

Abstract
The overall goal of this study was to provide evidence for the clinical validity of nine genetic variants in five genes previously associated with irinotecan neutropenia and pharmacokinetics. Variants associated with absolute neutrophil count (ANC) nadir and/ or irinotecan pharmacokinetics in a discovery cohort of cancer patients were genotyped in an independent replication cohort of 108 cancer patients. Patients received single-agent irinotecan every 3 weeks. For ANC nadir, we replicated UGT1A1*28, UGT1A1*93 and SLCO1B1*1b in univariate analyses. For irinotecan area under the concentration–time curve (AUC0-24), we replicated ABCC2 -24C>T; however, ABCC2 -24C>T only predicted a small fraction of the variance. For SN-38 AUC0-24 and the glucuronidation ratio, we replicated UGT1A1*28 and UGT1A1*93. In addition to UGT1A1*28, this study independently validated UGT1A1*93 and SLCO1B1*1b as new predictors of irinotecan neutropenia. Further demonstration of their clinical utility will optimize irinotecan therapy in cancer patients.
INTRODUCTION
Irinotecan is an anticancer agent commonly used for the treatment of metastatic colorectal cancer and other solid tumors. Irinotecan is a potent inhibitor of topoisomerase I, and is initially hydrolyzed to its active metabolite, SN-38, which is then subsequently inactivated through UGT1A1-mediated glucuronidation. A significant proportion of patients treated with irinotecan develop toxicities, including severe neutropenia. Neutropenia is a common, serious, dose-dependent and dose-limiting toxicity of irinotecan.1
A common, germline genetic variation in UGT1A1 predisposes patients to an increased risk of irinotecan-induced toxicities.2,3 The number of TA repeats in the UGT1A1 promoter is inversely proportional to the transcriptional efficiency of the gene,4 mRNA expression5 and protein levels.6 Patients with the UGT1A1*28 variant have seven TA repeats (compared with six repeats in patients with UGT1A1*1), have decreased SN-38 glucuronidation7 and experience increased systemic exposure to SN-38, which results in a higher risk of severe neutropenia.1 As a result, an FDA-approved UGT1A1*28 genotyping test has been made commercially available,8 and the irinotecan label has been revised to include UGT1A1*28 as a predisposing factor for severe neutropenia.9
Irinotecan-induced neutropenia is a complex, polygenic phenotype. There is significant interindividual variation in systemic exposure to both irinotecan and SN-38 that cannot be explained solely by UGT1A1*28. Several additional genetic variants contribute to both variability in irinotecan pharmacokinetics and the risk of severe neutropenia.10-16 The FDA-approved UGT1A1*28 genetic test has only moderate predictive power for severe toxicity due to its low positive predictive value,8 and therefore the genetic test has not been incorporated into routine clinical practice. The discovery of additional variants associated with neutropenia is needed to improve the utilization of irinotecan genetic testing.
Pharmacogenetic studies have identified a vast set of genetic variants as predictors of chemotherapy efficacy and toxicity. The majority of these proposed variants have failed to produce similar results across different studies, which has limited the clinical utility of pharmacogenetics.17,18 Therefore, prospective replication of pharmacogenetic findings in independent and external cohorts of patients is essential to hasten the implementation of pharmacogenetics into routine clinical practice.
In a previous study of cancer patients treated with single-agent irinotecan, novel gene variants that were associated with irinotecan disposition and toxicity were identified.16 In addition to UGT1A1*28, other variants, mostly in drug transporter genes, were associated with neutropenia and irinotecan pharmacokinetics. Therefore, we conducted a replication study to test the clinical validity of these variants in an external cohort of cancer patients treated with single-agent irinotecan.
MATERIALS AND METHODS
Study design
The overall goal of the study was to replicate genetic associations for irinotecan neutropenia and pharmacokinetics previously identified in a discovery cohort.16 The primary objective was to validate the associations between four genetic variants and absolute neutrophil count (ANC) nadir by testing them in an external replication cohort. The secondary objective was to validate the effects of eight genetic variants previously associated with pharmacokinetic parameters in the discovery cohort by analyzing them in the replication cohort. Thus, a total of nine common variants in five genes (ANC nadir and the pharmacokinetic phenotypes shared two variants) were genotyped in the replication cohort and tested for associations. Variants for replication testing were selected based on significant genotype–phenotype associations (P ≤ 0.05) observed in the discovery cohort. All patients in the replication cohort were White, and therefore only the previously genotyped White patients comprised the discovery cohort (n = 67).16
Patient characteristics
In the discovery cohort, advanced solid tumor patients were treated at the University of Chicago (Chicago, IL, USA) with a 90-min infusion of single-agent irinotecan every 3 weeks at 300 mg m−2 (n = 18) or 350 mg m−2 (n = 49). Eligibility criteria included adequate hematopoietic function (white blood cell count ≥ 3500 per μl, ANC ≥ 1500 per μl, platelets ≥100 000 per μl), normal renal and hepatic function (creatinine ≤ 1.5 mg dl−1, total bilirubin ≤ 1.25 × upper limit of normal (ULN), and AST/ALT < 5 × ULN), and adequate performance status (Karnofsky score ≥ 70%). Plasma pharmacokinetic parameters of irinotecan and metabolites were measured during and after the first cycle infusion of irinotecan. Forty-two genetic variants in twelve candidate genes of the irinotecan pathway were previously genotyped and tested for association with irinotecan pharmacokinetics and ANC nadir, measured during cycle 1.
In the replication cohort, 108 White advanced solid tumor patients were treated at the Erasmus University Medical Center, Erasmus MC Cancer Institute (Rotterdam, The Netherlands).19-21 Patients received a 90-min infusion of single-agent irinotecan every 3 weeks at 600 mg (flat dose, n = 58), 350 mg m−2 (n = 31), or 380–1060 mg (flat dose calculated according to an algorithm,19 n = 19). Eligibility criteria included adequate hematopoietic function (ANC ≥ 2000 per μl, platelets ≥ 100 000 per μl) and normal renal and hepatic function (creatinine clearance ≥ 60 ml min−1, total bilirubin ≤ 1.25 × ULN and AST/ALT ≤ 3 × ULN). Plasma pharmacokinetics of irinotecan and metabolites were measured during and after the first cycle infusion.
All patients in the discovery and replication cohorts provided written informed consent and the local institutional review boards approved the clinical protocols. Patient characteristics from the discovery and replication cohorts are provided in Table 1.
Table 1.
Baseline patient characteristics and pharmacokinetic data from the discovery and the replication cohorts
| Discovery cohort (n = 67) | Replication cohort (n = 108) | |||
|---|---|---|---|---|
| Dose | ||||
| 300 mg m−2 | 18 (26.9%) | — | ||
| 350 mg m−2 | 49 (73.1%) | 31 (29.7%) | ||
| Flat dose (600 mg) | — | 58 (53.7%) | ||
| Dose by algorithm (380–1060 mg) | — | 19 (17.1%) | ||
| Sex | ||||
| Male | 42 (62.7%) | 60 (55.6%) | ||
| Female | 25 (37.3%) | 48 (44.4%) | ||
| Median | Range | Median | Range | |
| Age (years) | 57 | 34–85 | 58 | 26–75 |
| BSA (m2) | 1.87 | 146–2.55 | 1.88 | 1.36–2.50 |
| Baseline ANC (cells per μl) | 5.27 | 2.18–14.36 | 5.12 | 1.30–13.60 |
| ANC Nadir (cells per μl) | 2.21 | 0.05–7.83 | 1.79 | 0.03–7.13 |
| Pharmacokinetic parameters | Mean | Range | Mean | Range |
| Irinotecan AUC0-24 (h ng ml−1) | 23251 | 8857–65305 | 22776 | 11422–67560 |
| SN-38 AUC0-24 (h ng ml−1) | 385 | 38–1957 | 364 | 79–1776 |
| SN-38G AUC0-24 (h ng ml−1) | 1824 | 360–8214 | 2238 | 396–6912 |
| SN-38G AUC0-24 to SN-38 AUC0-24 ratio (glucuronidation ratio) | 5.85 | 0.78–37.57 | 7.32 | 1–24.07 |
Abbreviations: ANC, absolute neutrophil count; AUC, area under the concentration–time curve. Flat dosing and dosing by algorithm19 were used only in the replication cohort. The distribution of the algorithm-derived doses includes: 380 mg (n = 1), 500 mg (n = 1), 520 mg (n = 2), 540 mg (n = 1), 560 mg (n = 1), 620 mg (n =2), 640 mg (n =1), 660 mg (n =1), 680 mg (n =1), 720 mg (n = 2), 740 mg (n = 3), 780 mg (n =1), 900 mg (n =1) and 1060 mg (n = 1).
Patient phenotyping: pharmacokinetic parameters and ANC nadir
In both cohorts, pharmacokinetic parameters included: irinotecan area under the concentration–time curve to the last time of sampling (AUC0-24), AUC0-24 of the active SN-38 metabolite, AUC0-24 of the inactive SN-38 glucuronide (SN-38G) and the ratio of SN-38G AUC0-24 to SN-38 AUC0-24 (glucuronidation ratio).
For the discovery cohort, samples were collected on day 1 of cycle 1 at baseline before irinotecan infusion, during the infusion (30, 60 and 90 min), and after the infusion (10, 20, 30 and 45 min, 1, 1.5, 2, 4, 6, 8, 12 and 24 h). Plasma concentrations of irinotecan and metabolites were measured, as previously reported.10 Pharmacokinetic parameters were calculated by non-compartmental analysis (WinNonlin, Pharsight, Cary, NC, USA).
For the replication cohort, samples were collected on day 1 of cycle 1 at baseline before infusion, during the infusion (30 and 90 min) and after the infusion (10, 20 and 30 min, and 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12 and 24 h). Plasma concentrations of irinotecan and metabolites were measured, as previously reported.20,22,23 Pharmacokinetic parameters were calculated by non-compartmental analysis (PK Solutions v2.0, Summit Research Services, Montrose, CO, USA).
In both cohorts, complete blood counts were taken at baseline, weekly throughout cycle 1, and then before the start of cycle 2 to obtain the measurements of the ANC nadir.
Genotype data
Nine common variants, previously associated with irinotecan pharmacokinetics and ANC nadir in the discovery cohort, were genotyped in the replication cohort: ABCB1 IVS9 44 A>G, ABCC1 1684 T>C, ABCC1 IVS11 -48C>T, ABCC2 3972C>T, ABCC2 -24C>T, SLCO1B1*1b, SLCO1B1*5, UGT1A1*28 and UGT1A1*93. DNA isolated from peripheral blood was used for genotyping. All genotyping assays were performed on an Applied Biosystems TaqMan 7500 (Life Technologies, Grand Island, NY, USA). UGT1A1*93 was genotyped by restriction fragment length polymorphism PCR, using 5′-ACCTCTAGTTACATAACCTGAA-3′ as the forward primer sequence and 5′-ATAAACCCGACCTCACCAC-3′ as the reverse primer sequence. UGT1A1*28 genotyping methods for the replication cohort have been previously described.20 All other variants were genotyped using TaqMan SNP genotyping assays (Life Technologies) as per the manufacturer’s instructions. Positive controls of known genotypes were used in the assays.
Statistics
Data for all phenotypes for both cohorts were log10 transformed. Hardy–Weinberg Equilibrium was evaluated for all nine variants genotyped in both the discovery and replication cohorts (Supplementary Table 1). In the discovery cohort, associations between genetic variants and clinical phenotypes were analyzed using linear regression, and were adjusted for sex, age and irinotecan dose (300 or 350 mg m −2). All ANC nadir analyses were also adjusted for baseline ANC.
In the replication cohort, we prospectively tested associations between the nine gene variants described above and phenotypes of ANC nadir and irinotecan pharmacokinetics. The same statistical methodologies employed for the discovery cohort were applied: linear regression adjusted for sex, age and irinotecan dose (350 mg m−2, 600 mg flat dose or dose by an algorithm19), with baseline ANC used to adjust the ANC nadir analysis. Flat doses were converted to mg m−2 according to the body-surface area of each patient. The same mode of inheritance (dominant, recessive or additive) used in the discovery cohort was also used in the replication cohort.
No general consensus exists to provide standardized criteria for replication cohort analyses. We considered a given variant’s association to be replicated based on direct comparison of the observed estimates of effect in the discovery and replication cohorts: an association’s estimate of effect in the replication cohort had to be in the same direction as in the discovery cohort (an increased or decreased estimate of phenotype change in both cohorts), and lie within the 95% confidence interval (CI) of the discovery cohort’s estimate. Two-sided P-values are reported for reference. Since comparisons between discovery and replication cohort estimates of effect were pre-specified and rely on 95% CIs from the discovery cohort, not on hypothesis testing in the replication cohort, issues related to multiplicity are not present. Therefore, no correction for multiple comparisons was performed.
RESULTS
This study sought to replicate, in an independent, external cohort of White cancer patients from the Netherlands, nine variants from five genes that had previously associated with ANC nadir or irinotecan pharmacokinetics.16 Baseline clinical patient characteristics and pharmacokinetic data (Table 1), as well as allele and genotype frequencies (Supplementary Table 1), were comparable between the two cohorts. Below we report the replication results of each variant for neutropenia and irinotecan pharmacokinetics (Table 2).
Table 2.
Univariate analyses of the associations between genetic variants and phenotypes
| Variant | Reference | Discovery cohort (n = 67)
|
Replication cohort (n = 74–103)
|
||||
|---|---|---|---|---|---|---|---|
| Estimate ± s.e. | 95% CI | P-value | n | Estimate ± s.e. | P-value | ||
| Log10 ANC Nadir | |||||||
| ABCC1 IVS11 -48 (C>T) | CC/CT | − 0489±0.201 | (−0.095, − 0.883) | 0.018 | 74 | 0.053± 0.117 | 0.652 |
| UGT1A1*28 (TA6>TA7) | *1*1=1 *1*28=2 | − 0.257±0.061 | (−0.137, − 0.377) | <0.001 | 75 | − 0.139±0.081 | 0.090 |
| *28*28=3 | |||||||
| UGT1A1*93 (G>A) | GG/AG | − 0.607±0.129 | (−0.354, − 0.860) | < 0.001 | 103 | − 0.417±0.170 | 0.016 |
| SLCO1B1*1b (A>G) | AA | 0.240±0.106 | (0.032, 0.448) | 0.027 | 84 | 0.278±0.107 | 0.012 |
| Log10 irinotecan AUC0-24 | |||||||
| ABCC2 –24 (C>T) | CC | 0.090±0.035 | (0.021, 0.159) | 0.012 | 94 | 0.067±0.030 | 0.040 |
| SLCO1B1*5(T>C) | TT | 0.084±0.036 | (0.013, 0.155) | 0.023 | 83 | − 0.010±0.035 | 0.769 |
| Log10SN-38 AUC0-24 | |||||||
| UGT1A1*28 (TA6>TA7) | *1*1= 1 | 0.140±0.046 | (0.050, 0.190) | 0.004 | 75 | 0.189±0.043 | <0.001 |
| *1*28=2 | |||||||
| *28*28= 3 | |||||||
| UGT1A1*93 (G>A) | GG= 1 | 0.130±0.047 | (0.038, 0.222) | 0.007 | 103 | 0.171±0.033 | <0.001 |
| AG=2 | |||||||
| AA=3 | |||||||
| ABCB1 IVS9 –44 (A>G) | AA | − 0.180±0.070 | (−0.043, -0.317) | 0.013 | 77 | 0.021±0.054 | 0.703 |
| Log10 SN-38G AUC0-24 | |||||||
| ABCC2 3972 (C> T) | CC/CT | 0.250±0.100 | (0.054, 0.446) | 0.019 | 105 | − 0.104±0.086 | 0.232 |
| Log10 SN-38G AUC0-24 /Log10 SN-38 AUC0-24 | |||||||
| ABCC1 1684 (T >C) | TT | − 0.316±0.153 | (−0.016, − 0.616) | 0.043 | 95 | − 0.052±0.059* | 0.382 |
| UGT1A1*28 (TA6>TA7) | *1*1= 1 | − 0.170±0.048 | (−0.076, − 0.264) | <0.001 | 103 | − 0.243±0.053 | <0.001 |
| *1*28=2 | |||||||
| *28*28= 3 | |||||||
| UGT1A1*93 (G > A) | GG= 1 | − 0.150±0.051 | (−0.050, − 0.250) | 0.004 | 75 | − 0.214±0.042 | <0.001 |
| AG=2 | |||||||
| AA=3 | |||||||
Abbreviations: ANC, absolute neutrophil count; AUC, area under the concentration–time curve; CI, confidence interval. Data were adjusted for age, sex and dose (mg m−2). ANC nadir was also adjusted for baseline ANC. The genotype reference groups were the same for all discovery and replication cohort analyses, with the exception of ABCC1 IVS11 -48C>T and ANC nadir. For ABCC1 IVS11 -48C>T and ANC nadir in the replication cohort, the reference genotype was only CC, as there were no TT genotypes. The estimates of effect of replicated variants are denoted in bold. The number of patients genotyped per variant in the replication cohort varied due to insufficient DNA quantity.
Although the association between ABCC1 1684T>C (dominant model) and glucuronidation ratio satisfies our criteria for replication, we are less convinced of the association, given the 84% reduction in the magnitude of the estimate as compared with that of the discovery cohort.
Replication of variants previously associated with ANC nadir
For ANC nadir, four variants that previously associated with ANC nadir in the discovery cohort were tested in the replication cohort. In the discovery cohort, UGT1A1*28 (additive model), UGT1A1*93 (recessive model) and ABCC1 IVS11 -48C>4 T (recessive model) were associated with decreased ANC nadir; SLCO1B1*1b (dominant model) was associated with increased ANC nadir. In the replication cohort, we considered UGT1A1*28, UGT1A1*93 and SLCO1B1*1b replicated, since the direction of the estimate of the effect for each variant was consistent between both cohorts (decreased ANC nadir for UGT1A1*28 and UGT1A1*93, as well as increased ANC nadir for SLCO1B1*1b) and each was within the 95% CIs for its respective discovery cohort estimate. ABCC1 IVS11 -48C>T failed to replicate (Table 2).
Replication of variants associated with the pharmacokinetic parameters of irinotecan
For irinotecan AUC0-24, two variants that were previously associated with irinotecan AUC0-24 in the discovery cohort were tested in the replication cohort. In the discovery cohort, ABCC2 -24C>T and SLCO1B1*5 (both dominant model) were associated with increased irinotecan AUC0-24. In the replication cohort, we considered ABCC2 -24C>T replicated since the direction of the estimate of the effect was consistent between both cohorts (increased AUC0-24 for both variants), and was within the 95% CIs for the discovery cohort estimate. SLCO1B1*5 failed to replicate (Table 2).
For SN-38 AUC0-24, three variants that were previously associated with SN-38 AUC0-24 in the discovery cohort were tested in the replication cohort. In the discovery cohort, UGT1A1*28 and UGT1A1*93 (both additive model) were associated with increased SN-38 AUC0-24, while ABCB1 IVS9> -44A>G (dominant model) was associated with decreased SN-38 AUC0-24. In the replication cohort, we considered UGT1A1*28 and UGT1A1*93 replicated since the direction of the estimate of the effect for each variant was consistent between both cohorts (increased AUC0-24 for both variants), and each was within the 95% CIs for its respective discovery cohort estimate. ABCB1 IVS9 -44A> G failed to replicate (Table 2).
For SN-38G AUC0-24, although ABCC2 3972C>T (recessive model) was associated with increased SN-38G AUC0-24 in the discovery cohort, it failed to replicate when tested in the replication cohort (Table 2).
For the glucuronidation ratio, three variants that associated with the glucuronidation ratio in the discovery cohort were tested in the replication cohort. In the discovery cohort, UGT1A1*28 (additive model), UGT1A1*93 (additive model) and ABCC1 1684T>C (dominant model) were associated with a decreased glucuronidation ratio. In the replication cohort, we considered UGT1A1*28 and UGT1A1*93, replicated, since the direction of the estimate of the effect for each variant was consistent between both cohorts (decreased glucuronidation ratio for all variants), and each was within the 95% CIs for its respective discovery cohort estimate. Although the association between ABCC1 1684 T>C (dominant model) and glucuronidation ratio also satisfies our criteria for replication, we are less convinced of the association, given the 84% reduction in the magnitude of the estimate as compared with that of the discovery cohort (Table 2).
DISCUSSION
In this replication study, we validated the clinical effects of new germline genetic variants for neutropenia and irinotecan pharmacokinetics using an independent, external cohort of White cancer patients treated with single-agent irinotecan.
The most important result of this study was the clinical validation of SLCO1B1*1b. To our knowledge, this provides the first replicated data implicating SLCO1B1*1b as a protective marker against irinotecan-induced neutropenia. SLCO1B1 encodes for organic anion transporter family member 1B1 (OATP1B1), and mediates hepatic uptake of both endogenous24,25 and xenobiotic compounds.26 OATP1B1 is a hepatic uptake transporter of SN-38,27,28 but not irinotecan.28 In this study, we have replicated results from the discovery cohort, and have shown that the variant *1b allele was associated with a higher ANC nadir compared with the reference sequence *1a allele (Figure 1a). Since SLCO1B1*1b is a non-synonymous variant (asparagine to aspartate amino-acid change), and SLCO1B1 is primarily expressed in the liver,29 we postulate this variant might associate with reduced neutropenia by altering systemic SN-38 exposure. The effect of SLCO1B1*1b on SN-38 AUC0-24 was − 0.083 ± 0.076 (mean ± s.e.) in the White patients of the discovery cohort (n = 67; P = 0.278), and because the P-value was >0.05, this association was not selected for analysis in the replication cohort. However, an exploratory univariate analysis (adjusted for dose (mg m −2), age and sex) revealed that SLCO1B1*1b was associated with decreased SN-38 AUC0-24 in the replication cohort (n = 84; − 0.128 ± 0.055, P = 0.023). These results support the hypothesis that the protective effect of SLCO1B1*1b against neutropenia could be due to increased hepatic uptake of SN-38, resulting in increased SN-38 elimination from the plasma after irinotecan infusion.
Figure 1.
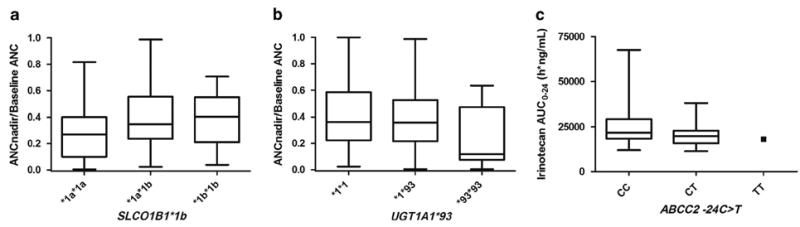
Associations between SLCO1B1*1b and absolute neutrophil count (ANC) nadir (a), UGT1A1*93 and ANC nadir (b) and ABCC2 -24C>T and log10 irinotecan area under the concentration–time curve (AUC0-24) (c) in the replication cohort. For the purpose of illustrating the replicated genetic associations, the data are not adjusted for the same factors used in the univariate analyses, and the differences among genotypes might not be the same as the ones reported in Table 2. ANC nadir is normalized to the baseline pretreatment ANC. Data are expressed as medians, 25th and 75th percentiles, minimums and maximums.
While the pharmacokinetic data are supportive of the protective effect of SLCO1B1*1b against neutropenia, the functional effect of this variant is less clear. Using RNA expression data from human livers,30 SLCO1B1*1b (as well as variants in linkage disequilibrium r2 ≥ 0.8) did not associate with changes in the mRNA expression of SLCO1B1 (results not shown). In oocyte studies, the uptake of SN-38 was higher for SLCO1B1*1b than SLCO1B1*1a (the reference sequence allele), but the observed difference was not statistically significant (see Figure 6a of Nozawa et al.28). Our results provide evidence that SLCO1B1*1b results in a gain of function, which leads to increased hepatic uptake of SN-38 from the plasma. Although this seems the most plausible hypothesis, other mechanisms related to the widespread functions of this transporter on several endogenous constituents cannot be ruled out.
Another important conclusion of this study is that UGT1A1*93 confers an increased risk of irinotecan-induced neutropenia. We replicated results from the discovery cohort, and have shown that the *93 variant was associated with a lower ANC nadir compared with the reference sequence *1 allele (Figure 1b). UGT1A1*93 is a − 3156G>A change discovered during a resequencing study of the region 5′ to the UGT1A exon 1.31 According to an analysis of more than 150 human livers where genome-wide genotyping data were available, UGT1A1*93 is a major determinant of decreased levels of the UGT1A1 protein (Pearson’s r = − 0.46, P = 3.5 × 10−9),30,32 and additional preliminary data corroborate these findings.33 Because UGT1A1*93 is in partial linkage disequilibrium with UGT1A1*28 among White patients (r2 = 0.68),34 our results suggest that UGT1A1*93, based on its greater estimate of effect for ANC nadir, may be a more robust marker for neutropenia than UGT1A1*28 (Table 2). While the UGT1A1*93 variant has not yet been included in the FDA-revised irinotecan label, we envision that recommendations supporting UGT1A1*93 genotyping could eventually replace UGT1A1*28 in the irinotecan drug label.
The association between ABCC2 -24C>T and increased irinotecan AUC0-24 was also replicated (Figure 1c). ABCC2 encodes for the multidrug resistant protein-2 and contributes to the biliary clearance of irinotecan, SN-38 and SN-38G.35,36 The -24C>T variant has been associated with a nearly 20% reduction in promoter activity.37 This observation is consistent with our results, where the variant T allele was associated with increased irinotecan AUC0-24, likely due to decreased biliary clearance. However, the estimate of effect size was relatively small (Table 2), and additional studies should be conducted to elucidate the extent of its clinical relevance.
Established criteria for conducting pharmacogenetic replication studies do not currently exist, but we provide a general framework for conducting such studies. Pharmacogenetic replication studies are beset with numerous challenges, including dosing and population heterogeneity between the discovery and replication cohorts. In our study, we attempted to control for population heterogeneity by comparing patients in the replication cohort to only the White patients from the original discovery cohort.16 Dosing heterogeneity between the two cohorts may have affected our ability to replicate some variants, but it did not confound all associations, as evidenced by the detection of associations serving as ‘positive controls’, such as UGT1A1*28 versus SN-38 AUC0-24 and UGT1A1*28 versus glucuronidation ratio (but not irinotecan AUC0-24). Moreover, we are confident that dosing heterogeneity did not significantly confound our replication results because irinotecan has been shown to demonstrate dose linear pharmacokinetics over a wide range of doses.38 Regarding our statistical approach, the assessment of replicated associations is not based on hypothesis testing, and therefore using P-values as our main criteria for replication would have been inappropriate. Moreover, given the influence of sample size on P-values, utilization of P-values as the main criteria for replication could have resulted in false negative results. We also cannot exclude the possibility that between-cohort differences limited our ability to detect phenotypic differences and replicate several variants.
This replication study allowed us to demonstrate the clinical validity of associations between UGT1A1*93 and SLCO1B1*1b and neutropenia. The effects of these two variants on neutropenia should be confirmed in studies where irinotecan is given in combination with other anticancer agents that have neutropenic effects (for example, with 5-fluorouracil). Additionally, the effects of these replicated variants can currently be applied only to White patients. Efforts should be made to validate these variants in patients from other races who receive irinotecan. Further validation of their clinical utility will aid in the implementation of routine irinotecan pharmacogenetic testing and optimization of personalized treatments for cancer patients.
Supplementary Material
Acknowledgments
This study was supported by funding from NIH/NIGMS U01GM61393, NIH/NCI K07CA140390-01, NIH/NIGMS T32GM086330 and the American Foundation for Pharmaceutical Education. We would like to acknowledge Dr Lana Crona and Mrs Anna Crollman for their help in editing this manuscript, as well as Dr Eric Seiser for his input on the analysis of the liver data in relation to SLCO1B1*1b.
Footnotes
CONFLICT OF INTEREST
Dr Federico Innocenti and Dr Mark J Ratain disclose that they receive royalties from UGT1A1 genotyping. Dr Gary L Rosner discloses that he owns stock in Pfizer. The remaining authors state no conflict of interest.
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website (http://www.nature.com/tpj)
References
- 1.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 2.Kim TW, Innocenti F. Insights, challenges, and future directions in irinogenetics. Ther Drug Monit. 2007;29:265–270. doi: 10.1097/FTD.0b013e318068623b. [DOI] [PubMed] [Google Scholar]
- 3.Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst. 2007;99:1290–1295. doi: 10.1093/jnci/djm115. [DOI] [PubMed] [Google Scholar]
- 4.Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucurono-syltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA. 1998;95:8170–8174. doi: 10.1073/pnas.95.14.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramirez J, Mirkov S, Zhang W, Chen P, Das S, Liu W, et al. Hepatocyte nuclear factor-1 alpha is associated with UGT1A1, UGT1A9 and UGT2B7 mRNA expression in human liver. Pharmacogenomics J. 2008;8:152–161. doi: 10.1038/sj.tpj.6500454. [DOI] [PubMed] [Google Scholar]
- 6.Ritter JK, Kessler FK, Thompson MT, Grove AD, Auyeung DJ, Fisher RA. Expression and inducibility of the human bilirubin UDP-glucuronosyltransferase UGT1A1 in liver and cultured primary hepatocytes: evidence for both genetic and environmental influences. Hepatology. 1999;30:476–484. doi: 10.1002/hep.510300205. [DOI] [PubMed] [Google Scholar]
- 7.Iyer L, King CD, Whitington PF, Green MD, Roy SK, Tephly TR, et al. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine dipho-sphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest. 1998;101:847–854. doi: 10.1172/JCI915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Innocenti F, Ratain MJ. Pharmacogenetics of irinotecan: clinical perspectives on the utility of genotyping. Pharmacogenomics. 2006;7:1211–1221. doi: 10.2217/14622416.7.8.1211. [DOI] [PubMed] [Google Scholar]
- 9.Product Information. Camptosar (irinotecan) New York: NPUC; Jul, 2012. http://labeling.pfizer.com/ShowLabeling.aspx?id=533. [Google Scholar]
- 10.Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 11.Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, et al. Influence of the organic anion-transporting polypeptide 1B1 (OATP1B1) polymorphisms on irinotecan-pharmacokinetics and clinical outcome of patients with advanced non-small cell lung cancer. Lung Cancer. 2008;59:69–75. doi: 10.1016/j.lungcan.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 12.Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY, et al. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer. 2007;110:138–147. doi: 10.1002/cncr.22760. [DOI] [PubMed] [Google Scholar]
- 13.Xiang X, Jada SR, Li HH, Fan L, Tham LS, Wong CI, et al. Pharmacogenetics of SLCO1B1 gene and the impact of *1b and *15 haplotypes on irinotecan disposition in Asian cancer patients. Pharmacogenet Genomics. 2006;16:683–691. doi: 10.1097/01.fpc.0000230420.05221.71. [DOI] [PubMed] [Google Scholar]
- 14.Mathijssen RH, Marsh S, Karlsson MO, Xie R, Baker SD, Verweij J, et al. Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin Cancer Res. 2003;9:3246–3253. [PubMed] [Google Scholar]
- 15.Rosner GL, Panetta JC, Innocenti F, Ratain MJ. Pharmacogenetic pathway analysis of irinotecan. Clin Pharmacol Ther. 2008;84:393–402. doi: 10.1038/clpt.2008.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Innocenti F, Kroetz DL, Schuetz E, Dolan ME, Ramirez J, Relling M, et al. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol. 2009;27:2604–2614. doi: 10.1200/JCO.2008.20.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coate L, Cuffe S, Horgan A, Hung RJ, Christiani D, Liu G. Germline genetic variation, cancer outcome, and pharmacogenetics. J Clin Oncol. 2010;28:4029–4037. doi: 10.1200/JCO.2009.27.2336. [DOI] [PubMed] [Google Scholar]
- 18.Innocenti F, Schilsky RL. Translating the cancer genome into clinically useful tools and strategies. Dis Model Mech. 2009;2:426–429. doi: 10.1242/dmm.004119. [DOI] [PubMed] [Google Scholar]
- 19.van der Bol JM, Mathijssen RH, Creemers GJ, Planting AS, Loos WJ, Wiemer EA, et al. A CYP3A4 phenotype-based dosing algorithm for individualized treatment of irinotecan. Clin Cancer Res. 2010;16:736–742. doi: 10.1158/1078-0432.CCR-09-1526. [DOI] [PubMed] [Google Scholar]
- 20.Mathijssen RH, de Jong FA, van Schaik RH, Lepper ER, Friberg LE, Rietveld T, et al. Prediction of irinotecan pharmacokinetics by use of cytochrome P450 3A4 phenotyping probes. J Natl Cancer Inst. 2004;96:1585–1592. doi: 10.1093/jnci/djh298. [DOI] [PubMed] [Google Scholar]
- 21.de Jong FA, Kehrer DF, Mathijssen RH, Creemers GJ, de Bruijn P, van Schaik RH, et al. Prophylaxis of irinotecan-induced diarrhea with neomycin and potential role for UGT1A1*28 genotype screening: a double-blind, randomized, placebo-controlled study. Oncologist. 2006;11:944–954. doi: 10.1634/theoncologist.11-8-944. [DOI] [PubMed] [Google Scholar]
- 22.de Bruijn P, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Determination of irinotecan (CPT-11) and its active metabolite SN-38 in human plasma by reversed-phase high-performance liquid chromatography with fluorescence detection. J Chromatogr B Biomed Sci Appl. 1997;698:277–285. doi: 10.1016/s0378-4347(97)00290-9. [DOI] [PubMed] [Google Scholar]
- 23.de Bruijn P, Willems EW, Loos WJ, Verweij J, Sparreboom A. Indirect determination of the irinotecan metabolite 7-ethyl-10-O-glucuronyl-camptothecin in human samples. Anal Biochem. 2004;328:84–86. doi: 10.1016/j.ab.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Abe T, Kakyo M, Tokui T, Nakagomi R, Nishio T, Nakai D, et al. Identification of a novel gene family encoding human liver-specific organic anion transporter LST-1. J Biol Chem. 1999;274:17159–17163. doi: 10.1074/jbc.274.24.17159. [DOI] [PubMed] [Google Scholar]
- 25.Hsiang B, Zhu Y, Wang Z, Wu Y, Sasseville V, Yang WP, et al. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999;274:37161–37168. doi: 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- 26.Oshiro C, Mangravite L, Klein T, Altman R. PharmGKB very important pharmacogene: SLCO1B1. Pharmacogenet Genomics. 2010;20:211–216. doi: 10.1097/FPC.0b013e328333b99c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iusuf D, Ludwig M, Elbatsh A, van Esch A, van de Steeg E, Wagenaar E, et al. OATP1A/1B transporters affect irinotecan and SN-38 pharmacokinetics and carboxylesterase expression in knockout and humanized transgenic mice. Mol Cancer Ther. 2014;13:492–503. doi: 10.1158/1535-7163.MCT-13-0541. [DOI] [PubMed] [Google Scholar]
- 28.Nozawa T, Minami H, Sugiura S, Tsuji A, Tamai I. Role of organic anion transporter OATP1B1 (OATP-C) in hepatic uptake of irinotecan and its active metabolite, 7-ethyl-10-hydroxycamptothecin: in vitro evidence and effect of single nucleotide polymorphisms. Drug Metab Dispos. 2005;33:434–439. doi: 10.1124/dmd.104.001909. [DOI] [PubMed] [Google Scholar]
- 29.Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–181. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
- 30.Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S, et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet. 2011;7:e1002078. doi: 10.1371/journal.pgen.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Innocenti F, Grimsley C, Das S, Ramirez J, Cheng C, Kuttab-Boulos H, et al. Haplotype structure of the UDP-glucuronosyltransferase 1A1 promoter in different ethnic groups. Pharmacogenetics. 2002;12:725–733. doi: 10.1097/00008571-200212000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Gillis N, Seiser E, Fallon J, Smith P, Innocenti F. Genome-wide analysis of the variation in hepatic protein expression of 22 key drug metabolizing enzymes. American Society of Clinical Pharmacology and Therapeutics (ASCPT) 2014 Annual Meeting; 14-16 March 2014; Abstract number 765 and poster number LBII-017. [Google Scholar]
- 33.Peterkin V, Bauman JN, Goosen TC, Paulauskis J, Williams JA, Myrand SP. Genetic variant UGT1A1*93 (-3156 G>A) is predictive of UGT1A1 enzyme activity and protein expression in human liver microsomes. 15th North American Regional International Society for the Study of Xenobiotics (ISSX) Meeting; 12-16 October 2008; Abstract number 331. [Google Scholar]
- 34.Cecchin E, Innocenti F, D’Andrea M, Corona G, De Mattia E, Biason P, et al. Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J Clin Oncol. 2009;27:2457–2465. doi: 10.1200/JCO.2008.19.0314. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen TD, Markova S, Liu W, Gow JM, Baldwin RM, Habashian M, et al. Functional characterization of ABCC2 promoter polymorphisms and allele-specific expression. Pharmacogenomics J. 2013;13:396–402. doi: 10.1038/tpj.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugiyama Y, Kato Y, Chu X. Multiplicity of biliary excretion mechanisms for the camptothecin derivative irinotecan (CPT-11), its metabolite SN-38, and its glucuronide: role of canalicular multispecific organic anion transporter and P-glycoprotein. Cancer Chemother Pharmacol. 1998;42:S44–S49. doi: 10.1007/s002800051078. [DOI] [PubMed] [Google Scholar]
- 37.Haenisch S, Zimmermann U, Dazert E, Wruck CJ, Dazert P, Siegmund W, et al. Influence of polymorphisms of ABCB1 and ABCC2 on mRNA and protein expression in normal and cancerous kidney cortex. Pharmacogenomics J. 2007;7:56–65. doi: 10.1038/sj.tpj.6500403. [DOI] [PubMed] [Google Scholar]
- 38.Chabot GG, Abigerges D, Catimel G, Culine S, de Forni M, Extra JM, et al. Population pharmacokinetics and pharmacodynamics of irinotecan (CPT-11) and active metabolite SN-38 during phase I trials. Ann Oncol. 1995;6:141–151. doi: 10.1093/oxfordjournals.annonc.a059109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.