement of the replication protein SSB in human DNA excision repair. Nature. 1991;347:538–541. doi: 10.1038/349538a0. [DOI] [PubMed] [Google Scholar]
- 19.Huang JC, Svoboda DL, Reardon JT, Sancar A. Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc Natl Acad Sci USA. 1992;89:3664–3668. doi: 10.1073/pnas.89.8.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shivji KK, Kenney MP, Wood RD. Proliferating cell nuclear antigen is required for DNA excision repair. Cell. 1992;69:367–374. doi: 10.1016/0092-8674(92)90416-a. [DOI] [PubMed] [Google Scholar]
- 21.Grossman L, Thiagalingam S. Nucleotide excision repair, a tracking mechanism in search of damage. J Biol Chem. 1993;268:16871–16874. [PubMed] [Google Scholar]
- 22.Satoh MS, Jones CJ, Wood RD, Lindahl T. DNA excision-repair defect of xeroderma pigmentosum prevents removal of a class of oxygen free radical-induced base lesions. Proc Natl Acad Sci USA. 1993;90:6335–6339. doi: 10.1073/pnas.90.13.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang JC, Hsu DS, Kazantsev A, Sancar A. Substrate specificity of human exinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc Natl Acad Sci USA. 1994;91:12213–12217. doi: 10.1073/pnas.91.25.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218:652–656. doi: 10.1038/218652a0. [DOI] [PubMed] [Google Scholar]
- 25.Painter RB, Cleaver JE. Repair replication, unscheduled DNA synthesis and the repair of mammalian DNA. Radiat Res. 1961;37:451–466. [PubMed] [Google Scholar]
- 26.Cleaver JE, Thomas GH. Measurement of unscheduled synthesis by autoradiography. In: Friedberg EC, Hanawalt PC, editors. DNA Repair, A Laboratory Manual of Research Procedures. I. Marcel Dekker; New York: 1981. pp. 277–287. [Google Scholar]
- 27.Michalopoulos G, Sattler GL, O’Connor L, Pitot HC. Unscheduled DNA synthesis induced by procarcinogens in suspensions and primary cultures of hepatocytes on collagen membranes. Cancer Res. 1978;38:1866–1871. [PubMed] [Google Scholar]
- 28.Williams GM, Mori H, McQueen CA. Structure–activity relationships in the rat hepatocyte DNA-repair test for 300 chemicals. Mutat Res. 1989;221:263–286. doi: 10.1016/0165-1110(89)90039-0. [DOI] [PubMed] [Google Scholar]
- 29.Killary AM, Fournier REK. A genetic analysis of extinction: trans-dominant loci regulate expression of liver-specific traits in hepatoma hybrid cells. Cell. 1984;38:523–534. doi: 10.1016/0092-8674(84)90507-5. [DOI] [PubMed] [Google Scholar]
- 30.Clarke R, Leonessa F, Brunner WN, Thompson EW. In vitro models. In: Harris JR, Lippman ME, Morrow M, Osborne CK, editors. Diseases of the Breast. Lippincott Williams and Wilkins; Philadelphia: 2000. pp. 347–348. [Google Scholar]
- 31.Engel LW, Young NA. Human breast carcinoma cells in continuous culture: a review. Cancer Res. 1978;38:4327–4339. [PubMed] [Google Scholar]
- 32.Stampfer MR, Bartley JC. Development of human mammary epithelial cell culture systems for studies on carcinogenesis and differentiation. In: Webber MM, editor. In Vitro Models for Cancer Research. CRC Press; Boca Raton, FL: 1986. pp. 11–29. [Google Scholar]
- 33.Latimer JJ. Epithelial Cell Cultures Useful For in Vitro Testing. #6,074,874. U.S. Patent. 2000
- 34.Latimer JJ, Hultner ML, Cleaver JE, Pedersen RA. Elevated DNA excision repair capacity in the extraembryonic mesoderm of the midgestation mouse embryo. Exp Cell Res. 1996;228:19–28. doi: 10.1006/excr.1996.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cleaver JE. DNA repair deficiencies and cellular senescence are unrelated in xeroderma pigmentosum cell lines. Mech Ageing Dev. 1984;27:189–196. doi: 10.1016/0047-6374(84)90044-7. [DOI] [PubMed] [Google Scholar]
- 36.Boyum A. Separation of leukocytes from blood and bone marrow. Scand J Clin Lab Invest. 1988;21(Supplement 97):7–106. [PubMed] [Google Scholar]
- 37.Forlenza MJ, Latimer JJ, Baum A. The effects of stress on DNA repair capacity. Psychol Health. 2000;15:881–891. doi: 10.1080/08870440008405589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergstraesser LM, Weitzman SA. Culture of normal and malignant primary human mammary epithelial cells in a physiologic manner stimulates in vivo growth patterns and allows discrimination of cell type. Cancer Res. 1993;53:2644–2654. [PubMed] [Google Scholar]
- 39.Band V, Sager R. Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc Natl Acad Sci USA. 1989;86:1249–1253. doi: 10.1073/pnas.86.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blatchford DR, Hendry KAK, Wilde CJ. Autocrine regulation of protein secretion in mouse mammary epithelial cells. Biochem Biophys Res Commun. 1998;248:761–766. doi: 10.1006/bbrc.1998.9057. [DOI] [PubMed] [Google Scholar]
- 41.Steier H, Cleaver JE. Exposure chamber for quantitative ultraviolet photobiology. Lab Practice. 1969;18:1295. [PubMed] [Google Scholar]
- 42.Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53:661–674. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schnitt SJ, Connolly JL. Pathology of benign breast disorders. In: Harris JR, Lippmann ME, Morrow M, Hellman S, editors. Diseases of the Breast. Lippincott-Raven; Philadelphia: 1996. pp. 27–41. [Google Scholar]
- 44.Freeman SE, Ryan SL. Excision repair of pyrimidine dimers in human peripheral blood lymphocytes: comparison between mitogen stimulated and unstimulated cells. Mutat Res. 1988;194:143–150. doi: 10.1016/0167-8817(88)90016-8. [DOI] [PubMed] [Google Scholar]
- 45.Barret JM, Calson P, Salles B. Deficient nucleotide excision repair activity in protein extracts from normal human lymphocytes. Carcinogenesis. 1995;16:1611–1616. doi: 10.1093/carcin/16.7.1611. [DOI] [PubMed] [Google Scholar]
- 46.Dell’Orco RT, Anderson LE. Unscheduled DNA synthesis in human diploid cells of different donor ages. Cell Biol Int Rep. 1981;5:359–364. doi: 10.1016/0309-1651(81)90005-9. [DOI] [PubMed] [Google Scholar]
- 47.Hennis HL, Braid HL, Vincent RA. Unscheduled DNA synthesis in cells of different shape in fibroblast cultures from donors of various ages. Mech Ageing Dev. 1981;16:355–361. doi: 10.1016/0047-6374(81)90019-1. [DOI] [PubMed] [Google Scholar]
- 48.Grossman L. The role of DNA damage and its repair in the aging process. Aging. 1992;4:252–257. doi: 10.1007/BF03324100. [DOI] [PubMed] [Google Scholar]
- 49.Cheng L, Guan Y, Li L, Legerski RJ, Einspahr J, Bangert J, Alberts DS, Wei Q. Expression in normal human tissues of five nucleotide excision repair genes measured simultaneously by multiplex reverse transcription-polymerase chain reaction. Cancer Epidemiol Biomarkers Prev. 1999;8:801–807. [PubMed] [Google Scholar]
- 50.Trainor KJ, Wigmore DJ, Chrysostomou A, Dempsey JL, Seshadri R, Morley AA. Mutation frequency in human lymphocytes increases with age. Mech Ageing Dev. 1984;27:83–86. doi: 10.1016/0047-6374(84)90084-8. [DOI] [PubMed] [Google Scholar]
- 51.Akiyama M, Kyoizumi S, Hirai Y, Kusunoki Y, Iwamoto KS, Nakamura N. Mutation frequency in human blood cells increases with age. Mutat Res. 1995;338:141–149. doi: 10.1016/0921-8734(95)00019-3. [DOI] [PubMed] [Google Scholar]
- 52.Moriwaki S, Ray S, Tarone RE, Kraemer KH, Grossman L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: reduced DNA repair capacity and increased DNA mutability. Mutat Res. 1996;364:117–123. doi: 10.1016/0921-8777(96)00029-8. [DOI] [PubMed] [Google Scholar]
- 53.Grossman L. Epidemiology of ultraviolet-DNA repair capacity and human cancer, Environ. Health Perspect. 1997;105:927–930. doi: 10.1289/ehp.97105s4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei Q. Effect of aging on DNA repair and skin carcinogenesis: a minireview of population-based studies. J Investig Dermatol Symp Proc. 1998;3:19–22. [PubMed] [Google Scholar]
- 55.Liber HL, Thilly WG. Mutation assay at the thymidine kinase locus in diploid human lymphoblasts. Mutat Res. 1982;94:467–485. doi: 10.1016/0027-5107(82)90308-6. [DOI] [PubMed] [Google Scholar]
- 56.Athas WF, Hedayati MA, Matanoski GM, Farmer ER, Grossman L. Development and field-test validation of an assay for DNA repair in circulating human lymphocytes. Cancer Res. 1991;51:5786–5793. [PubMed] [Google Scholar]
- 57.Kishimoto S, Tomino S, Inomata K, Kotegawa S, Saito T, Kuroki M, Mitsuya H, Hisamitsu S. Age-related changes in the subsets and functions of human T lymphocytes. J Immunol. 1978;121:1773–1780. [PubMed] [Google Scholar]
- 58.Song L, Kim YH, Chopra RK, Proust JJ, Nagel JE, Nordin AA, Adler WH. Age-related effects in T cell activation and proliferation. Exp Gerontol. 1993;28:313–321. doi: 10.1016/0531-5565(93)90058-l. [DOI] [PubMed] [Google Scholar]
- 59.Goukassian D, Gad F, Yaar M, Eller MS, Nehal US, Gilchrest BA. Mechanism and implications of the age-associated decrease in DNA repair capacity. FASEB J. 2000;14:1325–1334. doi: 10.1096/fj.14.10.1325. [DOI] [PubMed] [Google Scholar]
- 60.Yager JD, Liehr JG. Molecular mechanisms of estrogen carcinogenesis. Annu Rev Pharmacol Toxicol. 1996;36:203–232. doi: 10.1146/annurev.pa.36.040196.001223. [DOI] [PubMed] [Google Scholar]
- 61.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- 62.Cole J, Green MHL, James SE, Henderson L, Cole H. A further assessment of factors influencing measurements of thioguanine resistant mutant frequency in circulating lymphocytes. Mutat Res. 1988;204:493–507. doi: 10.1016/0165-1218(88)90044-4. [DOI] [PubMed] [Google Scholar]
- 63.Khaidakov M, Glickman BW. Possible factors leading to a misjudgement of mutant frequencies in HPRT assay. Mutat Res. 1996;354:9–14. doi: 10.1016/0027-5107(95)00253-7. [DOI] [PubMed] [Google Scholar]
- 64.Petersen OW, Lind Nielsen H, Gudjonssonn T, Villadsen R, Ronnov-Jessen L, Bissell MJ. The plasticity of human breast carcinoma cells is more than epithelial to mesenchymal conversion. Breast Cancer Res. 2001;3:213–217. doi: 10.1186/bcr298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savagner P. Leaving the neighborhood: molecular mechanisms involved in epithelial-mesenchymal transition. BioEssays. 2001;23:912–923. doi: 10.1002/bies.1132. [DOI] [PubMed] [Google Scholar]
- 66.Robbins JH, Kraemer KH. Prolonged ultraviolet-induced thymidine incorporation into xeroderma pigmentosum lymphocytes: studies on its duration, amount, localization and relationship to hydroxyurea. Biochim Biophys Acta. 1972;277:7–14. doi: 10.1016/0005-2787(72)90345-0. [DOI] [PubMed] [Google Scholar]
- 67.Andrews AD, Robbins JH, Kraemer KH, Buell DN. Xeroderma pigmentosum long-term lymphoid lines with increased ultraviolet sensitivity. J Natl Cancer Inst. 1974;53:691–693. doi: 10.1093/jnci/53.3.691. [DOI] [PubMed] [Google Scholar]
- 68.Langlois RG, Bigbee WL, Jensen RH. The glycophorin A assay for somatic cell mutations in humans. In: Mendelsohn ML, Albertini RJ, editors. Mutation and the Environment, Part C: Somatic and Heritable Mutation, Adduction and Epidemiology. Wiley-Liss; New York: 1990. pp. 47–56. (Prog. Clin. Biol. Res. 340C) [PubMed] [Google Scholar]
- 69.Martin GM, Ogburn CE, Colgin LM, Gown AM, Edland SD, Monnat RJ., Jr Somatic mutations are frequent and increase with age in human kidney epithelial cells. Hum Mol Genet. 1996;5:215–221. doi: 10.1093/hmg/5.2.215. [DOI] [PubMed] [Google Scholar]
- 70.Kovacs E, Stucki D, Weber W, Müller HJ. Impaired DNA-repair synthesis in lymphocytes of breast cancer patients. Eur J Cancer Clin Oncol. 1986;22:863–869. doi: 10.1016/0277-5379(86)90375-5. [DOI] [PubMed] [Google Scholar]
- 71.Kovacs E, Almendral A. Reduced DNA repair synthesis in healthy women having first degree relatives with breast cancer. Eur J Cancer Clin Oncol. 1987;23:1051–1057. doi: 10.1016/0277-5379(87)90358-0. [DOI] [PubMed] [Google Scholar]
- 72.Fong LY, Jensen DE, Magee PN. DNA methyl-adduct dosimetry and O6-alkylguanine-DNA alkyl transferase activity determinations in rat mammary carcinogenesis by procarbazine and N-methylnitrosourea. Carcinogenesis. 1990;11:411–417. doi: 10.1093/carcin/11.3.411. [DOI] [PubMed] [Google Scholar]
- 73.Hamdan MA, Guzman RC, Yang J, Beattie C, Mascharack P, Nandi S. Depletion of O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine in three-dimensional cultures of normal human breast epithelial cells. Carcinogenesis. 1992;13:1743–1749. doi: 10.1093/carcin/13.10.1743. [DOI] [PubMed] [Google Scholar]
- 74.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 75.Rosen EM, Fan S, Goldberg ID. BRCA1 and prostate cancer. Cancer Invest. 1998;19:396–412. doi: 10.1081/cnv-100103134. [DOI] [PubMed] [Google Scholar]
- 76.Welsch PL, Schubert EL, King MC. Inherited breast cancer: an emerging picture. Clin Genet. 1998;54:447–458. doi: 10.1111/j.1399-0004.1998.tb03764.x. [DOI] [PubMed] [Google Scholar]
- 77.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]