

Abstract
The transcription factor Miz1 negatively regulates TNF-induced JNK activation and cell death by suppressing TRAF2 K63-polyubiquitination; upon TNF stimulation, the suppression is relieved by Mule/ARF-BP1-mediated Miz1 ubiquitination and subsequent degradation. It is not known how Mule is activated by TNF. Here we report that TNF activates Mule by inducing the dissociation of Mule from its inhibitor ARF. ARF binds to and thereby inhibits the E3 ligase activity of Mule in the steady state. TNF induces tyrosine phosphorylation of Mule, which subsequently dissociates from ARF and becomes activated. Inhibition of Mule phosphorylation by silencing of the Spleen Tyrosine Kinase (Syk) prevents its dissociation from ARF, thereby inhibiting Mule E3 ligase activity and TNF-induced JNK activation and cell death. Our data provides a missing link in TNF signaling pathway that leads to JNK activation and cell death.
Keywords: Mule/ARF-BP1, tumor suppressor ARF, TNF, JNK, ubiquitination
Introduction
TNF is a pleiotropic cytokine that plays a critical role in apoptosis, inflammation and immune response. Dysregulation of the TNF signaling pathway has been implicated in the pathogenesis of a wide range of human diseases (1, 2). TNF binds to TNF-R1, resulting in the formation of TNF-R1 complex 1 that is composed of TNF-R1-associated death domain protein (TRADD), TNF-receptor-associated factor 2 and 5 (TRAF2 and 5), and receptor interacting protein 1 (RIP1), leading to activation of multiple downstream effectors including JNK (also known as stress-activated protein kinase; SAPK), as well as p38 and the IκB kinase (IKK) complex (3–8). When NF-κB activation is impaired, JNK activation becomes prolonged, which contributes to TNF-induced apoptosis (3, 7, 9–11). The non-receptor tyrosine kinase Spleen Tyrosine Kinase (Syk) is potently activated in response to TNF (12). Syk has been shown to be involved in adaptive immunity, cellular adhesion, osteoclast maturation, and platelet activation (13–18). However, the role of Syk in TNF-induced JNK activation is poorly understood.
Mule (also known as ARF-BP1, HUWE1, Ureb1, LASU1, and HECTH9) belongs to the E5-AP C terminus (HECT) domain-containing ubiquitin ligase (E3). It is ubiquitiously expressed in human tissues and is found to be overexpressed in certain types of cancers, including lung, breast, and colorectal carcinomas (19, 20). Mule contains three domains known to serve as protein-protein interaction sites: a UBA domain, a WWE domain, a well-conserved BH3 domain, as well as a HECT domain at its C-terminus. Unlike members of other E3 families, HECT E3s process an intrinsic enzymatic ubiquitin transfer activity, which plays a direct catalytic role in the final attachment of ubiquitin to the substrate proteins. It has been reported that Mule catalyzes the ubiquitination of a variety of substrates, such as p53, c-Myc, MyoD, HDAC2, E3Histone, Mcl-1, TopBP1, Cdc6 and Pol β, thereby playing an important role in cell cycle arrest, apoptosis, DNA replication, DNA damage response and repair (19–27).
The transcription factor Miz1 plays a critical role in cell cycle arrest, proliferation, differentiation, and apoptosis through transcriptional activation or repression of its target genes, including p15Ink4b, p21Cip1, Mad4, and Bcl-2 (22, 28–30). Miz1 is localized to both cytoplasm and nucleus (29). The role of Miz1 in the nucleus has been extensively investigated (31). However, the role of Miz1 in the cytoplasm was poorly understood. We recently reported that cytoplasmic Miz1 suppresses TNF-induced JNK1 activation and cell death through inhibition of TRAF2 K63-linked polyubiquitination (32). The suppression is relieved by Mule, which interacts with and catalyzes K48-linked polyubiquitination of Miz1 for proteasomal degradation in response to TNF (26). However, it is not known how Mule is activated by TNF.
The tumor suppressor ARF has been reported to directly bind to Mule and inhibit its E3 ligase activity toward p53 (33). ARF is part of an alternative transcript of the INK4b-ARF-INK4a locus. This locus encodes three proteins: p15INK4b, p16INK4a, and ARF. In human, ARF is known as p14ARF, and the mouse form is called p19ARF, sharing about 50% sequence homology. ARF is involved in fundamental cellular activities, including cell cycle arrest, ribosome biogenesis (34, 35), DNA damage response (36, 37), and autophagy (38, 39). It has been shown that ARF is a key activator of the p53 pathway, by inhibiting p53 ubiquitin ligases Mdm2 and Mule. ARF can also induce cell cycle arrest in an Mdm2- and p53-independent but Mule-dependent manner (33). It was not known, however, whether ARF inhibits Mule-mediated Miz1 ubiquitination, and if so, how the inhibitory effect of ARF is released in response to TNF. Here, we report that TNF induces Syk-mediated tyrosine phosphorylation of Mule, which results in the dissociation of Mule from ARF, thereby activating Mule to ubiquitinate Miz1 for proteasomal degradation, providing a missing link in TNF signaling pathway that leads to JNK activation and cell death.
Results
ARF inhibits Mule-mediated Miz1 ubiquitination
Previously, we reported that K48-linked polyubiquitination of Miz1 by Mule triggers its proteasomal degradation, thereby relieving Miz1 suppression on TNF-induced JNK activation and apoptosis (26, 32, 38, 40). Since ARF can directly bind to Mule and inhibit its E3 ligase activity toward p53 (19), we sought to determine whether ARF inhibits Mule-mediated Miz1 ubiquitination. HEK293 cells were transfected with expression vector encoding M2-Mule, which was then immunoprecipitated and used as the E3 ligase in an in vitro ubiqutination assay with GST-Xpress-Miz1 as the substrate in the absence or presence of recombinant ARF proteins. Ubiquitination of Miz1 was decreased in the presence of ARF in a dose-dependent manner, consistent with the previous report that ARF inhibits Mule ubiquitin ligase activity (Fig. 1A). In parallel, HEK293 cells were transfected with expression vectors encoding M2-Mule and Xpress-ARF or empty vector. In vitro ubiquitination assay showed that ubiquitination of Miz1 was reduced when Mule was immunoprecipitated from cells co-transfected with ARF under non-stimulated conditions (Fig. 1B). To determine whether ARF inhibits TNF-induced Mule-mediated Miz1 ubiquitination, we silenced ARF by its specific siRNA. Knockdown of ARF enhanced the ability of TNF-stimulated Mule to ubiquitinate Miz1 in vitro (Fig. 1C). Finally, in vivo ubiquitination assay showed that silencing of ARF increased TNF-induced K48-linked polyubiquitination of endogenous Miz1 (Fig. 1D and 1E). Taken together, ARF inhibits Mule- mediated Miz1 ubiquitination.
Figure 1. ARF inhibits Mule-mediated Miz1 ubiquitination in the steady state and in response to TNF.
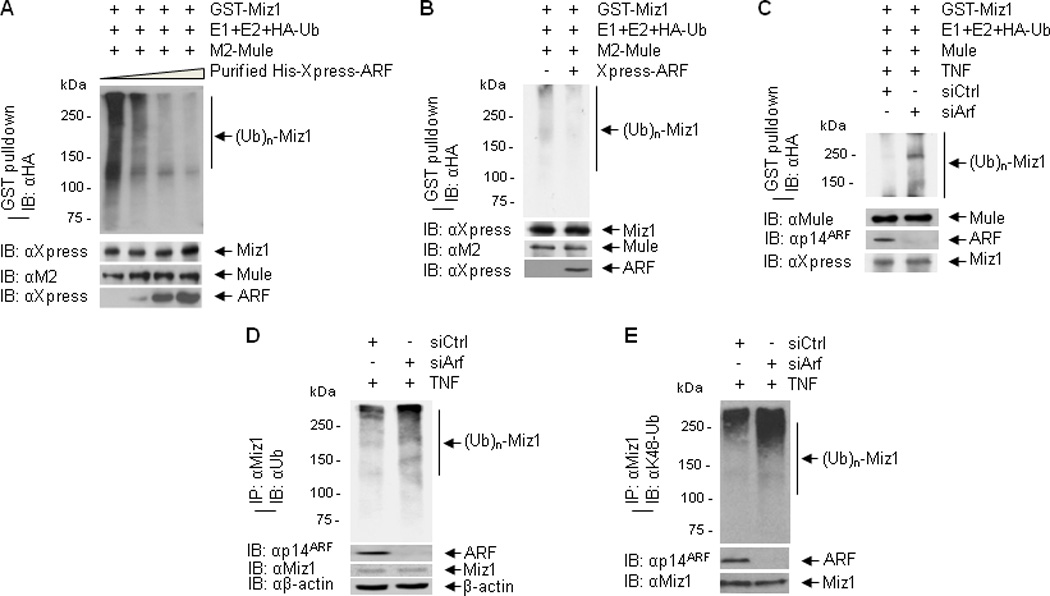
(A) GST-Xpress-Miz1 was incubated with immunoprecipitated M2-Mule and various concentrations of purified His-Xpress-ARF in an in vitro ubiquitination assay in the presence of HA-ubiquitin, ATP and E1/E2. Ubiqutination of GST-Xpress-Miz1 was analyzed by GST pulldown followed by immunoblotting with anti-HA antibody. (B) In vitro ubiquitination of GST-Miz1 by immunoprecipitated M2-Mule from HEK293 cells transfected with M2-Mule and Xpress-ARF or empty vector. (C) In vitro ubiquitination of GST-Miz1 by immunoprecipitated endogenous Mule from HEK293 cells transfected with control siRNA or ARF siRNA followed by treatment with TNF. (D, E) In vivo ubiquination assay. HEK293 cells were transfected with control siRNA or ARF siRNA, followed by treatment with TNF. Ubiquitination of immunoprecipitated Miz1 was analyzed by immunoblotting with ubiquitin antibody (D) or K48-ubiquitin antibody (E).
ARF does not interfere with Mule-Miz1 binding
Next we determined whether ARF also regulates the interaction between Mule and Miz1. In vitro binding assay showed that the interaction between Mule and Miz1 was not affected by the addition of purified recombinant ARF (Fig. 2A). The interaction between Mule and Miz1 was enhanced in response to TNF (Fig. 2B), consistent with our previous report (26). Silencing of ARF did not affect TNF-induced Mule-Miz1 interaction (Fig. 2B). These data suggest that ARF inhibits Mule-mediated Miz1 ubiquitination most likely through suppression of Mule E3 ligase activity instead of interfering with Mule-Miz1 binding.
Figure 2. ARF does not interfere with Mule-Miz1 binding.

(A) HEK293 cells were transfected with M2-Mule, which was then immunoprecipitated and subjected to in vitro binding assay with purified His-Xpress-Miz1 in the presence of various concentrations of purified ARF. (B) Cells were transfected with control siRNA or siRNA for ARF, followed by treatment without or with TNF in presence of MG-132. The interaction between Mule and ARF was determined by immunoprecipitation with Mule antibody followed by immunoblotting with Miz1 antibody.
TNF induces ARF-Mule dissociation
We previously reported that in response to TNF, Mule is activated and ubiquitinate Miz1 (19). It has been reported that ARF directly binds to Mule in the steady state (33). We tested whether ARF dissociates from Mule in response to TNF, resulting in the activation of Mule. HEK293 cells were stimulated with TNF for 0, 2, and 5 min, and ARF-Mule interaction was determined by co-immunoprecipitation with Mule antibody, followed by immunoblotting with ARF antibody. The interaction between ARF and Mule was readily detected in the steady state, consistent with the previous report (19). At 5 min after TNF stimulation, the association between ARF and Mule was dramatically decreased (Fig. 3A), which was timely correlated with the ubiquitin ligase activity of Mule and the ubiquitination status of endogenous Miz1, as demonstrated by in vitro and in vivo ubiquitination assays (Fig. 3B and 3C). Together, these data suggest that in the steady state, Mule is sequestered by ARF; after TNF stimulation, ARF dissociates from Mule, leading to Mule activation.
Figure 3. TNF induces ARF-Mule dissociation.
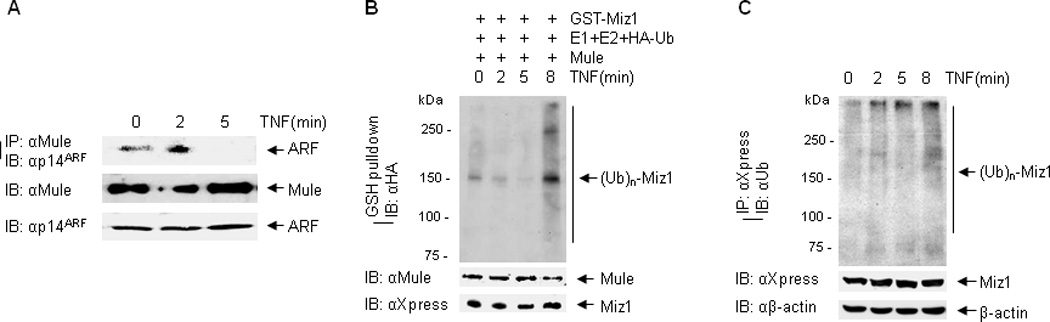
(A) HEK293 cells were treated with TNF for 0, 2, and 5 min. The interaction between Mule and ARF was determined by immunoprecipitation with Mule antibody, followed by immunoblotting with p14ARF antibody. (B) In vitro ubiquitination of GST-Miz1 by immunoprecipitated endogenous Mule from HEK293 cells treated with TNF for various times as indicated. (C) In vivo ubiquitination of Miz1 in cells treated with TNF for various times as indicated.
Tyrosine phosphorylation of Mule in response to TNF is required for its dissociation from ARF
An emerging theme regarding the regulation of the HECT E3 ligases is that phosphorylation of the HECT E3 ligases may increase their activity, probably by relieving the inhibitory intra- or extramolecular interactions (41–44). We sought to determine whether Mule is phosphorylated in response to TNF, thereby resulting in the release of the inhibitory ARF for Mule activation. Cells were treated with TNF for 0, 5, 8, and 10 min. Mule was immunoprecipitated followed by immunoblotting with phospho-Ser/Thr or phospho-Tyr antibody. Phosphorylation signals with the phospho-Tyr antibody were evident at 5 min, and increased by 8 and 10 min after TNF stimulation (Fig. 4A). By contrast, we were unable to detect phosphorylation signals with the phospho-Ser/Thr antibody (data not shown). These results suggest that Mule is tyrosine phosphorylated in response to TNF.
Figure 4. Mule is Tyrosine phosphorylated in response to TNF, which is required for ARF dissociation.
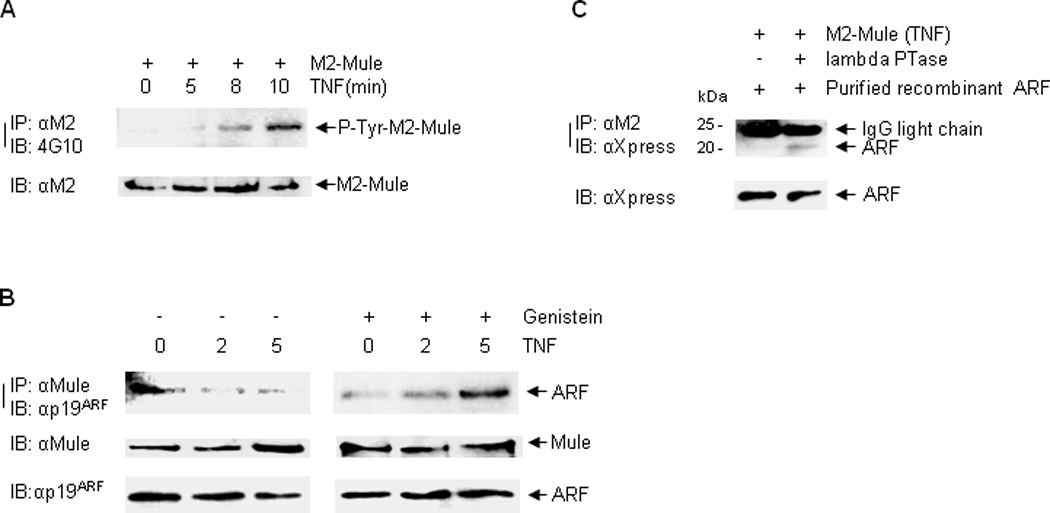
(A) HEK293 cells transfected with M2-Mule were pretreated with MG-132, Okadaic Acid, and Na3VO4, followed by treatment with TNF for various times as indicated. M2-mule was immunoprecipitated followed by immunoblotting with phospho-tyrosine antibody (4G10). (B) MLE12 cells were pretreated without or with Genistein, followed by treatment with TNF for 0, 2, and 5 min. The interaction between Mule and ARF was determined by immunoprecipitation with Mule antibody, followed by immunoblotting with p19ARF antibody. (C) Immunoprecipitated M2-Mule from TNF-treated cells was pre-incubated without or with Lambda phosphatase, followed by the addition of purified ARF. The interaction between M2-Mule and purified ARF was determined.
To determine whether Mule tyrosine phosphorylation is required for the dissociation of ARF from Mule, cells were pre-treated with Genistein, a tyrosine kinase inhibitor, followed by treatment with TNF at 0, 2 and 5 min. Co-immunoprecipitation assay showed that pre-treatment with Genistein blocked the ARF-Mule dissociation (Fig. 4B). In addition, in vitro binding assay showed that the interaction between TNF-stimulated Mule and purified recombinant ARF was enhanced when Mule was pre-incubated with lambda phosphatase, which releases phosphate groups from phosphorylated residues (Fig. 4C). These data suggest that tyrosine phosphorylation of Mule is required for ARF dissociation from Mule.
Mule tyrosine phosphorylation contributes to its E3 ligase activity
Next we sought to determine whether Mule tyrosine phosphorylation positively regulates its E3 ligase activity. Cells were pre-treated without or with Genistein, followed by treatment with TNF. In vitro ubiquitination assay showed that the ability of Mule to ubiquitinate Miz1 is decreased when Mule was immunoprecipitated from cells pre-treated with Genistein (Fig. 5A). It has been reported that Mule contains a consensus tyrosine phosphorylation site Y4271, and mutation of this site significantly attenuated the inhibitory effect of Mule on p53 (45, 46). We investigated whether mutation of Y4271 to non-phosphorylatable phenylalanine inhibited Mule E3 ligase activity. In vitro ubiquitination assay demonstrated that the Mule(Y4271F) mutant did not inhibit its E3 ligase activity (Fig. 5B), suggesting Y4271 of Mule is unlikely involved in the interaction of Mule with ARF.
Figure 5. Mule tyrosine phosphorylation contributes to its E3 ligase activity.
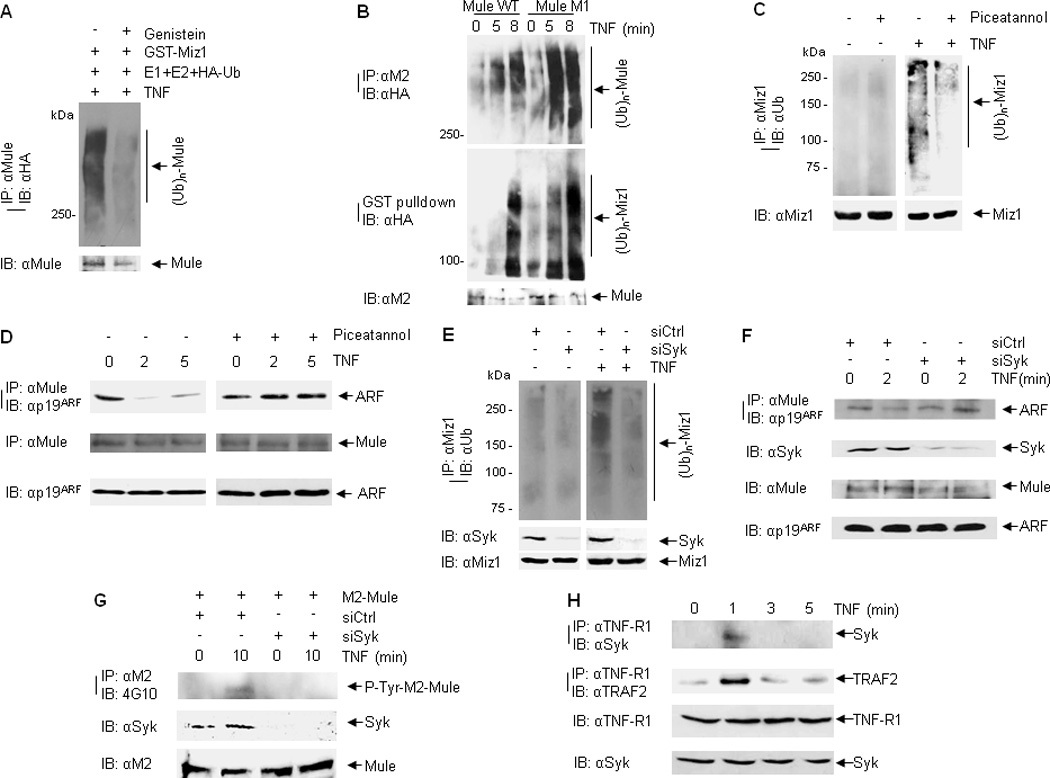
(A) In vitro self-ubiquitination of immunoprecipitated Mule from TNF-treated cells (without or with Genistein pre-treatment). (B) HEK293 cells were transfected with M2-Mule(WT) or M2-Mule(M1) plasmids, followed by stimulation with TNF for various times as indicated. M2-Mule was immunoprecipitated and used for the E3 ligase in an in vitro ubiquitination assay in the absence or presence of GST-Xpress-Miz1. Self-ubiquitination of Mule was analyzed by immunoblotting with HA (ubiquitin) antibody. Ubiquitination of GST-Xpress-Miz1 was determined by GST pulldown followed by immunoblotting with HA (ubiquitin) antibody. (C and D) WT MEFs were pre-treated without or with Piceatannol followed by TNF treatment. Ubiquitination of endogenous Miz1 was determined by immunoprecipitation with Miz1 antibody followed by immunoblotting with ubiquitin antibody (C). Mule-ARF interaction was analyzed by immunoprecipitation with Mule antibody followed by immunoblotting with ARF antibody (D). (E) In vivo ubiquitination of endogenous Miz1 from WT MEFs transfected with control siRNA or Syk siRNA followed by TNF treatment. (F) Mule-ARF interaction from WT MEFs transfected with control siRNA or Syk siRNA followed by TNF treatment. (G) WT MEFs cotransfected with M2-Mule and siRNA (control siRNA or siRNA for ARF) were pretreated with Okadaic Acid and Na3VO4, followed by treatment with TNF for various times as indicated. M2-mule was immunoprecipitated by M2 antibody, followed by immunoblotting with phospho-tyrosine antibody (4G10). (H) WT MEFs were treated with TNF for various times as indicated. The interaction between TNF-R1 and Syk was analyzed by immunoprecipitation with TNF-R1 antibody, followed by immunoblotting with Syk antibody.
We then searched for the potential tyrosine kinase that may phosphorylate Mule in response to TNF. A variety of tyrosine kinase inhibitors, including Piceatannol, which inhibits p56lck and Syk; PF-562271, which inhibits focal adhesion kinase (FAK) and Pyk2; and 1-Napthyl PP1, which inhibits c-Src family kinases v-Src, c-Fyn and c-Abl, were used. In vivo ubiquitination assay showed that TNF-induced Miz1 ubiquitination was decreased when cells were treated with Piceatannol (Fig. 5C). In addition, Piceatannol prevented the dissociation of ARF from Mule in response to TNF (Fig. 5D). Furthermore, silencing of Syk reduced Miz1 ubiquitination in response to TNF (Fig. 5E). Consistently, silencing of Syk abolished the dissociation of Mule from ARF (Fig. 5F) and Mule tyrosine phosphorylation (Fig. 5G). Collectively, these data suggest that TNF-induced tyrosine phosphorylation of Mule by Syk contributes to its E3 ligase activity. To understand how Syk is activated by TNF, we determined whether Syk is recruited to TNF-R1 complex I. WT MEFs were stimulated with TNF for 1, 3 and 5 min. Syk recruitment to TNF-R1 was determined by co-immunoprecipitation with TNF-R1 antibody, followed by immunoblotting with Syk antibody (Fig. 5H). The interaction between Syk and TNF-R1 was readily detected at 1 min after TNF stimulation. Under this condition, TRAF2 was recruited to TNF-R1, consistent with previous reports (47, 48). These data suggest that TNF may activate Syk by promoting Syk recruitment to TNF-R1, thereby contributing to JNK activation.
Mule tyrosine phosphorylation contributes to TNF-induced JNK activation and cell death
Previously we reported that in response to TNF, Mule ubiquitinates Miz1 for TNF-induced JNK activation and cell death. We determined whether inhibition of Mule tyrosine phosphorylation, which is required for its activation, suppressed TNF-induced JNK activation and cell death. As expected, treatment with Genistein inhibited TNF-induced JNK activation (Fig. 6A). However, Genistein treatment did not affect TNF-induced cell death (Supplementary Fig. 1A). It is possible that Genistein has a positive role in TNF-induced cell death independently of JNK, thereby neutralizing the effect of JNK on cell death. Piceatannol treatment suppressed TNF-induced JNK activation and cell death (Fig. 6B and D). Similarly, silencing of Syk inhibited TNF-induced JNK activation and cell death (Fig. 6C and E). These data suggest that Mule tyrosine phosphorylation promotes TNF-induced JNK activation and cell death.
Figure 6. Mule tyrosine phosphorylation contributes to TNF-induced JNK activation and cell death.
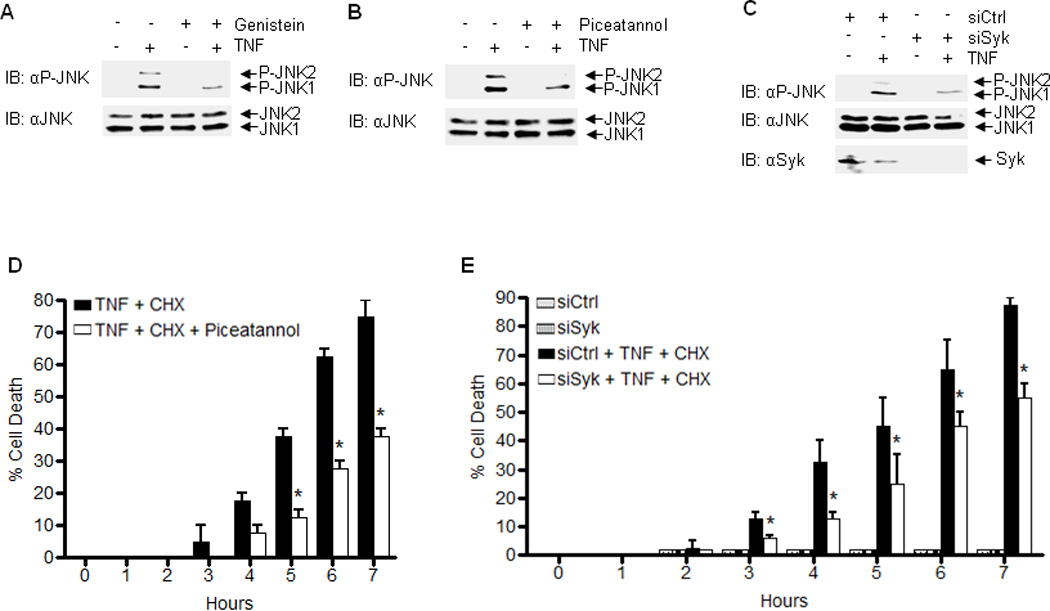
(A) WT MEFs were pretreated without or with Genistein (40uM) for 30 min followed by treatment without or with TNF for 15 min. Phophorylation of JNK was determined by immunoblotting. (B) WT MEFs were pretreated without or with Piceatannol, followed by treatment with TNF for 15 min. Phosphorylation of JNK was determined. (C) WT MEFs were transfected with control siRNA or Syk siRNA, followed by treatment with TNF for 15 min. Phosphorylation of JNK was determined. (D) WT MEFs were pretreated without or with Piceatannol, followed by treatment with TNF plus CHX for various times. Apoptotic cell death was determined. (E) WT MEFs were transfected with control siRNA or Syk siRNA, followed by treatment with TNF plus CHX for various times. Apoptotic cell death was determined. Results were presented as means ± s.e.m. and represent two independent experiments (D and E). *, P < 0.05 by two-way ANOVA.
The regulation of the Mule-Miz1-JNK axis by ARF promoted us to determine the role of ARF in TNF-induced cell death. Interestingly, ARF−/− MEFs were resistant to TNF-induced cell death (Supplementary Fig. 1B), and silencing of ARF also decreased TNF-induced cell death (Supplementary Fig. 1C); although under these conditions, TNF-induced JNK activation was augmented (Supplementary Fig. 1D and E). These data suggest that ARF positively regulates TNF-induced cell death independently of the Mule-JNK axis, which overrides the effect of ARF-regulated Mule-JNK axis on cell death. ARF has been reported to inhibit NF-kB activation while contributes to p53 stability (49, 50), which may account for its positive regulation of TNF-induced cell death. As ARF plays a fundamental role in many cellular activities, dissecting the ARF-Mule interaction to specifically manipulate TNF-induced JNK activation/cell death might provide the therapeutic target without affecting the other functions of ARF.
Discussion
The HECT-domain-containing E3 ligase Mule is the E3 ubiquitin ligase that ubiquitinates Miz1 and trigger Miz1 proteasomal degradation, thereby positively regulating TNF-induced JNK activation and cell death (26). How Mule itself is activated by TNF is not known. Here we report that TNF induces the dissociation of ARF from Mule through Syk-mediated tyrosine phosphorylation of Mule, thereby allowing activation of the Mule-Miz1-JNK signaling pathway and cell death (Fig. 7), based on the following observations.
Figure 7. A model of Mule activation by TNF.
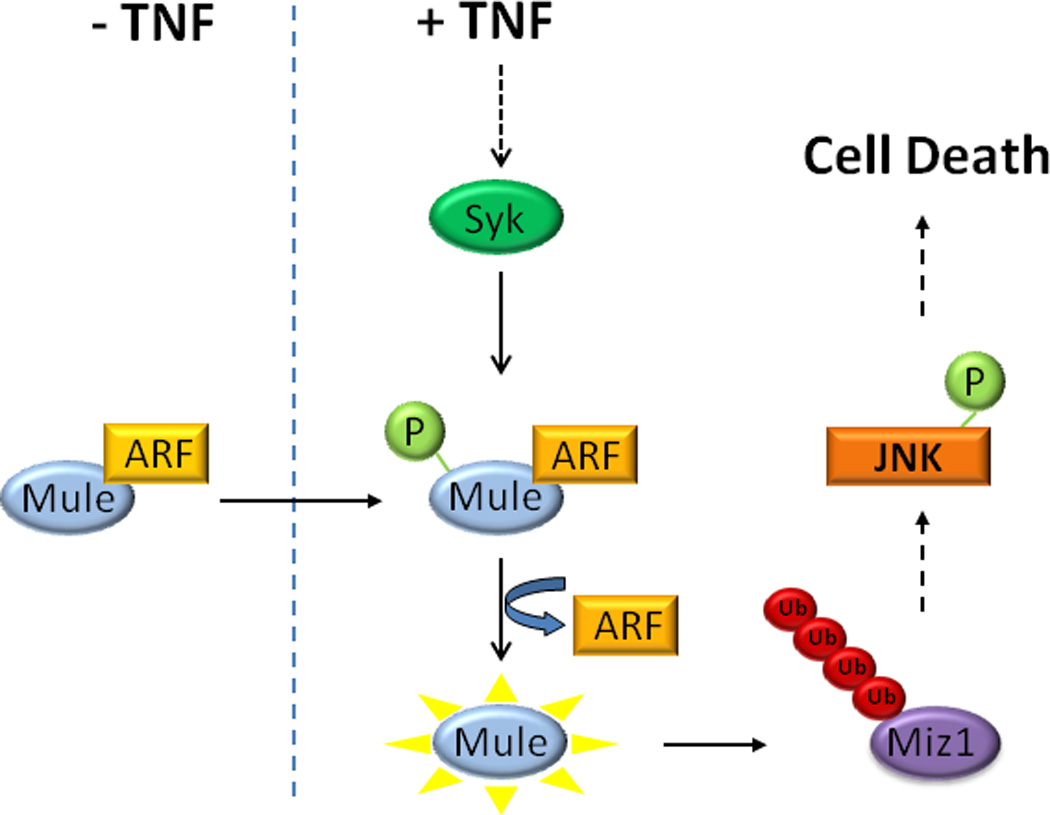
TNF induces tyrosine phosphorylation of Mule via Syk, which then leads to the dissociation of the inhibitory ARF from Mule. Mule becomes activated and catalyzes the K48-linked polyubiquitination of Miz1, promoting its degradation. Degradation of Miz1 relieves its suppression on JNK, allowing JNK to be activated and leading to cell death.
First, ectopic expression of ARF inhibited, while silencing of ARF enhanced Mule-mediated Miz1 ubiquitination in vitro and in vivo (Fig. 1). Second, TNF induced the dissociation of ARF from Mule, which was timely correlated with Mule activation (Fig. 3). Third, Mule was tyrosine phosphorylated in response to TNF (Fig. 4). Finally, Syk selective inhibitor Piceatannol or silencing of Syk prevented the dissociation of ARF, thereby suppressing Mule-mediated Miz1 ubiquitination, JNK activation, and cell death in response to TNF (Fig. 5–6).
A number of studies indicate that Mule has complex biological effects. Chen and colleagues conclude that Mule has an antiapoptotic function (via p53 ubiquitination and degradation), whereas Zhong et al. suggest that Mule is actually proapoptotic (via Mcl-1 ubiquitination and degradation). Our studies indicate that Mule promotes apoptosis through ubiquitination and degradation of Miz1 (26, 32). It is plausible that under different pathophysiological conditions, Mule may target different substrates, leading to different outcomes. This is supported by the fact that the Mule gene is highly expressed in lung and breast carcinomas (19), while in a recent study, Mule was shown to suppress Ras-mediate tumorigenesis by preventing c-Myc/Miz1 complexes accumulation, of which promoting cell-cycle arrest as a barrier to malignant transformation (51).
The tumor suppressor ARF is a critical component of tumor surveillance; it functions as a checkpoint to prevent unrestricted cellular proliferation in response to aberrant oncogenic signaling. The tumor suppressive function of ARF is mainly mediated through suppression of p53 ubiquitin ligases Mdm2 and Mule, which results in p53 stabilization and p53-dependent apoptosis or cell cycle arrest (33, 52). ARF also induce cell cycle arrest in a p53- and Mdm2-independent but Mule-dependent manner (33). Our results demonstrated the role of the ARF-Mule pathway in the regulation of Miz1 stability, thus, Miz1 may be a downstream effector involved in the Mule-mediated but p53-independent function of ARF.
Tyrosine phosphorylation is essential for activation of Mule by TNF, which mediates the dissociation of the inhibitory ARF from Mule. Previously, it has been reported that Mule contains a consensus tyrosine phosphorylation site in its C-terminal HECT domain (53, 54), However, our results showed that this site was not essential for TNF-induced, Mule-mediated Miz1 ubiquitination. Future studies are needed to identify the tyrosine phosphorylation site of Mule that is required for ARF dissociation and activation of Mule in response to TNF.
TNF controls the dynamic interaction between ARF and Mule. Previously, it has been shown that Mule binds to numerous partners, which often regulate Mule activity, under different pathophysiological conditions (20–27, 33). In the case of ARF and Mule interaction, it was reported that Mule E3 ubiquitin ligase activity is significantly inhibited by its binding partner ARF (19). However, whether ARF-Mule interaction can be regulated by extracellular signals and if so, how signal-induced dynamic interaction affects Mule activity was not known. Our data show that TNF controls the dynamic interaction between ARF and Mule through Syk-mediated tyrosine phosphorylation of Mule, thereby regulating Mule E3 ligase activity.
Regulation of ARF-Mule interaction by Syk-mediated tyrosine phosphorylation of Mule may have broad implications. Mule is involved in many important cellular activities, including transcriptional regulation, DNA replication, DNA damage response and repair, and apoptosis (20–27, 33). Syk-mediated tyrosine phosphorylation of Mule, which disrupts ARF-Mule interaction, might be the common activation step in Mule-mediated ubiquitination of other substrates. It will be of interest to investigate whether the ARF-Mule interaction is altered under different pathophysiological conditions. The ARF-Mule interaction may also have therapeutic implications; it might represent an attractive target for manipulation of TNF-JNK signaling pathway without affecting the other functions of TNF and/or ARF.
Materials and Methods
Reagents, Plasmids, and siRNAs
The antibody against Miz1 (sc-22837), ubiquitin (sc-8017), LaminA/C (sc-20681), TNF-R1 (H-5, sc-8436), TRAF2 (N-19, sc-877) and Xpress (sc-499) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies against phospho-JNK (4671) and Syk (2712P) were from Cell Signaling (Danvers, MA, USA). The antibody against JNK (554285) was from Pharmingen (San Diego, CA, USA). The antibody against K48-ubiquitin (05-1307) was from Millipore (Billerica, MA, USA). The antibody against Mule (4213) was from ProSci (Poway, CA, USA). The antibodies against p19ARF (ab26696) and P14ARF (ab11048), and the inhibitor PF-562271 (ab141360) were from Abcam (Cambridge, MA, USA). Glutathione-sepharose beads (95016-984) was from GE (Pittsburgh, PA, USA). Ubiquitin aldehyde (U-201), purified E1 (U5633), cycloheximide (CHX) (C7698), MG-132 (C2211), and the antibodies against M2 (F3165), β-actin (A1978) and HA (H9658) were from Sigma-Aldrich (St. Louis, MO, USA). TNF (410-MT-050) was from R&D Systems (Minneapolis, MN, USA). Recombinant E2 (UbcH5) (E2-622), and HA-ubiquitin (U-110) were from Boston Biochem (Cambridge, MA, USA). Genistein (345834), Okadaic Acid (495604), and Piceatannol (527948) were from Calbiochem (Billerica, MA, USA). 1-Naphthyl PP1 (10954) was from Cayman Chemical (Ann Arbor, MI, USA). λ-phosphatase (P0753S) was from New England Biolabs (Ipswich, MA, USA). Ni-NTA Magnetic Agarose beads (36111) were from QIAGEN (Valencia, CA, USA). GST-Miz1, Xpress-Miz1, HA-ubiquitin (WT), and M2-Mule (WT) were described previously (26, 32, 55). Y4271F (Mule-M1) mutation was introduced into M2-Mule (WT) using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, 200521-5, Santa Clara, CA, USA). His-tagged Xpress-ARF from HEK293 cells was purified under native conditions using NiNTA Agarose beads (Qiagen), according to the manufacturer’s instructions. All constructs were verified by DNA sequencing. The p14Arf siRNA oligos (GAACAUGGUGCGCAGGUUC), p19Arf siRNA oligos (AGGUGAUGAUGAUGGGCAA), and Syk SMARTpool siRNA (L-041084-00) were from Thermo Scientific (Waltham, MA, USA).
siRNA transfection
Cells were transfected with the scrambled siRNA control (siCtrl), or ARF siRNA (20 nM for WT MEFs and MLE12 cells; 100 nM for HEK293 cells), or Syk siRNA (200 nM for WT MEFs). Twenty-four hr later, cells were serum-starved for 24 hr followed by treatment with TNF.
Cell Culture, Transfection, and Apoptosis Assays
Cell culture conditions and transfections were described previously (55, 56). siRNAs were transfected using Lipofectamine 2000 (Invitrogen, 52887). Plasmids were transfected using Turbofect (Thermo Scientific, R0531). Apoptosis assays were performed as we described previously (32, 57, 58).
Coimmunoprecipitation
Cell extracts were prepared using the co-IP buffer (20 mM Tris, pH 7.6, 100 mM NaCl, 1 mM EDTA, 0.1% Nonidet P-40, 1 mM DTT, 10 mM p-nitrophenyl phosphate, 1 µg/mL aprotinin, and 1 mM Na3VO4) and incubated with specific antibody pre-coupled with Protein A Sepharose beads for overnight at 4 °C. Immune complexes were washed with co-IP buffer three times, and then analyzed by immunoblotting.
In Vitro Ubiquitination Assay
Purified GST-Xpress-Miz1 (100 ng) was incubated with E1 (100 ng), E2 UbcH5 (500 ng), ubiquitin (10 µg), and immunoprecipitated Mule, in an ATP-regenerating system (50 mM Tris, pH 7.6, 5 mM MgCl2, 2 mM ATP, 10 mM creatine phosphate, 3.5 U/mL creatine kinase, and 0.6 U/mL inorganic pyrophosphatase) in the presence of MG-132 (50 µM) at 37 °C for 1 hr.
In Vivo Ubiquitination Assay
Cells were pretreated with MG-132 (50 µM) for 4 hr before TNF stimulation. Cells were harvested with 150 µL M2 buffer (20 mM Tris, pH 7.6, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 0.5% Nonidet P-40, 1 mM DTT, 1 µg/mL aprotinin, 10 mM p-nitrophenyl phosphate, 1 mM Na3VO4, 0.8 mM NEM and 5 µM ubiquitin aldehyde), followed by the addition of SDS to a final concentration of 1% and boiled for 5 min. Cell extracts were then diluted 10-fold in M2 buffer before immunoprecipitation. Ubiquitinated proteins were analyzed by immunoblotting with ubiquitin antibody.
Statistical analysis
Data were analyzed by two-way ANOVA, P values as indicated, with the assumption of normal distribution of data and equal sample variance. Sample sizes were selected on the basis of preliminary results to ensure an adequate power. All cells used for the study were included for the statistical analysis, with no randomization or blinding involved. No exclusion of data points was used.
Supplementary Material
(A) WT MEFs were pre-treated with Genistein followed by treatment with TNF plus CHX for various times. Apoptotic cell death was determined. (B) WT or ARF−/−MEFs were treated with TNF plus CHX for various times. Apoptotic cell death was determined. (C) WT fibroblasts transfected with control siRNA or Syk siRNA were treated with TNF plus CHX for various times. Apoptotic cell death was determined. Results were presented as means ± s.e.m. and represent two independent experiments (A–C). *, P < 0.05 by two-way ANOVA. (D) WT or ARF−/− MEFs were stimulated with TNF for 15 min. Phosphorylation of JNK was determined. (E) WT MEFs were transfected with control siRNA or ARF siRNA, followed by treatment with TNF for 15 min. Phosphorylation of JNK was determined.
Acknowledgments
We thank Anning Lin (University of Chicago) for valuable reagents that made this work possible. We thank Jie Yan (University of Chicago), Hong Kuang (Northwestern University), and Chi Do (Northwestern University) for excellent technical assistance. This work was partially supported by National Institutes of Health (GM081603-03, HL114763-01A1) (J.L.), and the Lung Science Training Program (2T32HL076139-11A1) (C.L).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992 May;13(5):151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 2.Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med. 1994;45:491–503. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- 3.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998 Aug 28;281(5381):1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 4.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993 Nov;7(11):2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 5.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994 May 12;369(6476):156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Guyton KZ, Gorospe M, Xu Q, Lee JC, Holbrook NJ. Differential activation of ERK, JNK/SAPK and P38/CSBP/RK map kinase family members during the cellular response to arsenite. Free Radic Biol Med. 1996;21(6):771–781. doi: 10.1016/0891-5849(96)00176-1. [DOI] [PubMed] [Google Scholar]
- 7.Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J Biol Chem. 2001 Sep 28;276(39):36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 8.Yeh KW, Chen JC, Lin MI, Chen YM, Lin CY. Functional activity of sporamin from sweet potato (Ipomoea batatas Lam.): a tuber storage protein with trypsin inhibitory activity. Plant Mol Biol. 1997 Feb;33(3):565–570. doi: 10.1023/a:1005764702510. [DOI] [PubMed] [Google Scholar]
- 9.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996 Nov 1;87(3):565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 10.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003 Jul 25;114(2):181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 11.Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997 Nov;7(5):715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 12.Takada Y, Aggarwal BB. TNF activates Syk protein tyrosine kinase leading to TNF-induced MAPK activation, NF-kappaB activation, and apoptosis. J Immunol. 2004 Jul 15;173(2):1066–1077. doi: 10.4049/jimmunol.173.2.1066. [DOI] [PubMed] [Google Scholar]
- 13.Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006 Dec;7(12):1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, et al. The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A. 2004 Apr 20;101(16):6158–6163. doi: 10.1073/pnas.0401602101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002 Apr;16(4):547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 16.Obergfell A, Eto K, Mocsai A, Buensuceso C, Moores SL, Brugge JS, et al. Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol. 2002 Apr 15;157(2):265–275. doi: 10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poole A, Gibbins JM, Turner M, van Vugt MJ, van de Winkel JG, Saito T, et al. The Fc receptor gamma-chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. EMBO J. 1997 May 1;16(9):2333–2341. doi: 10.1093/emboj/16.9.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vines CM, Potter JW, Xu Y, Geahlen RL, Costello PS, Tybulewicz VL, et al. Inhibition of beta 2 integrin receptor and Syk kinase signaling in monocytes by the Src family kinase Fgr. Immunity. 2001 Oct;15(4):507–519. doi: 10.1016/s1074-7613(01)00221-7. [DOI] [PubMed] [Google Scholar]
- 19.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule Is a Critical Mediator of the ARF Tumor Suppressor. Cell. 2005;121(7):1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 20.Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005 Nov 4;123(3):409–421. doi: 10.1016/j.cell.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 21.Hall JR, Kow E, Nevis KR, Lu CK, Luce KS, Zhong Q, et al. Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Mol Biol Cell. 2007 Sep;18(9):3340–3350. doi: 10.1091/mbc.E07-02-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herold S, Hock A, Herkert B, Berns K, Mullenders J, Beijersbergen R, et al. Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J. 2008 Nov 5;27(21):2851–2861. doi: 10.1038/emboj.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Oughtred R, Wing SS. Characterization of E3Histone, a novel testis ubiquitin protein ligase which ubiquitinates histones. Mol Cell Biol. 2005 Apr;25(7):2819–2831. doi: 10.1128/MCB.25.7.2819-2831.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noy T, Suad O, Taglicht D, Ciechanover A. HUWE1 ubiquitinates MyoD and targets it for proteasomal degradation. Biochem Biophys Res Commun. 2012 Feb 10;418(2):408–413. doi: 10.1016/j.bbrc.2012.01.045. [DOI] [PubMed] [Google Scholar]
- 25.Parsons JL, Tait PS, Finch D, Dianova II, Edelmann MJ, Khoronenkova SV, et al. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009 Oct 21;28(20):3207–3215. doi: 10.1038/emboj.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Do H, Tian X, Zhang C, Liu X, Dada LA, et al. E3 ubiquitin ligase Mule ubiquitinates Miz1 and is required for TNFalpha-induced JNK activation. Proc Natl Acad Sci U S A. 2010 Jul 27;107(30):13444–13449. doi: 10.1073/pnas.0913690107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005 Jul 1;121(7):1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Kosan C, Saba I, Godmann M, Herold S, Herkert B, Eilers M, et al. Transcription factor miz-1 is required to regulate interleukin-7 receptor signaling at early commitment stages of B cell differentiation. Immunity. 2010 Dec 14;33(6):917–928. doi: 10.1016/j.immuni.2010.11.028. [DOI] [PubMed] [Google Scholar]
- 29.Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M. An alternative pathway for gene regulation by Myc. EMBO J. 1997 Sep 15;16(18):5672–5686. doi: 10.1093/emboj/16.18.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wanzel M, Kleine-Kohlbrecher D, Herold S, Hock A, Berns K, Park J, et al. Akt and 14-3-3eta regulate Miz1 to control cell-cycle arrest after DNA damage. Nat Cell Biol. 2005 Jan;7(1):30–41. doi: 10.1038/ncb1202. [DOI] [PubMed] [Google Scholar]
- 31.Do-Umehara HC, Chen C, Urich D, Zhou L, Qiu J, Jang S, et al. Suppression of inflammation and acute lung injury by Miz1 via repression of C/EBP-delta. Nat Immunol. 2013 May;14(5):461–469. doi: 10.1038/ni.2566. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Liu J, Zhao Y, Eilers M, Lin A, Herold S, Hock A, et al. Miz1 is a signal- and pathway-specific modulator or regulator (SMOR) that suppresses TNF-alpha-induced JNK1 activation. Proc Natl Acad Sci U S A. 2009 Oct 27;106(43):18279–18284. doi: 10.1073/pnas.0906328106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005 Jul 1;121(7):1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 34.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003 Nov;12(5):1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 35.Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 2004 Feb;24(3):985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, et al. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev. 1998 Aug 1;12(15):2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan SH, Moritsugu J, Wahl GM. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc Natl Acad Sci U S A. 2000 Mar 28;97(7):3266–3271. doi: 10.1073/pnas.050560997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008 Jan 15;68(2):352–357. doi: 10.1158/0008-5472.CAN-07-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006 May 19;22(4):463–475. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001 Nov 15;414(6861):313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 41.Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011 Jan 27;69(2):231–243. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 42.Gallagher E, Gao M, Liu YC, Karin M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci U S A. 2006 Feb 7;103(6):1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004 Oct 8;306(5694):271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 44.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009 Jun;10(6):398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 45.Gu J, Dubner R, Fornace AJ, Jr, Iadarola MJ. UREB1, a tyrosine phosphorylated nuclear protein, inhibits p53 transactivation. Oncogene. 1995 Nov 16;11(10):2175–2178. [PubMed] [Google Scholar]
- 46.Gu J, Ren K, Dubner R, Iadarola MJ. Cloning of a DNA binding protein that is a tyrosine kinase substrate and recognizes an upstream initiator-like sequence in the promoter of the preprodynorphin gene. Brain Res Mol Brain Res. 1994 Jul;24(1–4):77–88. doi: 10.1016/0169-328x(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 47.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996 Jan 26;84(2):299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi M, Rothe M, Goeddel DV. Anatomy of TRAF2. Distinct domains for nuclear factor-kappaB activation and association with tumor necrosis factor signaling proteins. J Biol Chem. 1996 Aug 16;271(33):19935–19942. doi: 10.1074/jbc.271.33.19935. [DOI] [PubMed] [Google Scholar]
- 49.Honda R, Yasuda H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999 Jan 4;18(1):22–27. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rocha S, Campbell KJ, Perkins ND. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol Cell. 2003 Jul;12(1):15–25. doi: 10.1016/s1097-2765(03)00223-5. [DOI] [PubMed] [Google Scholar]
- 51.Inoue S, Hao Z, Elia AJ, Cescon D, Zhou L, Silvester J, et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013 May 15;27(10):1101–1114. doi: 10.1101/gad.214577.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006 Sep;6(9):663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 53.Gu J, R K, Dubner R, Iadarola MJ. Cloning of a DNA binding protein that is a tyrosine kinase substrate and recognizes an upstream initiator-like sequence in the promoter of the preprodyorphin gene. Molecular Brain Research. 1994;24:77–88. doi: 10.1016/0169-328x(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 54.Gu Jun, D R, Fornace Albert J, Jr, Iadarola MJ. UREB1, a tyrosine phosphorylated nuclear protein, inhibits p53 transactivation. Oncogene. 1995;11:2175–2178. [PubMed] [Google Scholar]
- 55.Liu J, Yan J, Jiang S, Wen J, Chen L, Zhao Y, et al. Site-specific ubiquitination is required for relieving the transcription factor Miz1-mediated suppression on TNF-alpha-induced JNK activation and inflammation. Proc Natl Acad Sci U S A. 2012 Jan 3;109(1):191–196. doi: 10.1073/pnas.1105176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, Minemoto Y, Lin A. c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis. Mol Cell Biol. 2004 Dec;24(24):10844–10856. doi: 10.1128/MCB.24.24.10844-10856.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, et al. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995 Apr 14;268(5208):286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 58.Liu J, Yang D, Minemoto Y, Leitges M, Rosner MR, Lin A. NF-kappaB is required for UV-induced JNK activation via induction of PKCdelta. Mol Cell. 2006 Feb 17;21(4):467–480. doi: 10.1016/j.molcel.2005.12.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) WT MEFs were pre-treated with Genistein followed by treatment with TNF plus CHX for various times. Apoptotic cell death was determined. (B) WT or ARF−/−MEFs were treated with TNF plus CHX for various times. Apoptotic cell death was determined. (C) WT fibroblasts transfected with control siRNA or Syk siRNA were treated with TNF plus CHX for various times. Apoptotic cell death was determined. Results were presented as means ± s.e.m. and represent two independent experiments (A–C). *, P < 0.05 by two-way ANOVA. (D) WT or ARF−/− MEFs were stimulated with TNF for 15 min. Phosphorylation of JNK was determined. (E) WT MEFs were transfected with control siRNA or ARF siRNA, followed by treatment with TNF for 15 min. Phosphorylation of JNK was determined.