

Abstract
Autoinflammatory disease (AID) is a newly proposed category of disorders characterized by unprovoked episodes of inflammation without any infectious or autoimmune evidence. We aimed to characterize the clinical and genetic features of patients who had recurrent fever and multi-system inflammation but remain unclassified for any established AIDs. Medical records of 1,777 patients who visited our Rheumatology Clinic between March 2009 and December 2010 were reviewed to identify those who met the following criteria; 1) presence of fever, 2) inflammation in two or more organ systems, 3) recurrent nature of fever or inflammation, 4) no evidence of infection or malignancy, 5) absence of high titer autoantibodies, and 6) failure to satisfy any classification criteria for known AIDs. Genotyping was performed for common missense variants in MEFV, NOD2/CARD15, and TNFRSF1A. A small number of patients (17/1,777, 0.95%) were identified to meet the above criteria. Muco-cutaneous and musculoskeletal features were most common, but there was a considerable heterogeneity in symptom combination. Although they did not satisfy any established classification criteria for AIDs, substantial overlap was observed between the clinical spectrum of these patients and known AIDs. According to the newly proposed Eurofever criteria for periodic fevers, eleven of them were classified as TNF receptor-associated periodic syndrome and two as mevalonate kinase deficiency. However, no examined genetic variants including those in TNFRSF1A were found in these patients. A new set of classification criteria needs to be developed and validated for Asian patients with unclassified AIDs.
Keywords: Autoinflammatory Disease, Fever, Classification
Graphical Abstract
INTRODUCTION
Autoinflammatory disease (AID) is a newly proposed category of disorders characterized by unprovoked episodes of inflammation without high titer autoantibodies or antigen-specific T cells (1). The concept of AID stemmed from the understanding of the genetic and molecular basis for inherited systemic inflammatory diseases such as tumor necrosis factor (TNF) receptor- associated periodic syndrome (TRAPS) and familial Mediterranean fever (FMF), both of which represent a prototype of illnesses marked by spontaneous febrile episodes and multi-system inflammation without hallmarks of autoimmunity. Since the connection was discovered between hereditary periodic fever syndromes called as cryopirin-associated periodic syndromes (CAPS) and gain-of-function mutation in NLRP3 (also known as NALP3 that encodes cryopyrin) (2), the biological concept has been formulated for AID that it encompasses a wide clinical spectrum of abnormally increased inflammation mediated by the cells and molecules of innate immune system.
Subsequently, the other disorders and syndromes were added into the category of AID based more on their clinical presentation than on the underlying biology to embrace genetically complex and acquired diseases and to widen the phenotypic scope of AID. Many well-known rheumatic diseases, including crystal- induced arthritis, palindromic rheumatism, vasculitic syndromes such as Behçet's disease (BD), systemic idiopathic juvenile arthritis, adult onset Still's disease (AOSD), Crohn's disease (CD), and spondyloarthropathy (SpA), are currently within the realm of AID (3). Despite the progress in understanding the concept and etiology of AIDs, there has been lack of systematic classification criteria for those who have recurrent febrile or inflammatory episodes without of any infectious or autoimmune causes. Moreover, the diagnostic criteria for inherited periodic fevers have been formulated only in the context of particular ethnic backgrounds (4,5). The individual classification criteria for acquired AIDs (BD, SpA, AOSD, or CD) are developed based on the typical features of a single disease. Thus, there are many patents who present with recurrent febrile or inflammatory episodes but remain unclassified under current classification systems (6).
Genetic variants of MEFV, NOD2/CARD15, and TNFRSF1A are known to transmit monogenic AIDs such as FMF, Blau syndrome, and TRAPS (7). However, they have been implicated in polygenic AIDs as well. For example, in Turkey where the prevalence of MEFV variants is high, missense mutations in MEFV are not only associated with FMF, but were also found to associate with BD (8), SpA (9), enthesitis-related arthritis (10), and PFAPA syndrome (periodic fever, aphthous stomatitis, pharyngitis, and adenitis) (11). Likewise, NOD2/CARD15 involves CD and other recurrent febrile illnesses (12,13). TNFRSF1A is associated with multiple sclerosis besides TRAPS (14). These findings may indicate that a broad clinical spectrum of inflammatory illnesses is associated with the genetic variants of these genes. In addition, clinical characteristics of a given AID are often influenced by the associated genetic or ethnic backgrounds (15).
In Korea, FMF is rare, but an adult onset case of FMF with no family history but with two point mutations in MEFV has been reported (16). Unlike Mediterranean populations, Korean population is more commonly affected by acquired or polygenic AIDs such as BD or CD than by hereditary or monogenic AIDs described above. In addition, enhanced utilization of NLRP3 in active BD compared with inactive or healthy subjects were reported (17), suggesting that autoinflammatory mechanism is one of the features of BD.
Hence, we aimed to investigate the frequency and clinical manifestations of Korean patients who had been afflicted by recurrent fever and multisystem inflammation without any infectious or autoimmune evidence but who failed to fit current classification systems for AIDs (3,7) and to examine the prevalence of MEFV, NOD2/CARD15, and TNFRSF1A variants in these patients.
MATERIALS AND METHODS
Patients
Medical records of 1,777 patients who visited or were referred to our Rheumatology Clinic at the Seoul National University Bundang Hospital between March 2009 and December 2010 were reviewed to identify patients who met all of the following criteria (13); 1) presence of fever of unknown origin, 2) inflammatory involvement in two or more organ systems including skin, mucous membrane, muscle, joint, vessels, and any internal organs during the course of illness, 3) recurrent nature of either fever or organ system inflammation (not necessarily the same organ), 4) no evidence suggesting microbial infection or malignancy, 5) absence of high titer autoantibodies, and 6) failure to satisfy any particular disease classification. HLA-B*51 and HLA-B*27 carriers were excluded.
Laboratory examination
Febrile patients free of any infectious, malignant, or autoimmune causes were enrolled in this study after extensive work-up including culture studies, serological tests, imaging studies, genetic tests, and pathological examinations. All patients underwent serological tests for systemic autoimmune diseases and/or vasculitis based on their clinical symptoms and signs, including but not limited to, anti-nuclear antibodies, rheumatoid factors, anti-U1 ribonucleoprotein antibodies, anti-dsDNA antibodies, anti-Ro and anti-La antibodies, anti-citrullinated protein antibodies, and anti-neutrophil cytoplasmic antibodies. Tests such as Coomb's tests, immunoglobulin levels, or complement 3 and 4 levels were also performed whenever antibody dependent process was suspected or the illness was diagnostically challenging.
Application of the provisional Eurofever clinical classification criteria
Although attempts to provide clinical guidelines and flowcharts to identify appropriate candidates for the testing have previously been tried, molecular or genetic testing could be misleading or inconclusive when performed in unselected patients (6,18). The provisional Eurofever classification criteria (19) are the recent attempt to develop evidence-based classification criteria for the four main periodic fevers (FMF, TRAPS, CAPS, and mevalonate kinase deficiency or MKD) based on the clinical manifestations. The criteria are distinguished from the previous clinical criteria in that they have been developed based on the large international registry of patients with different AIDs and of heterogeneous geographic and ethnic distribution. We applied these criteria to our 17 patients to examine if our unclassified AIDs satisfy this new clinical classification criteria dedicated to the four major inherited periodic fever syndromes.
Genetic study
Genomic DNA was extracted from peripheral blood from 16 patients (all except patient #13) and 5 healthy controls using QIAamp blood kits (Qiagen, Valencia, CA, USA). TaqMan® predesigned primers and probes (Applied Biosystems, Foster city, CA, USA) were used for rs28940579 (V726A), rs28940578 (M694I), rs28940580 (M680I), and rs37433930 (E148Q) in MEFV, rs2066844 (R702W) and rs2066845 (G908R) in NOD2/CARD15, rs4149584 (R92Q), rs34751757 (T90N), and rs1419637 (P46L) in TNFRSF1A. Restriction fragment length polymorphism (RFLP) method was used to genotype rs61752717 (M694V) in MEFV (20) and rs2066847 (3020InsC) in NOD2/CARD15 as previously described (21).
Ethics statement
This study was approved by the institutional review board of Seoul National University Bundang Hospital (B-1207-164-104) and informed patient consent was obtained.
RESULTS
Clinical features of the patients
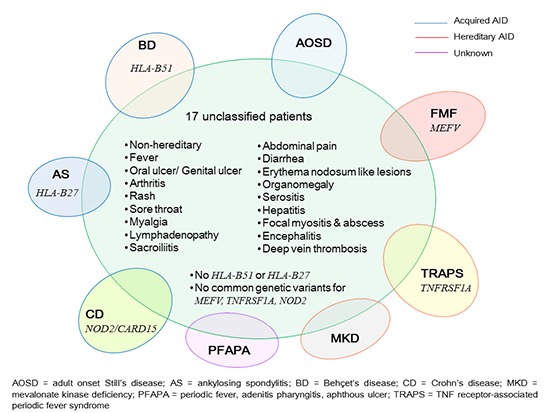
Seven hundred and thirty six patients (736/1,777, 41.5%) presented with inflammatory signs or symptoms of non-infectious and non-cancer origin. Rheumatoid arthritis (n=198), crystal-induced arthropathy (n=173), SpA (n=110), BD (n=58), palindromic rheumatism (n=55), systemic lupus erythematosus (SLE) (n=22), and primary Sjogren's syndrome (n=18) were the most common diagnoses. Among 44 patients who had fever during their illness without evidence of infection or malignancy, we identified 17 patients (17/1,777, 0.95%) who met the study enrollment criteria as defined above. None of them had family history of similar illness. Median age (range) at symptom onset was 37 (18-55) years. Female-to-male ratio was 10:7. The clinical features of these patients are summarized in Table 1. The clinical spectrum of these 17 "unclassified" patients included inflammatory symptoms and signs across various organ systems. Mucocutaneous inflammation such as rash (any rash in 9/17, 52.9%) and oral ulcers (10/17, 58.8%), musculoskeletal inflammation such as arthritis (8/17, 47.1%) and focal myositis/abscess (2/17, 11.8%), and non-specific symptoms such as myalgia (7/17, 41.2%) and headache (6/17, 35.3%) were common features. Organomegaly, encephalitis, serositis, and venous thrombosis were also observed in some patients. However, patient-to-patient diversity in symptom combination made it difficult to bundle up those with shared features. One patient (#8) who initially presented with fever, sore throat, and generalized myalgia had a previous history of oral and genital ulcers and later developed oligoarthritis and then EN-like skin lesions. She was finally diagnosed as having BD.
Table 1. Clinical features of 17 patients identified to have recurrent fever and multi-system inflammation without infectious or autoimmune evidence.
| Patient | Sex | Age | Clinical symptoms or signs except fever | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OU/GU | Arthritis | Macular rash | Sore throat | Myalgia | HA | LAP | Sacroiliitis | EN | Abd pain & diarrhea | HSM | Serositis | Others | |||
| 1 | F | 19 | +/- | + | + | + | + | ||||||||
| 2 | M | 18 | Monophasic, hip | + | +/C | +* | Gluteal myositis & sterile abscess | ||||||||
| 3 | F | 38 | +/+ | +/C | |||||||||||
| 4 | M | 37 | +/U | + | + | + | + | + | Hepatitis, encephalitis | ||||||
| 5 | F | 55 | +/P | + | + | + | |||||||||
| 6 | M | 50 | +/- | DVT, encephalitis | |||||||||||
| 7 | M | 37 | Cyclic, PL | + | + | + | |||||||||
| 8 | F | 47 | +/+ | Cyclic, OG | + | + | + | + | Found to have BD | ||||||
| 9 | F | 37 | +/+ | + | + | +/C | |||||||||
| 10 | M | 37 | + | + | + | + | |||||||||
| 11 | F | 28 | +/- | +/C | + | ||||||||||
| 12 | F | 22 | +/- | Monophasic, PL | + | + | + | + | |||||||
| 13 | F | 40 | +/- | Cyclic, OG | + | + | + | +* | |||||||
| 14 | F | 37 | +/+ | Cyclic, OG | + | ||||||||||
| 15 | M | 43 | Cyclic, PL | ||||||||||||
| 16 | M | 22 | +/- | Cyclic, OG | + | ||||||||||
| 17 | F | 53 | + | +* | Gluteal myositis & sterile abscess | ||||||||||
| Total frequency | 10/4 | 8 | 7/2 | 7 | 7 | 6 | 5/4 | 3 | 3 | 3 | 3 | 2 | |||
*No chronic inflammatory back pain during the course of illness; sacroiliitis confirmed in magnetic resonance imaging. Abd, abdominal; BD, Behcet's disease; C, cervical lymphadenopathy; DVT, deep vein thrombosis; EN, erythema nodosum-like lesions; F, female; GU, genital ulcers; HA, non-organic headache; HSM, hepatosplenomegaly; LAP, lymphadenopathy; M, male; OG, oligoarthritis; OU, oral ulcers; P, pruritic; U, Urticarial; PL, polyarthritis.
The initial therapy included oral corticosteroids to all patients with or without non-steroidal anti-inflammatory drugs (n=8), methotrexate (n=3), and sulfasalazine (n=2). For those with oral or genital ulcers, colchicine (n=6) and/or dapsone (n=5) were added from the beginning. Tacrolimus (n=3), mycophenolate mophetil (n=2), cyclophosphamide (n=1), and azathioprine (n=1) were added to refractory patients. Most patients eventually improved with treatment except for three patients who required biological agents. Of the three, one patient is free of symptom with tocilizumab monotherapy (patient #15), the other patient died of disseminated herpes zoster infection during anti-TNF-α therapy (patient #6) combined with immunosuppressives. The third patient did not respond to anti-TNF therapy and is still on low dose steroid with intermittent symptom attacks (patient #5). No patient was given anti-IL-1 agent.
Scores of 17 patients according to the provisional Eurofever clinical classification criteria
Thirteen patients showed positive scores above cut-off values (Table 2). Eleven were positive for TRAPS and the other two for MKD.
Table 2. Scores according to the provisional Eurofever clinical classification criteria.
| Patient | Scores | Features of other AIDs | |||
|---|---|---|---|---|---|
| FMF Cut off ≥60 |
TRAPS Cut off ≥43 |
CAPS Cut off ≥52 |
MKD Cut off ≥42 |
||
| 1 | 34 | 33 | 25 | 50 | |
| 2 | 24 | 48 | 15 | 24 | Sacroiliitis |
| 3 | 15 | 33 | 0 | 35 | OU, GU |
| 4 | 19 | 54 | 40 | 19 | |
| 5 | 34 | 72 | 15 | 11 | |
| 6 | 25 | 33 | 40 | 22 | OU, DVT |
| 7 | 34 | 66 | 15 | 11 | |
| 8 | 47 | 20 | 40 | 22 | Found to have BD |
| 9 | 15 | 33 | 15 | 35 | OU, GU |
| 10 | 34 | 72 | 40 | 19 | |
| 11 | 15 | 48 | 40 | 43 | |
| 12 | 25 | 66 | 15 | 22 | |
| 13 | 25 | 72 | 15 | 22 | Sacroiliitis |
| 14 | 34 | 48 | 40 | 22 | OU, GU |
| 15 | 43 | 48 | 40 | 11 | |
| 16 | 43 | 29 | 25 | 42 | |
| 17 | 34 | 54 | 40 | 11 | Sacroiliitis |
AIDs, autoinflammatory diseases; BD, Behcet's disease; CAPS, cryopirin-associated periodic syndrome; DVT, deep vein thrombosis; FMF, familial Mediterranean fever; GU, genital ulcer; MKD, mevalonate kinase deficiency; OU, oral ulcer; TRAPS, tumor necrosis factor receptor-associated periodic syndrome.
Genetic features of the patients
Table 3 shows the genotyping results for SNPs in MEFV, NOD2/CARD15, and TNFRSF1A. TaqMan® assay failed to genotype rs37433930 (E148Q) in MEFV for any of the samples. The examined variants (V726A, M694I, M680I, and M694V in MEFV, R702W, G908R, and 3020InsC in NOD2/CARD15, R92Q, T90N, and P46L in TNFRSF1A) were not found in all study subjects (Table 3).
Table 3. Genetic features of 17 patients identified to have recurrent fever and multi-system inflammation without infectious or autoimmune evidence.
| Genes | SNP ID | Coding annotations | Alleles (reference > risk) | Genotyping method | Genotype results* |
|---|---|---|---|---|---|
| MEFV | rs28940579 | V726A | A>G | TaqMan® assay | A/A |
| rs28940578 | M694I | G>A | TaqMan® assay | G/G | |
| rs28940580 | M680I | G > A/C | TaqMan® assay | G/G | |
| rs3743930 | E148Q | G>C | TaqMan® assay | Failed | |
| rs61752717 | M694V | A>G | RFLP | A/A | |
| TNFRSF1A | rs4149584 | R92Q | G>A | TaqMan® assay | G/G |
| rs34751757 | T90N | C>A | TaqMan® assay | C/C | |
| rs4149637 | P46L | C>T | TaqMan® assay | C/C | |
| NOD2/CARD15 | rs2066844 | R702W | C>T | TaqMan® assay | C/C |
| rs2066845 | G908R | G>C | TaqMan® assay | G/G | |
| rs2066847 | 3020InsC | -/C | RFLP | -/- |
*No polymorphism in any of the variants was found.
DISCUSSION
Only a small proportion of patients (0.95%) were identified to have "unclassified" AIDs, showing recurrent fever and inflammation without any infectious or autoimmune evidence. Considering that this study was performed in a tertiary-referral-hospital, the population-based rate is likely to be even rare. Clinical features of these "unclassified" AID patients showed a striking overlap with those of other well-established AIDs such as BD, SpA, or AOSD although they failed to satisfy any particular classification criteria for AIDs. Lack of uniqueness in the clinical features or the wide patient-to-patient variation in symptom combination might be attributed to potential heterogeneity of these patients. There could be several reasons why these patients have remained "unclassified". First, insufficient sensitivity of current classification criteria that do not intend to classify patients with atypical and/or limited expression of a given disease could have failed to identify these patients. Second, one of the noticeable findings in these patients is that clinical features in some patients showed a mixture of two or more well-established AIDs particularly BD, AOSD, or SpA (e.g., patients #2,7.9,12,13,16). Third, certain clinical features of AIDs could be indistinguishable from those of other AIDs, autoimmune diseases, or even viral infection probably due to shared abnormalities in innate immune compartment between these disorders. In this sense, significant part of the symptoms and signs seen in these patients lacked specificity to a certain disease. Forth, viral activation of innate immune system might be important to trigger AIDs in genetically susceptible patients. Frequently observed sore throat and cervical lymphadenopathy might suggest that part of their symptoms (particularly initial symptoms) could be of viral origin. Moreover, it has been shown that clinical phenotype can be variable despite a common genetic abnormality (8). Thus, classification of systemic AIDs based on clinical phenotype seems to have limits and could be misleading.
MEFV, NOD2/CARD15, and TNFRSF1A mutations/polymorphisms have been implicated in more than one AID although they are best known for FMF, CD, or TRAPS, respectively (3,8,9,10,11,12,13,14). The AIDs known to be associated with these genes include BD, SpA, enthesopathy, and PFAPA syndrome, whose clinical features show a striking overlap with those of our patients. Therefore, we attempted to claim a plausible contribution of these genes in our patients but were not able to find the genetic variants in MEFV, NOD2/CARD15, and TNFRSF1A.
MEFV polymorphisms have been investigated in 96 Korean patients with AOSD and 165 healthy controls (22); V726A, M694V, M680I were not found, and E148Q were found 46.7% of controls and 45.8% of patients. No R702W, G908R, or 3020InsC in NOD2/ CARD15 were found in 40 Korean pediatric patients with CD and 30 healthy controls (23). In addition, none of 205 Korean patients with SpA and 200 healthy controls carried R702W, G908R, or 3020InsC except one patient having G908R (24). R92Q in TNFRSF1A is known to be associated with TRAPS and multiple sclerosis but this variant was not found in 178 Korean patients with inflammatory demyelinating disease and in 237 healthy controls (25). These negative results suggest that common variants in MEFV, NOD2/CARD15, and TNFRSF1A seen in Mediterranean populations are rare in Koreans.
The Eurofever criteria have been validated using four genetically proven patient groups with FMF, TRAPS, CAPS, or MKD (19). Although their extended application to genetically uncertain patients with periodic fevers or patients with a chronic disease course than a periodic pattern showed successful performance, their performance in unselected patients with presumable AIDs is still unclear. Furthermore, the populations in the Eurofever registry included only a minority of Asians. Eleven patients were classified as TRAPS and two as MKD according to the Eurofever criteria in our study. However, none of the eleven patients possessed the common genetic variants of TNFRSF1A. Therefore, our finding might suggest that the specificity of the criteria is in fact low in unselected patients with recurrent fever or inflammation. Alternatively, it might suggest that other culprit variants in TNFRSF1A exist in Korean patients with TRAPS. In addition, part of the patients with positive scores for TRAPS had a key clinical finding of the already known AIDs such as SpA or BD. Thus, the Eurofever criteria need to be further validated in Asian populations where the disease associated genetic variants frequently found in Mediterranean region are rare and where other AIDs such as BD are prevalent.
We restricted patients who possessed HLA-B*51 to exclude those who presented partial expression of BD. However, considering frequent oral and genital ulcers in the examined patients, it would have been interesting to estimate the proportion of HLA-B*51 in these unclassified patients and to see their predisposition to BD.
Most patients (88.2%, 15/17) achieved clinical remission with treatment using systemic steroids, NSAIDs, secondary immunosuppressives, and/or biologics. None of our patients including patient #5 suffered amyloidosis, possibly due to effective treatment, adult onset of disease, and relative short follow-up period. However, vigilant monitoring for such potential complications will be mandatory in refractory cases.
Although the present study is limited due to a small sample size, it shows several messages. Inflammatory signs and symptoms of 17 Korean patients with unclassified AIDs often showed a mixture of two or more well-established AIDs. The symptom combination of these patients was widely heterogeneous. Although majority of our patients were classified as TRAPS according to the newly proposed Eurofever clinical classification criteria, no common variants in TNFRSF1A were found in these patients. Thus, a new set of classification criteria should be developed and validated in the context of Asian population.
Footnotes
Funding: This work was supported by the research grant from Seoul National University Bundang Hospital (Grant No. 11-2010-033).
DISCLOSURE: The authors declare that they have no competing conflicts of interest.
AUTHOR CONTRIBUTION: Conception and design: Kang EH. Acquisition of data: Yang JA. Genetic study: Choi JY. Analysis and interpretation of data and manuscript preparation: all authors.
References
- 1.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784–790. doi: 10.1016/j.cell.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, Migdal A, Padeh S, Pras M. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40:1879–1885. doi: 10.1002/art.1780401023. [DOI] [PubMed] [Google Scholar]
- 5.Gattorno M, Sormani MP, D’Osualdo A, Pelagatti MA, Caroli F, Federici S, Cecconi M, Solari N, Meini A, Zulian F, et al. A diagnostic score for molecular analysis of hereditary autoinflammatory syndromes with periodic fever in children. Arthritis Rheum. 2008;58:1823–1832. doi: 10.1002/art.23474. [DOI] [PubMed] [Google Scholar]
- 6.Simon A, van der, Vesely R, Myrdal U, Yoshimura K, Duys P, Drenth JP; Approach to genetic analysis in the diagnosis of hereditary autoinflammatory syndromes. Rheumatology (Oxford) 2006;45:269–273. doi: 10.1093/rheumatology/kei138. [DOI] [PubMed] [Google Scholar]
- 7.Grateau G, Hentgen V, Stojanovic KS, Jéru I, Amselem S, Steichen O. How should we approach classification of autoinflammatory diseases. Nat Rev Rheumatol. 2013;9:624–629. doi: 10.1038/nrrheum.2013.101. [DOI] [PubMed] [Google Scholar]
- 8.Kirino Y, Zhou Q, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, Özyazgan Y, Ugurlu S, Erer B, Abaci N, et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behçet disease. Proc Natl Acad Sci USA. 2013;110:8134–8139. doi: 10.1073/pnas.1306352110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akkoc N, Gul A. Familial Mediterranean fever and seronegative arthritis. Curr Rheumatol Rep. 2011;13:388–394. doi: 10.1007/s11926-011-0191-9. [DOI] [PubMed] [Google Scholar]
- 10.Gülhan B, Akkuş A, Ozçakar L, Beşbaş N, Ozen S. Are MEFV mutations susceptibility factors in enthesitis-related arthritis patients in the eastern Mediterranean. Clin Exp Rheumatol. 2014;32:S160–S164. [PubMed] [Google Scholar]
- 11.Taniuchi S, Nishikomori R, Iharada A, Tuji S, Heike T, Kaneko K. MEFV Variants in Patients with PFAPA Syndrome in Japan. Open Rheumatol J. 2013;7:22–25. doi: 10.2174/1874312901307010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao Q, Zhou L, Cusumano P, Bose N, Piliang M, Jayakar B, Su LC, Shen B. A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis Res Ther. 2011;13:R148. doi: 10.1186/ar3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Jager, PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Migita K, Agematsu K, Yazaki M, Nonaka F, Nakamura A, Toma T, Kishida D, Uehara R, Nakamura Y, Jiuchi Y, et al. Familial Mediterranean fever: genotype-phenotype correlations in Japanese patients. Medicine (Baltimore) 2014;93:158–164. doi: 10.1097/MD.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim AL, Jang HJ, Han JW, Song YK, Song WJ, Woo HJ, Jung YO, Kae SH, Lee J. Familial Mediterranean fever: the first adult case in Korea. J Korean Med Sci. 2012;27:1424–1427. doi: 10.3346/jkms.2012.27.11.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim EH, Park MJ, Park S, Lee ES. Increased expression of the NLRP3 inflammasome components in patients with Behçet’s disease. J Inflamm (Lond) 2015;12:41. doi: 10.1186/s12950-015-0086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Federici L, Rittore-Domingo C, Koné-Paut I, Jorgensen C, Rodière M, Le Quellec A, Touitou I. A decision tree for genetic diagnosis of hereditary periodic fever in unselected patients. Ann Rheum Dis. 2006;65:1427–1432. doi: 10.1136/ard.2006.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Federici S, Sormani MP, Ozen S, Lachmann HJ, Amaryan G, Woo P, Koné-Paut I, Dewarrat N, Cantarini L, Insalaco A, et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015;74:799–805. doi: 10.1136/annrheumdis-2014-206580. [DOI] [PubMed] [Google Scholar]
- 20.Brik R, Shinawi M, Kepten I, Berant M, Gershoni-Baruch R. Familial Mediterranean fever: clinical and genetic characterization in a mixed pediatric population of Jewish and Arab patients. Pediatrics. 1999;103:e70. doi: 10.1542/peds.103.5.e70. [DOI] [PubMed] [Google Scholar]
- 21.Baruch Y, Dagan E, Rosner I, Fiorilli M, Gershoni-Baruch R, Rozenbaum M. MEFV, TNFRSF1A and CARD15 mutation analysis in Behçet’s disease. Clin Exp Rheumatol. 2011;29:S24–S27. [PubMed] [Google Scholar]
- 22.Kim JJ, Kim JK, Shim SC, Choe JY, Kim TH, Jun JB, Yoo DH. MEFV gene mutations and their clinical significance in Korean patients with adult-onset Still’s disease. Clin Exp Rheumatol. 2013;31:60–63. [PubMed] [Google Scholar]
- 23.Jang JY, Song SM, Kim KM, Oh SH, Lee YJ, Rhee KW. Lack of common NOD2 mutations in Korean pediatric patients with inflammatory bowel disease. Pediatr Int. 2010;52:888–889. doi: 10.1111/j.1442-200X.2010.03269.x. [DOI] [PubMed] [Google Scholar]
- 24.Kim TH, Rahman P, Jun JB, Lee HS, Park YW, Im HJ, Snelgrove T, Peddle L, Hallett D, Inman RD. Analysis of CARD15 polymorphisms in Korean patients with ankylosing spondylitis reveals absence of common variants seen in western populations. J Rheumatol. 2004;31:1959–1961. [PubMed] [Google Scholar]
- 25.Park TJ, Kim HJ, Kim JH, Bae JS, Cheong HS, Park BL, Shin HD. Associations of CD6, TNFRSF1A and IRF8 polymorphisms with risk of inflammatory demyelinating diseases. Neuropathol Appl Neurobiol. 2013;39:519–530. doi: 10.1111/j.1365-2990.2012.01304.x. [DOI] [PubMed] [Google Scholar]