

Abstract
We present an approach toward dynamic nanoimaging: live fluorescence of cells encapsulated in a bionanoreactor is complemented with in situ scanning electron microscopy (SEM) on an integrated microscope. This allows us to take SEM snapshots on-demand, that is, at a specific location in time, at a desired region of interest, guided by the dynamic fluorescence imaging. We show that this approach enables direct visualization, with EM resolution, of the distribution of bioconjugated quantum dots on cellular extensions during uptake and internalization.
Keywords: scanning electron microscopy, live cell imaging, fluorescence microscopy, quantum dots, epidermal growth factor
Graphical abstract
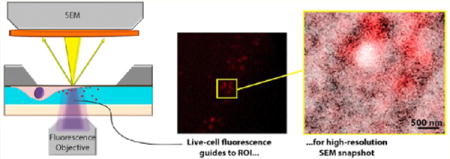
Structural images can be obtained at nanometer resolution with electron microscopy (EM). Also, designated nanoreactors allow structural dynamics to be followed with EM under atmospheric conditions or even in liquid.1–8 However, the invasive character of the electron irradiation precludes this dynamic EM on soft bio- and nanomaterials, such as live cells. Nevertheless, the nanostructural context and high-resolution localization of biomolecules within this context is crucial for understanding function and malfunction in cellular and biomolecular processes.
Fluorescence microscopy (FM) is the key technology for live imaging of dynamics of molecules in nanostructured environments, but it lacks the capability to image underlying structural context and also falls short in reaching the required molecular resolution. Super-resolution techniques improve FM resolution by an order of magnitude, but often compromise temporal resolution, for example, due to sequential switching and recording of fluorescent proteins.9–12 Also, super-resolution FM does not image the underlying, unlabeled ultrastructural context as can be observed with EM, even on cells in liquid.1–3,5,6 We bridge this gap by physically integrating13–16 live-cell fluorescence microscopy into a scanning electron microscope, where we use FM observations of cells encapsulated in a bionanoreactor to determine both region and time of interest for high-resolution scanning electron microscopy (SEM) snapshots.
We use the presented method to study the internalization and cellular distribution of epidermal growth factor (EGF) in filopodia of fibroblasts possessing EGF receptors (EGFR). EGF is known to trigger several signaling cascades responsible for cellular motility, DNA replication, and cell proliferation.17 EGF binding the EGFR leads to dimerization and activation of the EGFR.18 EGFR then undergo retrograde transport on cell edge protrusions via the F-actin cytoskeleton.19 Activated EGFR are internalized through clathrin-dependent endocytosis, and they are either sorted into the recycling endosomes and returned to the plasma membrane, or centripetally transported from peripheral early endosomes to perinuclear multivesicular bodies (MVBs)/late endosomes.20 EGFR that do not recycle follow the degradative trafficking pathway and are ultimately dumped into lysosomes. Microtubules are associated with the regulation of EGFR internalization, intracellular trafficking and targeting to lysosomes.21 EGFR-mediated internalization and transport in filopodia has been visualized with FM using EGF-conjugated quantum dots (QDOTs).19 For FM, concentrations are typically low to allow tracing single complexes (see also the Supporting Information and Figure S1). However, this precludes observation of the full distribution of internalizing EGF-QDOT’s and their precise location on the filopodia. QDOT’s are well suited for prolonged dynamic imaging due to their brightness and photostability,22 while their electron-dense core allows visibility in EM.23,24
We show that our method enables direct visualization, with EM resolution, of the distribution of EGF-conjugated quantum dots on cellular extensions during uptake and internalization. The SEM snapshots also reveal positions of quantum dot complexes on the cellular transport network including temporary arrested positions close to structural bottlenecks such as track junctions and lipid inclusions.
RESULTS
Bionanoreactor Design and On-Demand Electron Microscopy
Our bionanoreactor design allowing integrated FM and SEM inspection is described in Figure 1a, b. The main concept behind this design is to monitor live-cell dynamics with FM in situ in the SEM after fluorescent labeled reagents have been added to a cell culture medium. Electron-beam exposure can now be delayed until reaction sites have been identified. Thus, there is no need for prior electron exposure that may disturb reaction kinetics or adversely affect cellular viability before the process of interest has started. The SEM image is then directed only at the site of interest identified in FM, thus embedding the fluorescence dynamics in a structural context with 30-fold increase in resolution. Using fluorescent nanoparticle labels allows identification of biomolecular positions within this structural context.
Figure 1.
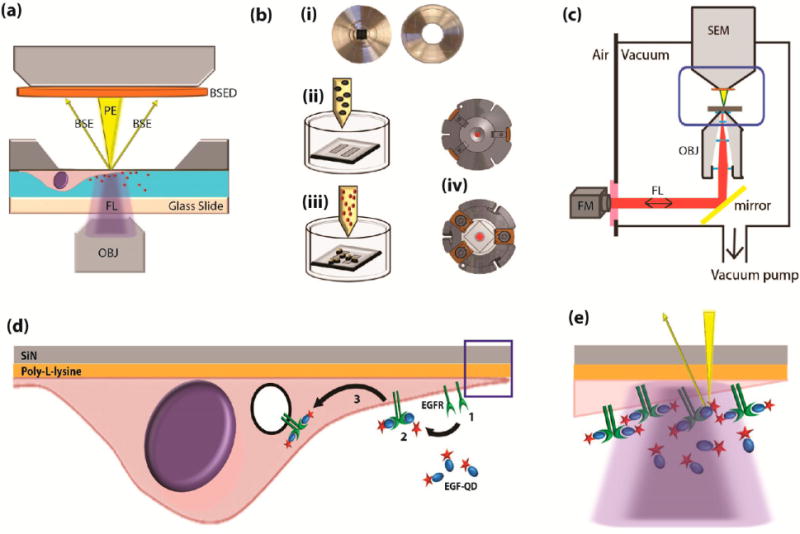
Integrated fluorescence and scanning electron microscopy with live-cell bionanoreactor (a) Schematic of a bionanoreactor for keeping live cells in an integrated fluorescence and scanning electron microscope. Cells are kept in liquid separated from the SEM vacuum by an electron-transparent silicon-supported SiN membrane (dark gray) and a light-transparent glass slide. OBJ, objective lens; FL, fluorescence excitation and emission; PE, primary electron beam; BSE, backscattered electrons; BSED, BSE detector. (b) Procedure for culturing cells and mounting the bionanoreactor: (i) The silicon microchip and the glass slide are glued to separate aluminum plates; (ii) the plate containing the microchip is coated with a thin layer of poly-L-lysine, placed in a Petri-dish and cell culture medium is added; (iii) after cells adhered to the membranes, reagent (EGF-conjugated quantum dots) are added; (iv) the two metal plates are then clamped together in a vacuum-tight holder. (c) Schematic of the integrated fluorescence and scanning electron microscope, with the holder placed in between both microscopes. Blue boxed area corresponds to (a). FM, fluorescence microscope; SEM, scanning electron microscope. (d) Illustration of EGF-mediated uptake of quantum dots (QDOT) in a cell cultured on the SiN membrane. (1) EGF receptors (EGFR) are present on the cell membrane. (2) EGFR dimerize upon binding the EGF-QDOT. (3) The EGFR are taken up and further transported in endosomes within the cell. (e) Particularly high concentration of EGFR occur at tips of cellular extensions (boxed area from (d)), within the light diffraction limit (purple), but discernible with electron microscope resolution (yellow, not to scale).
The bionanoreactor is designed for use with one of the recently presented integrated light and electron microscopes.13–16 Our methodology to take SEM snapshots guided by live-cell FM is equally applicable to other systems,14,15 but may require slight changes in reactor or experimental design. Our bionanoreactor consists of two parts, one of which contains the (upper) silicon nitride (SiN) membrane that serves as electron transparent window for SEM and onto which cells are cultured (Figure 1a). The distance between this SiN membrane and the lower glass plate, a standard light microscopy slide, is chosen such (0.2 mm) that FM can be carried out with a high-NA objective lens on dynamics taking place at the SiN-water interface. Glass slide and SiN membrane are attached on distinct metal platelets (Figure 1b), so that the SiN membrane can be placed in a Petri-dish for cell culture. After successful culture and identification of active cells on the membrane window area, the two plates are clamped together in a leak-free holder (Figure 1b) with 70 μL of buffer solution. Specific fluorescent labeled reagents can be added to this liquid medium prior to closing. The holder is then positioned in the integrated FM-SEM microscope (Figure 1c). Here, FM-SEM axial alignment is within 5 μm13 so that the SEM imaging area can be selected anywhere within the FM field of view with ~100 nm registration accuracy.
To monitor cellular uptake mediated by epidermal growth factor (EGF) and EGF receptors (EGFR) on the cell membrane, we used streptavidin-conjugated quantum dots (QDOTs) complexed to biotinylated EGF (EGF-QDOT) (Figure 1d). This system has been previously used for FM demonstration of EGFR transport in cellular extensions and filopodia,19 typically at low EGF-QDOT concentrations to allow tracing single complexes (see also the Supporting Information and Figure S1). Due to their brightness and photostability, QDOTs are well suited for prolonged dynamic imaging,22 while the electron-dense core also allows visibility in EM.23,24 Here, we use high concentrations of EGF-QDOTs to show that our SEM snapshots image the full distribution of internalizing EGF-QDOT complexes including the particularly dense accumulation at the filopodia tip (Figure 1e).
Intracellular Quantum Dot Transport and Structural Bottlenecks
Figure 2a shows an overlay of reflection image and the fluorescent image of the cells recorded in the integrated microscope, after the addition of EGF-QDOTs. The positioning of the cells on the silicon nitride membrane and the distribution of QDOT clusters are clearly visible. Live-cell dynamics are monitored with the fluorescence microscope to identify regions active in EGF-QDOT uptake and transport (see Supporting Information Video S1). Figure 2b shows a time-lapse series of such an active transport region, also depicted with a white rectangle in Figure 2a (see also Supporting Information Video S2). At some locations, clusters of QDOTs appear temporarily trapped (denoted with an asterisk), whereas at other regions clusters continuously move along cellular structure (see also Supporting Information Video S2). We use these dynamic observations to select this region for a SEM snapshot, which is presented in Figure 2c. Cellular structure and the cortex are clearly visible. Outside the area covered by the cell, large clusters of QDOTs attach to the bare window. The bright white spot visible within the cellular structure probably represents a lipid droplet or lipid rich domain on the cell membrane.2 Individual QDOTs cannot be identified at this magnification, but inside the cell darker tracks can be seen reminiscent of the trajectories followed by internalized EGF-QDOT conjugates. Figure 2d and e shows enlarged images of the regions depicted with blue and green rectangles in Figure 2c, respectively. Here, single EGF-QDOTs as well as clusters of particles are clearly visible as dark spots on the tracks identified in Figure 2c. To confirm that the single dark spots correspond to a single EGF-QDOT complex, intensity profiles were taken over the two spots indicated in Figure 2d and 2e respectively. The full width half-maximum (fwhm) of these profiles are 22 and 23 nm, respectively (Figure 2f). Considering the electron beam broadening25 in the SiN membrane, the poly L-lysine layer, and the cell membrane, these values are consistent with the producer mentioned size for the QDOT particles. The calculated 25%–75% edge resolution values for 10 particles using both left and right side of the line profiles vary between 7 and 8.5 nm. This variation may well result from the presence of QDOTs at different depths inside the cell which alters the effective beam broadening per QDOT. These values constitute an ~30-fold increase in resolution compared to wide-field FM, as also highlighted in the overlay image in Figure 2g.
Figure 2.
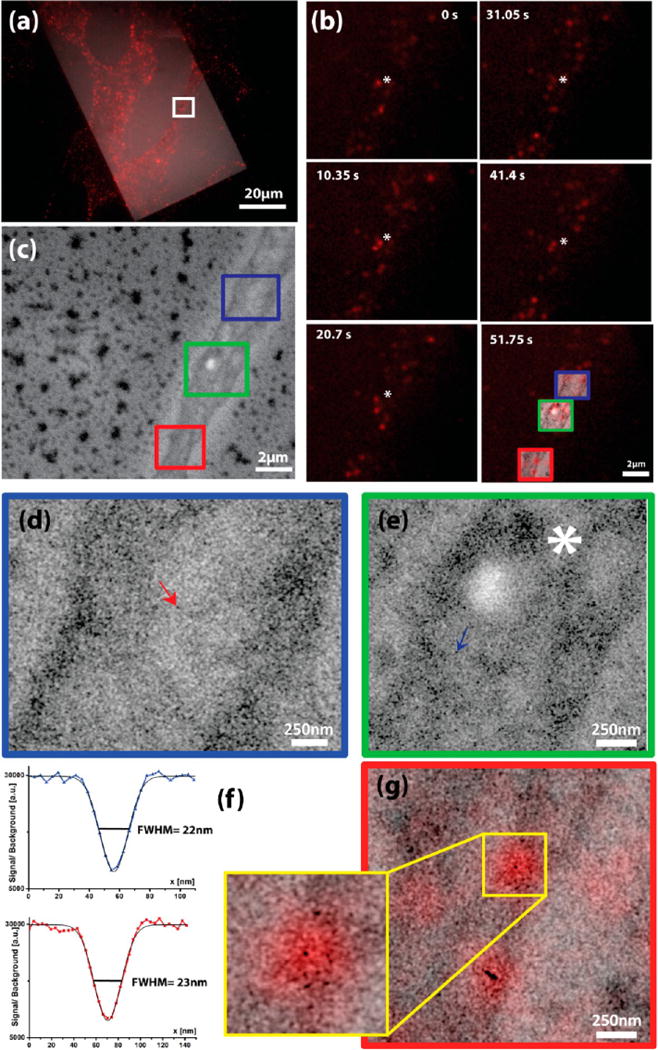
Live fluorescence microscopy identifies a region of interest for high-resolution SEM (a) Overlay of reflection and fluorescence images of CV1 cells on SiN membranes labeled with EGF-QDOTs (b) Time-lapse images of the white rectangular area in (a), which shows highly dynamic EGF-QDOT transport in the live-cell FM (see also Supporting Information Video S2). Some EGF-QDOT signals, like the one denoted with an asterisk, appear quasi-static while others are rather dynamic. The last panel shows the overlay of the higher magnification SEM images with the fluorescent image of the same regions. (c) SEM snapshot of the area depicted in (a) and (b). (d) Higher magnification image of the blue boxed area in (c). (e) Higher magnification image of the green boxed area in (c). A higher concentration of QDOT clusters is visible around the cellular cortex and at crossings of QDOT tracks. (f) Line profiles of the two EGF-QDOT complexes indicated with arrows in (d) and (e), with fwhm of 22 and 23 nm, respectively. (g) FM-SEM overlay of the red boxed area in (c) highlighting resolution difference between QDOT in FM (red) and EM contrast (black).
In the last panel in Figure 2b, the higher magnification SEM images are overlaid to the last frame of the fluorescence dynamics. Again, the FM-SEM resolution gap is clearly observable. The fluorescence signal is seen to mainly originate from dense clusters of QDOTs visualized in the SEM images. There is particular overlap between the fluorescence and SEM images for (quasi-)static clusters of QDOTs (Figure 2e). The localization of the (quasi-)static QDOT clusters clearly coincides with structural components such as the intersections between trajectories and the lipid droplet indicated with an asterisk (Figure 2d, e, and g). We note that the number of QDOTs visible in the SEM images is certainly higher than the amount that can be extrapolated from the fluorescence images. This suggests either a lot of QDOT particles are in a nonfluorescent state or their fluorescence is too low to be detected in the current (wide-field FM) setup. It should be considered that highly dynamic regions appear weaker in fluorescence than the static regions, as the QDOTS are likely being transported individually at the dynamic regions, whereas an accumulation of QDOTS will cause bright intensities at the static regions. We note that it is an important benefit that both fluorescent and nonfluorescent QDOTs are visualized, as each QDOT depicts the position of an internalized EGF(R) molecule.
Our SEM results are consistent with stand-alone confocal fluorescence observations about the directional transport of EGF-QDOT conjugates after their internalization (Supporting Information and Figure S1). The tracks followed by internalized QDOTs can be clearly distinguished, and as an example Figure 2g shows a magnification of the red boxed region in the last panel in Figure 2b, with fluorescence and SEM overlaid. A crossing of tracks is clearly observed in the SEM image, colocalizing with bright QDOT fluorescence. Again, a higher density of QDOTs is visible around this crossing point and also at the flattened extension around the cortex. These regions also predominantly colocalize with the strong and (quasi-)static fluorescence spots, suggesting an accumulation and slower transport of QDOT clusters around these structural “bottlenecks”.
Imaging Active Uptake Regions and EGFR Distributions on Cellular Extentions
Besides relating FM recorded transport dynamics to the cellular structure, our approach also allows direct inspection of the regions active in EGF-QDOT uptake around the cell boundary. The dynamics of EGF-QDOTs observed with live-cell fluorescence imaging provides a means to identify the active regions for EGF-QDOT uptake (see Supporting Information Video S1). One such uptake region is indicated with a square in Figure 3a and a time-lapse series of this region is shown in Figure 3b (see also Supporting Information Video S3 for the full movie). The bright fluorescent spot in the upper right part of the figure, indicated with box 1 in the last panel of Figure 3b, shows sequential events of attachment of QDOTs from the upper part, and detachment into the cell interior at the lower end. Similar dynamics, but with less overall fluorescence is observed in the area marked with square 2. The last panel of the timelapse series shows an overlay of FM image with the corresponding SEM image, taken just following the fluorescent imaging. Contours of the cell and intracellular structures, like lipid droplets and internal tracks, are readily visible in the SEM image. The FM-identified active uptake regions labeled as 1 and 2 as mentioned above (see also Supporting Information Video S3), are found to directly colocalize with filopodial structures in the SEM images. Higher magnification images of these regions are represented in Figure 3c and d, respectively. In correspondence with FM observations, a high density of QDOT clusters is visible along the filopodia in Figure 3c and d, especially at the tip of the filopodial structure visible in Figure 3d. The docking of EGF-QDOT conjugates at the cell contour is also visible in Figure 3d and is consistent with previous observations that the conjugates wait at the microtubule frontier before being further transported into the cellular matrix (Supporting Information).
Figure 3.
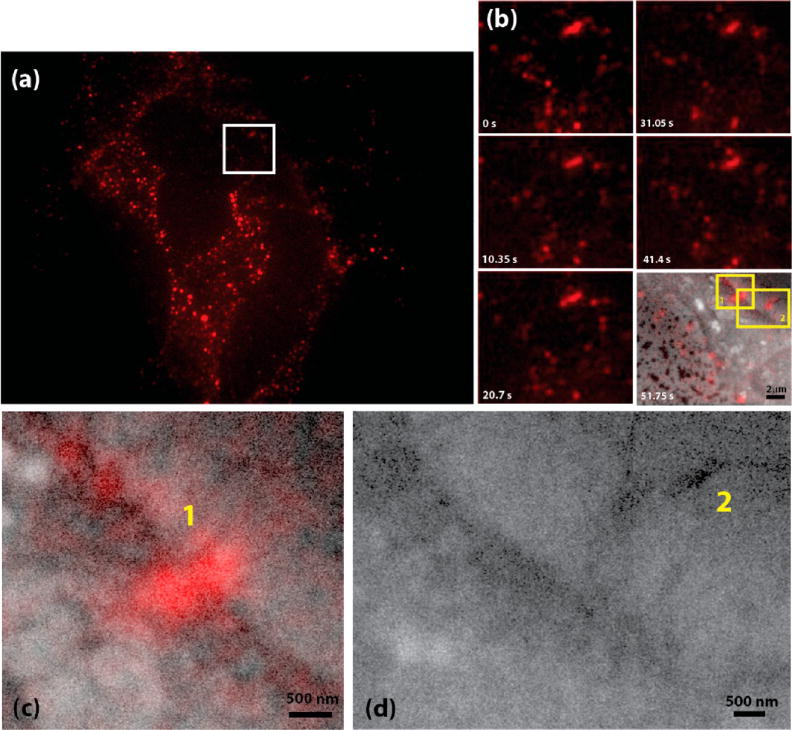
SEM snapshots of EGF-QDOT uptake. (a) Fluorescence image of CV1 cells on SiN membranes labeled with EGF-QDOTs. Live cell fluorescence microscopy observations highlight some regions as active sites for EGF(R) uptake. (b) Time-lapse series of the, white boxed area in (a); see also Supporting Information Video S3 imaged in the bionanoreactor in the integrated microscope with FM. Numbered regions appeared as highly dynamic uptake regions in FM observations, definig the region of interest for a SEM snapshot. The last panel of the time-lapse series shows the overlay of the higher magnification SEM images with the fluorescent image of the same regions. (c) Higher magnification overlay of fluorescent and SEM images of the area in the timelapse series depicted as 1. Similarly, panel (d) shows an overlay of fluorescent and SEM images of the labeled as 2. EGF-QDOT conjugates are visible as dark spots and localize especially around the plasma membrane and around filopodial structures.
Live cell FM observations enable us to pinpoint several of these active uptake regions. Another highly active uptake region, at a longer cellular extension, is presented in Figure 4a. Here, we can identify a so-called docking region, where EGF-QDOTs accumulate for further transport into the cell interior, and a tip region. In the FM observations (see also Supporting Information Video S1), we observe QDOTs attaching and transporting to the bright fluorescent docking region. Both docking and tip region are indicated in the image. At this position, we record a SEM snapshot, shown in Figure 4b directly overlaid with the FM image. Three regions in Figure 4b, marked as c–e are represented at higher magnification in Figure 4c–e, respectively. The images shown in Figure 4c and d also show the QDOTs present along the filopodia, especially at the tip region. The region represented in Figure 4e, which is the tip of the cellular extension, contains a high density of QDOTs indicating the tip as an active uptake site for EGF-QDOT conjugates, with particularly high local concentration of EGFR. This is also consistent with our standalone confocal fluorescence observations about the uptake of EGF-QDOT conjugates through cellular extensions and filopodial structures (Supporting Information and Figure S1).
Figure 4.

SEM reveals uptake and EGFR accumulation at filopodia. (a) Magnified fluorescence image showing an EGF-QDOT docking region with uptake at an extension tip in the live-cell observations. (b) Overlay of fluorescence and corresponding SEM snapshot of the area depicted in (a). Regions shown with white squares appeared as highly active uptake regions in FM dynamic observations. (c–e) SEM images of the regions in (b). Filopodial structures are present in (c) and (d), and especially the tip of the cellular extension in (e) shows a dense accumulation of EGF-QDOTs, indicating the tip as a highly active uptake site with dense EGF(R) accumulation.
DISCUSSION
The above results demonstrate SEM imaging of individual biomarkers in connection to cellular structure during a live process. A single snapshot reveals QDOT distributions within the light optical diffraction limit, highlighting filopodial and especially cellular extension tips as active sites for EGF(R) accumulation. Within the cellular transport process, we see arrested movement and increased concentrations of EGF-QDOT complexes at structural bottlenecks like junctions and lipid drops. This is in correspondence with previous FM observations but with visualization of higher label densities. The resolution achieved in the presented data is estimated to be ~7.5 nm, about 30 times higher than confocal fluorescence microscopy. Superresolution light microscopy can reach similar resolution, but live cell imaging is usually only possible at the expense of resolution. Additionally, this method does not require data postprocessing, and the SEM image adds cellular structural detail like cellular boundary, protrusions, and lipid-rich regions to the FM images without additional labeling.
The SEM resolution depends on the broadening of the electron beam in the silicon nitride and poly-L-lysine layers, and thus for optimal resolution, the sample should be placed directly beneath the membrane.26 Because of the limited electron penetration depth, the SEM image naturally sections the upper, adhesion part of the cell. Our images were obtained at an electron energy of 14 keV, which we found optimal for observing cellular structure. Higher electron energies might give the possibility to image nanoparticles at slightly larger depth, but this remains to be investigated. In general, cellular processes like adhesion, motility, cell to cell connections, and cellular signaling are well-suited to study with this methodology, but deep intracellular or nuclear processes cannot be targeted. Scanning transmission electron microscopy (STEM), operated typically at 100–300 keV, has been shown to image intracellular nanoparticles with nanometer resolution throughout the entire volume of the sample holder.27 However, integrated in situ high-resolution FM in STEM has so far not been reported and may be technically hard to achieve.
The SEM signal can be further improved by using a thinner SiN membrane, but the strength and rigidity of SiN membranes should be assured, as they seal the liquid sample in the high vacuum SEM chamber. Even though we have presented the use of 20 nm SiN membranes previously,26 we chose to work with 30 nm SiN membranes as they provide a good compromise between strength and resolution. The expected broadening of the primary beam after passing through a 30 nm membrane is less than 2 nm. This resolution would be satisfactory for most biological imaging applications. Another option would be to use other materials like recently reported graphene,28 but the quenching of fluorescence by graphene should be considered.29 We would like to note that the ideal landing energy for imaging through membranes of different materials and different thicknesses would be different, and should be optimized for each condition. Additionally, signal can be further improved using in-lens positioned and/or other BSE detectors with higher collection efficiency.
Sample preparation for the approach presented here is similar to standard sample preparation for FM without the use of cryo- or chemical fixation and heavy metal staining commonly applied for EM. Thus, nanoscale-resolution EM visualization is restricted to electron-dense labels, but the EM images of the unlabeled structure reveal detail beyond what is observable with nonlabeling light optical approaches such as phase contrast microscopy. Structural elements like actin, microtubules could be visualized by adding multiple labels in diffraction-limited FM, which could aid correlative recognition in the SEM snapshot. Superresolution microscopy can overcome diffraction, however it is still technically challenging to do multicolor live cell imaging, and to obtain the data in a single snapshot as with our FM-guided SEM. As EM label, besides QDOTs, for example, FluoroNanoGold30 or second harmonic nanoprobes,31 and recent photoconvertible probes or metal-tagging proteins32–35 could be used. In the latter case, it is particularly interesting to note that live cells could be exposed to low concentrations of gold salt for tagging metallothionein without signs of morphological or viability changes for up to 1 h.35 Although temporal correlation between FM and EM images would be lost, the possibility to maintain the region of interest in view and under FM monitoring throughout the EM labeling procedure could be an important benefit.
As an imaging buffer, we chose to work with Tyrode’s buffer after trying distilled, demineralized waters and phosphate buffered saline (PBS) solutions. For live cell imaging, it is essential that cells are maintained as closely as possible to physiological pH, oxygen, temperature, and so forth. The media should be also optically transparent for fluorescence imaging. On the other hand, for SEM imaging of samples in liquid, it is important that the imaging medium is conductive enough to avoid charging effects on the sample, and has a backscattering coefficient close to that of water. It was reported that saline solutions can have backscattering coefficients 10% higher than water alone,36 which would generate a high background signal during imaging. We also observed a high background with PBS. Also Good’s buffers reported to form radical species upon photon or electron radiation, like HEPES,37 were avoided, and Tyrode’s buffer was chosen as the optimal imaging buffer as also used by others.38
Even though present implementation is with wide-field fluorescence microscopy, incorporation of confocal optical sectioning could improve imaging quality in the upper part of the sample. With a wide field setup, the signal from the QDOTs on the membrane, which are observable with SEM, can be overwhelmed by the lower positioned QDOTs (see, e.g., Figure 4a). Therefore, a confocal FM setup may lead to improved correlation between FM and SEM signals. Optical resolution can be further improved by using water immersion lenses and vacuum-compatible water-type immersion oil bringing the maximum NA from 0.95 up to ~1.3.
Radiation damage is critical for electron microscopy of cells. We use FM to monitor and select regions of interest limiting electron-beam exposure to the recorded SEM snapshots. Therefore, damage occurring after exposure would be more of a concern if repetitive SEM imaging is aimed at the same region. The typical electron dose used for the images in this study was ~103 e−/nm2, and we did not observe morphological changes in the cells with this dose. However, dissociation of the cells from the SiN membrane was observed after recording a series of consecutive images from the same region (see Supporting Information Video S4). On the other hand, the possibility of repeated imaging of the same area in liquid STEM imaging of fixed cells without structural damage was previously reported for electron doses which are 2 magnitudes higher than that used in this study.39 Radiation damage of biological samples in EM has mostly been addressed for fixed, dehydrated samples in conventional or environmental EM.40,41 For samples in liquid, charge accumulation is prevented by grounding through the conductive fluid, but radical species and aqueous electrons are generated in water by irradiation with electron energy higher than 10 eV, the bond energy of a valence electron in water.42,43 Thus, damage to the sample will largely be a secondary effect due to the radical species generated in the fluid. In situ FM during electron exposure can provide a novel means to quantitatively study electron-beam induced radiation damage in cells, e.g., by monitoring cell viability through fluorescence reporters. Ultimately, the effects of radiation damage could also be further minimized by using scanning strategies that keep the accumulation of radical species below a critical level.
CONCLUSIONS
We have presented a novel approach to nanoimaging, bridging the gap between dynamic FM and high resolution ultrastructural EM by imaging the cells in their near-native, liquid environment, encapsulated in a bionanoreactor, placed in an integrated FM-SEM. Fluorescence microscopy observations of cellular dynamics allow us to determine both region and time of interest for a single high-resolution SEM snapshot of the region of interest. Our results show how integration of live FM into EM complements the dynamic view on labeled molecules with a snapshot that provides structural context and single label positions at ~30-fold increased resolution. FM-guided EM delays electron exposure until taking the snapshot, directed only at a region of interest. This enables us to directly visualize the dense accumulation of Epidermal Growth Factor (EGF) Receptors at filopodia and especially tips of cellular extensions during uptake of EGF-conjugated quantum dots. Intracellular SEM snapshots reveal positions of EGF-quantum dot complexes on the transport network including temporary arrested positions close to structural bottlenecks such as track junctions and lipid inclusions. This approach may not only open entirely novel perspectives for imaging biological dynamics, but could also be useful for studying other processes typically taking place in liquid, such as in soft matter science, nanoparticle synthesis and (self-)assembly, and (photo-)catalysis.
MATERIALS AND METHODS
SCLEM Setup
The experiments were performed on a home-built integrated high NA light-electron microscope where an inverted epifluorescence microscope is integrated into a commercial SEM (FEI Quanta FEG 200). The integrated microscope is explained in detail elsewhere.13 The setup is schematically indicated in Figure 1c. In brief, the field of view of the inverted FM coincides with the SEM field of view. The optical axes of the FM and the SEM are aligned to within 5 μm in the integrated microscope by translating the FM objective lens below the sample and electron beam column.13 The ~5 μm axial alignment ensures that the e-beam can be positioned anywhere within the FM field of view. The sample and objective stages are mounted on a vacuum door for the SEM which allows retrofitting of the FM into the commercial SEM system. Light is guided in and out of the vacuum chamber through a window and all FM components except for the objective lens and a mirror are placed outside of the vacuum on a breadboard attached to the door. The alignment between FM and SEM was further improved by using electron beam deflectors. A corner of one SiN membrane was brought in the center of FM field of view and the electron beam was finely deflected to bring the corner also in the center of SEM field of view. In this way, an overlay accuracy of sub-100 nm was achieved between the FM and the SEM images.
For SEM imaging of the CV1 cells with QDOTs, the SEM was operated under high vacuum with a 14 keV primary beam energy and a working distance of 8 mm. The images were recorded with 4096 × 3775 pixels with a pixel dwell time of 100 μs.
For the fluorescence imaging of QDOT uptake in CV1 cell, the integrated microscope was equipped with a Nikon CFI PLAN APO, 40×, NA 0.95 dry objective lens and a Nikon 1.5× tube lens. A 470 nm Thorlabs LED source was used for illumination. A dichroic mirror (Semrock FF506-Di03), which has an edge wavelength of 506 nm, separates excitation and emission light. The detection path consisted of a 655 nm bandpass filter (Semrock 655/40 nm BrightLine) and a CCD camera (Andor Clara). Images were recorded using μ-Manager1.2.
Preparation of SiN Membranes and the Bionanoreactor
The chips were manufactured by depositing low stress silicon nitride (30 nm) on silicon wafers (300 μm) by low-pressure chemical vapor deposition. Two windows (300 × 100 μm) were opened in each chip by anisotropic etching in a KOH heat bath. The wafers were then coated with photoresists and diced. To remove the chips from the wafers, they were first kept in 50 °C acetone for approximately 15 min. The chips were then washed in 100% ethanol and finally rinsed with ddH2O. They were cleaned with RCA1 (125 mL of ddH2O; 50 mL of H2O2; 25 mL of NH4; 15 min) and rinsed with ddH2O. Prior to seeding, the chips were plasma cleaned for 15 min (250 W) and coated with poly-L-lysine.
The BNR was designed in order to contain samples immersed in liquid, leak-free in the SEM vacuum chamber. The basic scheme for a holder for liquid samples in simultaneous CLEM is given in Figure 1a and b. To facilitate simultaneous correlative light and electron microscopy, the holder has both an electron transparent and a light transparent window. The electron transparent window, a 40 nm thick SiN membrane, is placed at the top. The light transparent window is placed at the bottom side and consists of glass to enable high-NA microscopy using immersion oils. For ease of use and to allow cell culturing, we chose to have the two windows on two separate metal plates that are clamped together in a leak-free holder. The top and bottom view of the designed holder is presented in Figure 1b. The glass slide and the microchips (μ-chip) bearing the SiN membranes are each bonded to a distinct metal plate. For our application, we chose to work with μ-chips bearing two 100 × 300 μm2 membranes per chip. The two metal plates, housing the μ-chip and the glass slide respectively, and the clamping holder are all constructed from stainless steel (1.4401/316) to ensure biocompatibility of the holder. The bottom plate comprises a notch for a 170 μm (no. 1.5 light microscopy standard) glass slide to be bonded in. It also has a hole with a diameter of 10 mm in the center to bring the light objective lens in close proximity for high NA low working distance imaging. The top plate has two designated grooves of which one hosts the fabricated μ-chip and the glue to bond the μ-chip. The other groove is for an O-ring, which is used for leak-free sealing (Figure 2c). A total volume of 10−7 m3 (100 μL) is left free in between the μchips and the glass slides, when mounted together in the clamping holder. The holder can be easily mounted on the piezo-stage of the integrated microscope.
Culturing CV1 Cells
Monkey kidney fibroblast (CV1) cell line (Cell Lines Service, Germany), was maintained in DMEM supplemented with 10% fetal bovine serum, penicillin/streptomycin, and 2 mM glutamine at 37° under 5% CO2. The cells were washed with phosphate buffered saline (PBS) (pH 7.4), trypsinized with 1× TrypLE Express (Invitrogen), and then seeded onto sterile, poly-L-lysine coated, 50 nm thick Si3N4 membranes, and they were grown overnight. Prior to quantum dot (QDOT) labeling, the cells were incubated in serum free medium for 4 h at 37° under 5% CO2.
EGF-QDOT Labeling of CV1 Cells
For QDOT-EGF labeling, EGF-QDOT conjugates were formed by incubating EGF-biotin (Invitrogen) with streptavidin-QDOT655 (Invitrogen) for 2 h at room temperature in 50 mM borate buffer, pH 8.3. A microcentrifuge purification column (Ultracel-100YM, Millipore) was used to remove unbound EGF-biotin. Then the cells were incubated with 5 nM EGF-QDOT655 in Tyrode’s buffer (Sigma), supplemented with 0.1% BSA (Sigma) and 50 mM D-glucose (Sigma-Aldrich) for 5 min at room temperature.38 After the staining, the cells were washed three times. Then 70 μL of previously stated Tyrode’s buffer was added in the enclosure, and the sample was imaged in liquid in the integrated microscope.
Supplementary Material
Acknowledgments
We would like to thank A.C. Zonnevylle and DELMIC BV for assistance and providing a prototype of their liquid-sample holder, and B. Giepmans for discussions on the manuscript. This work was partly carried out within the Microscopy Valley research program funded by STW – Perspectief voor de Topsectoren. DSBvO acknowledges support from NanoNextNL, a consortium of the Dutch government and 130 partners.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b03970.
Imaging the uptake and transport of EGF bound QDOTs in a stand alone confocal fluorescence and differential interference contrast (DIC) microscope, and supplemental image presenting imaging stages of EGF-QDOT uptake and transport (PDF)
Fluorescent imaging of fibroblasts in liquid enclosure: the whole membrane area (AVI)
Fluorescent imaging of fibroblasts in liquid enclosure: the selected region in Figure 2, also shown in the time-lapse images (AVI)
Fluorescent imaging of fibroblasts in liquid enclosure: the selected region in Figure 3, also shown in the time-lapse images (AVI)
SEM video showing the detachment of cells after multiple repeated exposures with the electron beam indicating the need for a single FM-guided snapshot (AVI)
Author Contributions
J.P.H. and P.K. conceived and initiated research; N.L., D.W.P., and J.P.H. designed experiments; N.L., D.S.B.v.O.S. and J.P.B. performed experiments; N.L., D.S.B.v.O.S., J.P.B., and J.P.H. analyzed data; N.L and J.P.H. wrote the manuscript with input from J.P.B, P.K., and D.W.P.
Notes
The authors declare the following competing financial interest(s): The integrated microscope and the liquid-sample holder served as prototypes for commercial products by Delmic BV. P.K. and J.P.H. are shareholders in Delmic BV.
References
- 1.De Jonge N, Ross FM. Electron Microscopy of Specimens in Liquid. Nat Nanotechnol. 2011;6:695–704. doi: 10.1038/nnano.2011.161. [DOI] [PubMed] [Google Scholar]
- 2.Thiberge S, Nechushtan A, Sprinzak D, Gileadi O, Behar V, Zik O, Chowers Y, Michaeli S, Schlessinger J, Moses E. Scanning Electron Microscopy of Cells and Tissues under Fully Hydrated Conditions. Proc Natl Acad Sci U S A. 2004;101:3346–3351. doi: 10.1073/pnas.0400088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Jonge N, Peckys DB, Kremers GJ, Piston DW. Electron Microscopy of Whole Cells in Liquid with Nanometer Resolution. Proc Natl Acad Sci U S A. 2009;106:2159–2164. doi: 10.1073/pnas.0809567106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng H, Smith RK, Jun Y-W, Kisielowski C, Dahmen U, Alivisatos AP. Observation of Single Colloidal Platinum Nanocrystal Growth Trajectories. Science. 2009;324:1309–1312. doi: 10.1126/science.1172104. [DOI] [PubMed] [Google Scholar]
- 5.Nishiyama H, Suga M, Ogura T, Maruyama Y, Koizumi M, Mio K, Kitamura S, Sato C. Atmospheric Scanning Electron Microscope Observes Cells and Tissues in Open Medium through Silicon Nitride Film. J Struct Biol. 2010;169:438–449. doi: 10.1016/j.jsb.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Klein KL, Anderson IM, de Jonge N. Transmission Electron Microscopy with a Liquid Flow Cell. J Microsc. 2011;242:117–123. doi: 10.1111/j.1365-2818.2010.03484.x. [DOI] [PubMed] [Google Scholar]
- 7.Yuk JM, Park J, Ercius P, Kim K, Hellebusch DJ, Crommie MF, Lee JY, Zettl a, Alivisatos aP. High-Resolution EM of Colloidal Nanocrystal Growth Using Graphene Liquid Cells. Science. 2012;336:61–64. doi: 10.1126/science.1217654. [DOI] [PubMed] [Google Scholar]
- 8.Park J, Zheng H, Lee WC, Geissler PL, Rabani E, Alivisatos AP. Direct Observation of Nanoparticle Superlattice Formation by Using Liquid Cell Transmission Electron Microscopy. ACS Nano. 2012;6:2078–2085. doi: 10.1021/nn203837m. [DOI] [PubMed] [Google Scholar]
- 9.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 10.Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-Color Super-resolution Imaging of Genetically Expressed Probes within Individual Adhesion Complexes. Proc Natl Acad Sci U S A. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hell SW. Toward Fluorescence Nanoscopy. Nat Biotechnol. 2003;21:1347–1355. doi: 10.1038/nbt895. [DOI] [PubMed] [Google Scholar]
- 12.Rust MJ, Bates M, Zhuang X. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zonnevylle AC, van Tol RFC, Liv N, Narvaez AC, Effting APJ, Kruit P, Hoogenboom JP. Integration of a High-NA Light Microscope in a Scanning Electron Microscope. J Microsc. 2013;252:58. doi: 10.1111/jmi.12071. [DOI] [PubMed] [Google Scholar]
- 14.Maruyama Y, Ebihara T, Nishiyama H, Suga M, Sato C. Immuno EM-OM Correlative Microscopy in Solution by Atmospheric Scanning Electron Microscopy (ASEM) J Struct Biol. 2012;180:259–270. doi: 10.1016/j.jsb.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Morrison IEG, Dennison CL, Nishiyama H, Suga M, Sato C, Yarwood A, O’Toole PJ. Atmospheric Scanning Electron Microscope for Correlative Microscopy. Methods Cell Biol. 2012;111:307–324. doi: 10.1016/B978-0-12-416026-2.00016-9. [DOI] [PubMed] [Google Scholar]
- 16.Liv N, Zonnevylle aC, Narvaez AC, Effting APJ, Voorneveld PW, Lucas MS, Hardwick JC, Wepf Ra, Kruit P, Hoogenboom JP. Simultaneous Correlative Scanning Electron and High-NA Fluorescence Microscopy. PLoS One. 2013;8:e55707. doi: 10.1371/journal.pone.0055707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole NB, Lippincott-Schwartz J. Organization of Organelles and Membrane Traffic by Microtubules. Curr Opin Cell Biol. 1995;7:55–64. doi: 10.1016/0955-0674(95)80045-x. [DOI] [PubMed] [Google Scholar]
- 18.Schlessinger J. Ligand-Induced, Receptor-Mediated Dimerization and Activation of EGF Receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 19.Lidke DS, Lidke Ka, Rieger B, Jovin TM, Arndt-Jovin DJ. Reaching out for Signals: Filopodia Sense EGF and Respond by Directed Retrograde Transport of Activated Receptors. J Cell Biol. 2005;170:619–626. doi: 10.1083/jcb.200503140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonard D, Hayakawa A, Lawe D, Lambright D, Bellve KD, Standley C, Lifshitz LM, Fogarty KE, Corvera S. Sorting of EGF and Transferrin at the Plasma Membrane and by Cargo-Specific Signaling to EEA1-Enriched Endosomes. J Cell Sci. 2008;121:3445–3458. doi: 10.1242/jcs.031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Y, Hubbert CC, Yao T-P. The Microtubule-Associated Histone Deacetylase 6 (HDAC6) Regulates Epidermal Growth Factor Receptor (EGFR) Endocytic Trafficking and Degradation. J Biol Chem. 2010;285:11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lidke DS, Nagy P, Heintzmann R, Arndt-Jovin DJ, Post JN, Grecco HE, Jares-Erijman Ea, Jovin TM. Quantum Dot Ligands Provide New Insights into erbB/HER Receptor-Mediated Signal Transduction. Nat Biotechnol. 2004;22:198–203. doi: 10.1038/nbt929. [DOI] [PubMed] [Google Scholar]
- 23.Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and Electron Microscopic Imaging of Connexin Trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- 24.Giepmans BNG, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH. Correlated Light and Electron Microscopic Imaging of Multiple Endogenous Proteins Using Quantum Dots. Nat Methods. 2005;2:743–749. doi: 10.1038/nmeth791. [DOI] [PubMed] [Google Scholar]
- 25.Thiberge S, Zik O, Moses E. An Apparatus for Imaging Liquids, Cells, and Other Wet Samples in the Scanning Electron Microscopy. Rev Sci Instrum. 2004;75:2280. [Google Scholar]
- 26.Liv N, Lazić I, Kruit P, Hoogenboom JP. Scanning Electron Microscopy of Individual Nanoparticle Bio-Markers in Liquid. Ultramicroscopy. 2014;143:93–99. doi: 10.1016/j.ultramic.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Dukes MJ, Ramachandra R, Baudoin J-P, Gray Jerome W, de Jonge N. Three-Dimensional Locations of Gold-Labeled Proteins in a Whole Mount Eukaryotic Cell Obtained with 3nm Precision Using Aberration-Corrected Scanning Transmission Electron Microscopy. J Struct Biol. 2011;174:552–562. doi: 10.1016/j.jsb.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park J, Park H, Ercius P, Pegoraro AF, Xu C, Kim JW, Han S-H, Weitz Da. Direct Observation of Wet Biological Samples by Graphene Liquid Cell Transmission Electron Microscopy. Nano Lett. 2015;15:4737–4744. doi: 10.1021/acs.nanolett.5b01636. [DOI] [PubMed] [Google Scholar]
- 29.Gaudreau L, Tielrooij KJ, Prawiroatmodjo GEDK, Osmond J, de Abajo FJG, Koppens FHL. Universal Distance-Scaling of Nonradiative Energy Transfer to Graphene. Nano Lett. 2013;13:2030–2035. doi: 10.1021/nl400176b. [DOI] [PubMed] [Google Scholar]
- 30.Takizawa T, Suzuki K, Robinson JM. Correlative Microscopy Using FluoroNanogold on Ultrathin Cryosections: Proof of Principle. J Histochem Cytochem. 1998;46:1097–1102. doi: 10.1177/002215549804601001. [DOI] [PubMed] [Google Scholar]
- 31.Pantazis P, Maloney J, Wu D, Fraser SE. Second Harmonic Generating (SHG) Nanoprobes for in Vivo Imaging. Proc Natl Acad Sci U S A. 2010;107:14535–14540. doi: 10.1073/pnas.1004748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grabenbauer M, Geerts WJC, Fernadez-Rodriguez J, Hoenger A, Koster AJ, Nilsson T. Correlative Microscopy and Electron Tomography of GFP through Photooxidation. Nat Methods. 2005;2:857–862. doi: 10.1038/nmeth806. [DOI] [PubMed] [Google Scholar]
- 33.Diestra E, Fontana J, Guichard P, Marco S, Risco C. Visualization of Proteins in Intact Cells with a Clonable Tag for Electron Microscopy. J Struct Biol. 2009;165:157–168. doi: 10.1016/j.jsb.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A Genetically Encoded Tag for Correlated Light and Electron Microscopy of Intact Cells, Tissues, and Organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risco C, Sanmartín-Conesa E, Tzeng W-P, Frey TK, Seybold V, de Groot RJ. Specific, Sensitive, High-Resolution Detection of Protein Molecules in Eukaryotic Cells Using Metal-Tagging Transmission Electron Microscopy. Structure. 2012;20:759–766. doi: 10.1016/j.str.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joy DC, Joy CS. Scanning Electron Microscope Imaging in Liquids – Some Data on Electron Interactions in Water. J Microsc. 2006;221:84–88. doi: 10.1111/j.1365-2818.2006.01548.x. [DOI] [PubMed] [Google Scholar]
- 37.Grady JK, Chasteen ND, Harris DC. Radicals from “Good’s” Buffers. Anal Biochem. 1988;173:111–115. doi: 10.1016/0003-2697(88)90167-4. [DOI] [PubMed] [Google Scholar]
- 38.Ring Ea, Peckys DB, Dukes MJ, Baudoin JP, de Jonge N. Silicon Nitride Windows for Electron Microscopy of Whole Cells. J Microsc. 2011;243:273–283. doi: 10.1111/j.1365-2818.2011.03501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dukes MJ, Peckys DB, de Jonge N. Correlative Fluorescence Microscopy and Scanning Transmission Electron Microscopy of Quantum-Dot-Labeled Proteins in Whole Cells in Liquid. ACS Nano. 2010;4:4110–4116. doi: 10.1021/nn1010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Egerton RF, Li P, Malac M. Radiation Damage in the TEM and SEM. Micron. 2004;35:399–409. doi: 10.1016/j.micron.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Royall CP, Thiel BL, Donald AM. Radiation Damage of Water in Environmental Scanning Electron Microscopy. J Microsc. 2001;204:185–195. doi: 10.1046/j.1365-2818.2001.00948.x. [DOI] [PubMed] [Google Scholar]
- 42.Swallow AJ. Radiation Chemistry An Introduction. Wiley; New York: 1973. [Google Scholar]
- 43.Schneider NM, Norton MM, Mendel BJ, Grogan JM, Ross FM, Bau HH. Electron–Water Interactions and Implications for Liquid Cell Electron Microscopy. J Phys Chem C. 2014;118:22373–22382. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.