

Abstract
The extremes of exercise capacity and health are considered a complex interplay between genes and the environment. In general, the study of animal models has proven critical for deep mechanistic exploration that provides guidance for focused and hypothesis-driven discovery in humans. Hypotheses underlying molecular mechanisms of disease and gene/tissue function can be tested in rodents to generate sufficient evidence to resolve and progress our understanding of human biology. Here we provide examples of three alternative uses of rodent models that have been applied successfully to advance knowledge that bridges our understanding of the connection between exercise capacity and health status. First we review the strong association between exercise capacity and all-cause morbidity and mortality in humans through artificial selection on low and high exercise performance in the rat and the consequent generation of the “energy transfer hypothesis.” Second we review specific transgenic and knockout mouse models that replicate the human disease condition and performance. This includes human glycogen storage diseases (McArdle and Pompe) and α-actinin-3 deficiency. Together these rodent models provide an overview of the advancements of molecular knowledge required for clinical translation. Continued study of these models in conjunction with human association studies will be critical to resolving the complex gene-environment interplay linking exercise capacity, health, and disease.
Keywords: genetic loci, mice, rats, humans, disease, exercise, performance
the strong statistical association between exercise capacity (aerobic or strength performance) and all-cause morbidity and mortality shape a natural linkage for wanting to understand how the extremes of performance can be a determinant of health and disease. Study of animal models has proven critical for deep exploration that provides guidance for focused and hypothesis driven discovery in humans (71). Here we provide three views on the use of rodent models that have been applied successfully to advance knowledge that bridges our understanding of the connection between exercise capacity and health status.
Rat Models Link Exercise Capacity and Health
Since the 1980s, Koch and Britton have been developing theoretical ideas and subsequently rat models, to provide substrate to resolve the connection between the capacity to convert stored energy into movement (energy transfer) and health. For theory, we employ ideas from evolution as it travels a nonequilibrium thermodynamic path of energy transfer. Given that an individual's exercise capacity can be considered the sum of both genetic (intrinsic) and environmental (acquired) variables, we made models for both of these features via two-way artificial selection.
Human health and clinical observations lead to the energy transfer hypothesis.
Large-scale clinical studies have demonstrated poor aerobic exercise capacity (peak rate of oxygen uptake per unit of body mass, V̇o2 peak, in ml O2·kg−1·min−1) to be a better predictor of morbidity and mortality compared with health risk factors such as high blood pressure, Type 2 diabetes, obesity, and smoking (38, 45, 59). Dysfunctional energy metabolism has been implicated in essentially all age-related disease conditions including cardiac arrhythmias and sudden cardiac death (2). Regular physical activity can reduce the risk of chronic disease and is beneficial in the treatment of numerous age-related diseases (18). Thus, it appears that the capacity for energy transfer (as measured typically through aerobic capacity, V̇o2 peak) could in fact be the central mechanistic determinant underlying the divide between complex disease and health (41): that is to say, the Energy Transfer Hypothesis. Simply stated, if capacity for energy transfer is directly linked to disease, then, via two-way artificial selective breeding of genetically heterogeneous rats, disease should segregate with low V̇o2 peak (as reflected by low running capacity), and resistance to disease would segregate with high V̇o2 peak (high capacity of running) (42).
Theoretical explanation.
Koch and Britton considered it critical to formulate a theoretical explanation that could mechanistically link the capacity for energy transfer (that is, the level of V̇o2 peak) with morbidity and mortality. Presently, three major views help to formulate a principle-based approach and account for the high complexity of multicellular life forms. In 1978, Ilya Prigogine (75) argued that systems will organize with greater complexity if they can dissipate energy faster than the individual parts operating independently (order from disorder). Prigogine declared that “Nonequilibrium thermodynamics leads to general results independent of any specific molecular model.” Contemporary with Prigogine's thesis, Baldwin and Krebs (8) outlined the concept that metabolic cycles evolved for greater energy transfer, and Peter Mitchell (55) discovered the chemiosmotic mechanism of ATP formation. Combined, Krebs and Mitchell defined central features for the molecular specification of energy transfer for motion of mass at the atomic, molecular, cellular, tissue, and organismal levels of organization. The combined ideas of these three investigations formulate two statements to support the energy transfer hypothesis: 1) evolution was underwritten by obligatory energy dissipation mechanisms (entropy) and 2) emergence of complexity was coupled to the high energetic nature afforded by atmospheric oxygen. The central importance of oxygen can be attributed to its ranking high in both abundance and electronegativity (second only to fluorine) compared with all other stable elements. Thus, reduction of oxygen yields a high transfer of energy for each electron exchange reaction. Combined, these ideas support a fundamental explanation for the strong association between low oxygen metabolism (i.e., low V̇o2 peak levels) and poor health that can help to guide interpretation and further hypothesis building.
Reduced intrinsic exercise capacity identifies increased multisystemic disease risk.
Almost two decades ago, Koch and Britton (42) began artificial selection for the first generation of rats to develop strains that contrasted for intrinsic (i.e., untrained) aerobic treadmill running capacity at peak exhaustion. The founder population consisted of ∼200 (male and female) genetically heterogeneous rats (N:NIH stock). Running capacity was assessed in rats at 11 wk of age on 5 consecutive days using an incremental velocity-ramped treadmill running protocol; the single best day of running was used as the criterion for selection. The rats showing lowest (LCR, n = 13) and highest capacity of running (HCR, n = 13) of each sex were selectively grouped and paired for mating. The goal at each generation was to select on trait and to use appropriate rotational breeding methods to maintain genetic diversity (40).
By generation 35 of breeding (2014) the LCR rats exhausted on average after 205 m and the HCR exhausted after 2,293 m of distance run (Fig. 1A). Progress with selection continues for both lines: significance tests for the regression of distance (dependent variable) with generation (independent variable) indicated a very high association (P < 2.2 × 10−16; regression coefficient different from zero) between these variables for both LCR and HCR rat lines. The narrow-sense heritability (h2) of running distance, which estimates the proportion of total phenotypic variance explained by additive effects of genes, was calculated across generations for each line (4). h2 was 0.43 in LCR and 0.47 in HCR rats (77).
Fig. 1.

A: selection for intrinsic running capacity. A violin plot for individual generations for females and males combined. The yellow oval to the left denotes the founder population (NIH:H, n = 153 phenotyped), while green and red ovals are for highest-capacity runners (HCR) and lowest-capacity runners (LCR) over 35 generations of selection. Each of the 71 “violins” conveys the density (via width) of the distribution for maximal distance run for each generation. The white dots indicate the median values, and the black vertical lines extending from the dots represent the range of the data that is within 1.5 times the interquartile range. A violin plot is essentially a box plot to which a rotated kernel density plot is added to each side of the box. B: progressive genetic differentiation revealed by 10K single nucleotide polymorphism genotyping data. A multidimensional scaling (MDS) plot (dimension 1 vs. 2) for 142 genotyped animals in 2 lines (LCR and HCR) and 3 generations, as indicated by different symbols. While the MDS plot is generated in an unsupervised fashion from the 142-by-142 pairwise distance matrix, dimension 1 reflects the HCR/LCR difference, and dimension 2 reflects the difference between generations. These patterns show that the 2 lines formed separate clusters at generation 5 (G5), and diverged further in G14 and G26. [B from Ren et al. (77) as permitted by PLoS One].
In a recent study a total of 142 LCR and HCR rats from three nonadjacent generations were genotyped over 10K single nucleotide polymorphism loci distributed throughout the genome. The data over these 10K markers were used to calculate an overall “genetic distance” among all pairs of animals, and such a distance matrix revealed six clusters corresponding to the two lines of animals for three generations each. When the pairwise distances were visualized in a two-dimensional plot, the HCR/LCR difference is reflected in one direction and the between-generation difference in another direction [Fig. 1B, reproduced from Ren et al. (77) with permission by PLoS One]. The HCR/LCR difference increased from generation 5 (G5) to G14, and further at G26.
Disease-related phenotypes also segregated. Consistent with the energy transfer hypothesis, susceptibility to disease emerged in rats with low capacity running and rats selected for high capacity running were relatively disease resistant (table 1). Clinically relevant changes related to the heart, liver, and skeletal muscle affected cancer susceptibility, cognition, and longevity (aging) in LCR relative to HCR.
Table 1.
Disease risks separate with exercise capacity (LCR relative to HCR)
| ↑ Susceptibility to cancer* Impaired lipid oxidation (71) |
↑ pulmonary hypertension* Alzheimer's neurodegeneration (19) |
↓ levels of spontaneous physical activity (29) ↑ hepatic steatosis (57) |
| Metabolic syndrome (94) | ↑ susceptibility to intracerebral hemorrhage (32) | ↓ Oxphos (81) |
| Disordered sleep (58) | ↑ vulnerability to ventricular fibrillation (35) | premature aging (43) |
| Impaired cognition (92) | ↑ risk of obesity (88) | diminished longevity (43) |
Not yet published; HRC, highest capacity of running; LCR, lowest capacity of running; Oxphos, oxygen phosphorylation rate.
Interestingly, behavior-related traits are also altered between LCR and HCR. In discrimination-reversal and T-maze tasks, the HCR rats significantly outperformed the LCR rats, particularly in phases requiring flexible cognition (67, 92). This involvement of the central nervous system (CNS) and health has recently been highlighted in humans. The CNS-related genes were the strongest contributors to individual variance to body mass index in the largest human genome-wide association study to date (>300,000 subjects) (49). This demonstrates independent support for the involvement of the CNS and significant links with multiple health and disease traits for which further exploration is warranted.
Of major clinical relevance, however, is the response of the LCR/HCR model system to positive and negative health environments. Both exercise training and caloric restriction can reverse the higher health risk features of the LCR, supporting environmental treatment and prevention for these above noted diseases (12).
Reduced training response identifies altered cardiovascular and signaling adaptations.
While exercise training is an effective prescription for a wide array of complex chronic diseases including Type 2 diabetes, coronary heart disease, osteoarthritis, cancer, anxiety, and depression (reviewed in Ref. 73), a key observation in human training studies is that up to 20% of subjects demonstrate little change in aerobic exercise capacity (V̇o2 peak) and can be considered exercise resistant (11).
To explore the inherited components of acquired exercise capacity Koch and Britton (44) developed low response trainers (LRT) and high response trainers (HRT) via two-way artificial selection starting in the year 2002. Maximal treadmill running capacity (distance) was tested before (DIST1) and after (DIST2) standardized aerobic treadmill training over 8 wk. Response to training was calculated as the change in exercise capacity (ΔDIST = DIST2 − DIST1). Both lines were provided the same absolute training. After 15 generations of selection, HRT rats improved on average 223 m run as a result of exercise training, while exercise capacity declined 65 m in LRT rats. The h2 for ΔDIST was 0.10 (Fig. 2, A and B). This lower h2 (0.1) for the response to training models relative to h2 for the above intrinsic models (∼0.4) likely relates to a larger environmental exposure of the training model in estimating the phenotype. The response to training phenotype requires measures over 66 days, whereas the intrinsic phenotype is captured in 5 days. Recall that h2 = additive genetic variance/phenotypic variance and that phenotypic variance = genotypic variance + environmental variance. Thus, the duration of environmental exposure is 13.2 times longer for the estimate of phenotype for the trainer model selection relative to the intrinsic.
Fig. 2.

Selection for response to exercise training in genetically heterogeneous rat populations as part of a large-scale selective breeding program for low (LRT) and high response to training (HRT). A: frequency distribution for the ΔDIST for 152 nonselected N/NIH rats after 8 wk treadmill training shown in ascending order. The brackets indicate the lowest and highest 10th percentile animals that were used as founders to start the LRT and HRT selected lines. Dotted line indicates the mean change in running capacity for the population. B: percentile rank score for the ΔDIST for 178 rats from generation 15 of selection arranged from lowest to highest. Light bars indicate LRT animals, and dark bars indicate HRT animals. Dotted lines indicate the mean change in running capacity for the LRT (light) and HRT (dark) selected lines. With equal training V̇o2 max measures pre and post show no significant changes in LRT group (C) but significant increases in HRT group (D). [A and B from Koch et al. (44) with permission from of American Physiological Society; C and D from Wisløff et al. (93) with permission from the American College of Cardiology.]
At generation 7 of selection, Wisløff et al. (93) evaluated the LRT and HRT with a high-intensity aerobic interval training (HIT) protocol proven superior for increasing V̇o2 peak and cardiac function in rats with low exercise tolerance. HRT and LRT did not differ in V̇o2 peak before training. After HIT, HRT rats demonstrated a 40% increase in V̇o2 peak, whereas it remained unchanged in LRT rats (Fig. 2). Cardiomyocytes from the left ventricle were isolated and prepared for confocal microscopy measurements. HIT produced adaptive changes for 12 morphometric and dynamic measures in cardiomyocytes from HRT rats. HIT uniformly resulted in either nonadaptive or maladaptive changes for these same measures in LRT. A microarray experiment of the LV free wall identified 360 differentially expressed genes (DEGs) between HRT-sedentary relative to LRT-sedentary and 324 DEGs between HRT-trained relative to LRT-trained. Of those, osteoglycin, an extracellular matrix protein, ranked as the greatest DEG and was lower in HRT relative to LRT in both the sedentary and trained conditions (−2.3- and −4.6-fold, respectively).
In generation 15 rats, Lessard et al. (47) discovered that LRT exhibit pronounced dysfunction characterized by insulin resistance, increased adiposity, and impaired exercise-induced angiogenesis in muscle. Mitochondrial capacity of the LRT, however, was intact and increased normally with exercise training, suggesting that mitochondria are not responsible for metabolic dysfunction in low responders. LRT also had increased stress/inflammatory signaling and altered transforming growth factor-β signaling, characterized by hyperphosphorylation of an exercise-regulated phosphorylation site on SMAD2.
In summary, models of the energy transfer hypothesis via rat selection using both the response to training and intrinsic capacity represent tools with which to investigate the link between performance and disease rather than via human correlations alone. This mode of unbiased, heterogeneous animal research is helping to prioritize and dissect the complex molecular mechanisms that determine the divide between health and disease.
Mouse Models of Glycogen Storage Diseases
Inbred mouse models are widely used as an approach to model primary-cause conditions (single gene-disorders) and gene function. Genetically modified mice can be generated and compared under equal environmental and genetic background to model exact phenotypes, mechanism, and treatment. This homogenous, in vivo approach to gene or gene-variant function has made them extremely resourceful for human research. One of the “extreme phenotype models” for studying the biology of human sport performance and human muscle function in health/disease conditions is McArdle disease [glycogenosis or glycogen storage disease (GSD) type V; MIM #232600].
McArdle disease in humans.
This autosomal recessive disorder is caused by deficiency in a key enzyme of muscle glycogen metabolism, the muscle isoform of glycogen phosphorylase or myophosphorylase. This enzyme liberates glucose-1-P from muscle glycogen, thereby generating the substrate for glycolysis (50). Impairment in glycolytic flux leads to a syndrome known as “exercise intolerance,” which typically manifests as acute episodes (or “crises”) of undue muscle fatigue and stiffness (usually upon start of exercise). Such crises frequently cause severe muscle damage or “rhabdomyolysis,” as reflected by high serum levels of the intramuscle protein creatine kinase (CK) sometimes accompanied by myoglobinuria or “dark urine” following exertion. Episodes of exercise intolerance are commonly triggered by isometric muscle contractions (e.g., carrying weights) or by dynamic vigorous activities involving larger muscle mass (e.g., brisk walking, running).
Generation of mouse model of McArdle disease recapitulates the human phenotype.
Despite the existence of naturally occurring models of McArdle disease in Charolais cattle (7) or sheep (87), they are of little practical use owing to the intrinsic difficulty of working with big animals. A “knock-in” mouse model of McArdle disease was therefore generated (64). These mice carry a stop-codon mutation, p.R50X (or p.Arg50*), in exon 1 of the myophosphorylase-encoding gene (pygm). Besides being an ideal model for gene therapy (see below), the reason for introducing the p.R50X variant into the mouse genome was that this is the commonest PYGM pathogenic mutation among Caucasians (62). Given the high homology in the myophosphorylase molecule among mammalian species (36), the corresponding arginine residue in codon 50 of the mouse genome was changed to a stop codon, to mimic the exact genetic variant found in humans.
McArdle (pygm p.R50X/p.R50X) mice recapitulate most phenotypic traits that are characteristic of patients (64) (Fig. 3). They lack myophosphorylase activity in their muscles, and, owing to the block in the first step of glycogenolysis, they have massive muscle glycogen stores (20–30 times above normal). Yet another hallmark of the disease, high serum CK levels accompanied by myoglobinuria following vigorous exercise, is also present. Exercise intolerance was also evident in both wire grip and treadmill tests. In both evaluations, p.R50X/p.R50X mice showed very poor performance compared with their age (2 mo old) and sex-matched wild-type (WT) counterparts, i.e., maximal capacity of running and grip strength (wire hold time) were <10% and ∼24% of normal, respectively. To assess whether there was any evidence of an effect of being a pygm p.R50X carrier, heterozygous mice (p.R50X/WT) were assessed. Despite performing considerably better than their p.R50X/p.R50X counterparts, they had 50% of normal myophosphorylase activity and reduced maximal capacity of running with some showing very poor (“McArdle-like”) grip strength. Humans who are heterozygous for pathogenic PYGM mutations are thought to be free of major disease symptoms, at least during daily life activities. (5) Research using the mouse model suggests that virtually 100% of myophosphorylase function is required to reach normal levels in both aerobic or more muscle strength/power oriented tasks.
Fig. 3.
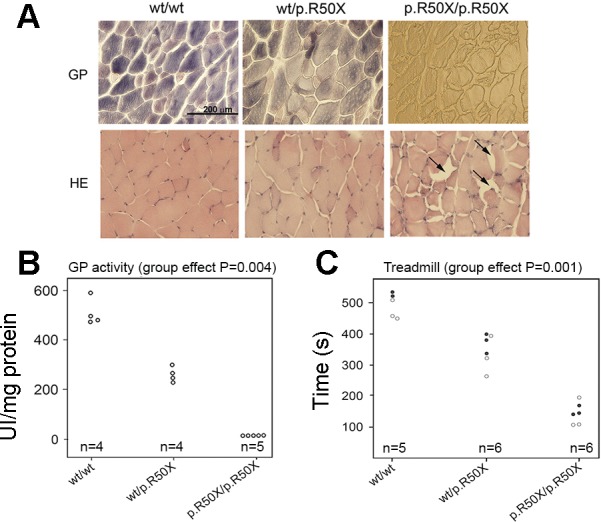
McArdle mouse phenotype. A: representative images of the gastrocnemius muscle indicate gross differences in glycogen. GP, glycogen phosphorylase activity staining; H&E, hematoxylin and eosin staining; wt, wild type. Arrows indicate glycogen accumulation in the homozygote (scale bar 200 μm, all images). B: glycogen phosphorylase activity is reduced with each copy of the p.R50X allele. C: treadmill performance is reduced in a similar dose-dependent manner with the p.R50X allele. [Figure adapted from Nogales-Gadea et al. 2012 (64) used with permission from Oxford University Press.]
To examine the downstream impact of p.R50X on disease, metabolic enzymes and both fast and slow fiber dominant muscles were analyzed (15). The glycolytic/fast-twitch muscles (extensor digitorum longus) of p.R50X/.p.R50X mice showed higher glycogen accumulation compared with more oxidative muscles (gastrocnemius and especially soleus). p.R50X/p.R50X mice exhibited dysregulation in other enzymes of glycogen catabolism/anabolism, including a compensatory downregulation of glycogen synthesis. This is a result of increased activity of the enzyme glycogen synthase in all types of muscles, which is consistent with findings in patients (63), coupled with an upregulated breakdown of glycogen into smaller molecules (i.e., increased activity of the glycogen debranching enzyme, at least in oxidative muscles).
Translational advancements and limitations of the McArdle mouse.
The McArdle mouse model not only provides mechanistic insight on the pathophysiology of the disease and the importance of muscle glycogen availability for proper muscle function, but it also can be used to assess potential therapies that aim to rescue myophosphorylase activity or treat the genetic defect. In contrast to more common knockout (KO) models (where a whole gene is disrupted or artificially removed), the McArdle knock-in mouse has a premature termination codon (PTC) in the pygm gene. Over ∼90% of McArdle patients have mutations generating PTC, which makes it an ideal model to assess gene-correction technologies. This includes the assessment of “read-through” therapies, which are compounds that can synthesize full proteins from short transcripts containing PTCs (65, 66, 80). Other therapies that can now be tested in vivo (22) are deacetylase inhibitors (e.g., valproate), which may help McArdle muscles express the two other isoforms (brain, liver) of glycogen phosphorylase. Although they are not naturally expressed in the adult muscle, the re-expression of alternative isoforms could theoretically compensate for the deficiency of muscle tissue-specific myophosphorylase. Exercise training studies in mice are also underway, with the purpose of understanding the molecular adaptations explaining the exercise benefits shown in patients after moderate-intensity aerobic (31, 53) or low-load resistance (weight lifting) exercise interventions (79).
Since exercise capacity is a main determinant of the clinical severity of the patients, it is important to determine mouse exercise capacity in depth. Some of the tests used to assess the exercise performance in mice were mentioned above, i.e., the wire grip test to assess muscle strength and the treadmill test to evaluate maximal capacity of running (a surrogate of V̇o2 peak) and are traditionally applied in research in the field. A unique hallmark of the disease in adult patients is the “second wind,” i.e., decrease in early exertional tachycardia after ∼10 min of constant-load, submaximal dynamic exercise (brisk walking, bicycling) accompanied by a decrease in muscle pain (50). In mice, the breathing rate during exercise can be detected with specifically designed devices (25); however, the stress-associated heart rate (HR) increase during treadmill exercise is difficult to measure (6). While cardiovascular adaptations can be measured using resting HR as an index of cardiovascular improvement (25), the second wind is the only feature of the disease that has not yet been reported in the McArdle mouse.
Mouse model Pompe disease: clinical translation.
The generation of a mouse model to understand mechanisms and treatment of McArdle disease has been influenced by another glycolytic defect: GSD II or Pompe disease (MIM #232300). Owing to inherited deficiency in the lysosomal enzyme acid maltase (or α-glucosidase) (61), glycogen accumulates not only in muscle, but also in other tissues to result in damage to the heart, liver, and nervous system. Without early diagnosis and enzyme replacement therapy (ERT) the disease can be fatal (10, 30, 48). The development of ERT (intravenous administration of recombinant produced enzyme) involved the use of clinically relevant mouse models of the disease. Further research has involved combination treatment of α-glucosidase replacement and aerobic training in a KO mouse model of Pompe disease. (61) Training for endurance, either with or without ERT, resulted in improved capacity of running, grip strength, motor function, and lean mass but did not reduce glycogen content beyond ERT. Working by mechanisms other than a reduction in glycogen content, this suggests endurance training maybe beneficial as an adjunctive therapy to enzyme replacement in Pompe disease. More recently these exercise benefits have been suggested to be mediated by a rejuvenation of the mitochondrial-lysosomal axis and a reactivation of the cellular clearance pathways. (60)
Mouse Model of α-Actinin-3 Deficiency
α-Actinin-3 deficiency is common in the human population.
The α-actinin-3 (ACTN3) gene is one of the most widely studied genes associated with human skeletal muscle performance. The sarcomeric α-actinins (encoded by ACTN2 and ACTN3) form major structural components of the muscle Z-line where they cross-link the thin filaments (reviewed in Ref. 46). In human skeletal muscle, α-actinin-2 is expressed in all fibers, while the expression of α-actinin-3 is specialized to fast muscle fibers (all fast glycolytic type 2X fibers and 50% of fast oxidative type 2A fibers) (54). In humans, a nonancestral version of the ACTN3 gene exists, which results in conversion of the codon for arginine (R) at position 577 to a premature stop codon (X) (69). While the frequency varies across different ethnic populations, ∼1.5 billion people worldwide are homozygous for the X allele (577XX genotype) and are completely α-actinin-3 deficient.
Answering questions of genetic redundancy with evidence from humans and mice.
With the discovery of this common loss-of-function polymorphism it was thought likely that this was a case of genetic redundancy, particularly as α-actinin-2 and α-actinin-3 were known to have high sequence homology and form homo- and heterodimers (17). However, experimental data in both mice and humans challenged this hypothesis. Sequence comparison of human, mouse, and chicken ACTN genes suggested early divergence and conservation of ACTN2 and 3. The ACTN3 expression pattern did not completely overlap (spatially or temporally) with ACTN2 during embryonic development (54), and most intriguingly, the variation in the frequency of ACTN3 null allele correlated with human migration (Fig. 4) (52). There was unusually low sequence diversity and high long-range linkage disequilibrium among X allele-containing haplotypes compared with the R allele in Europeans and Asians, which is consistent with strong, recent positive selection on X allele in these populations (52). It was therefore possible that the presence of the ACTN3 null genotype was advantageous to some human populations under specific environmental conditions (such as species richness or temperature tolerance).
Fig. 4.
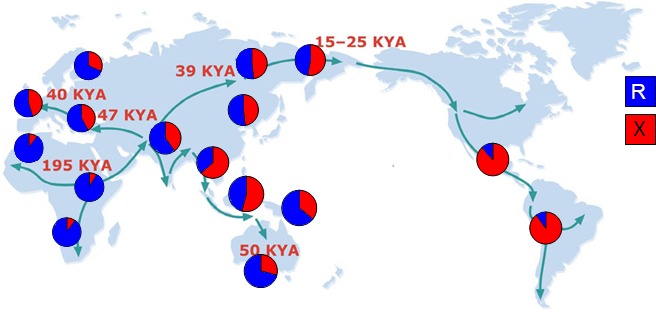
Global frequencies of R577X ACTN3 alleles in native populations. Pie charts indicate the percentage of the ACTN3 577R (blue) and the null 577X (red) alleles in native populations from 16 different regions. Arrows depict human migration out of Africa ∼50 thousand years ago (KYA) [data from MacArthur et al. 2007 (52)].
Since any effect on muscle function was hypothesized to be most readily observable at the extremes of human performance, ACTN3 genotypes were analyzed in elite athletes compared with controls. It was hypothesized that with the ACTN3 genotype the loss of a fast muscle protein could alter the capacity of the individual to perform sprint or endurance activities (27, 89). This initial study demonstrated that the frequency of α-actinin-3 deficiency (577XX genotype) was lower in 107 sprint/power athletes (5%) compared with 436 nonathlete controls (18%; P < 0.0001) (97) (Fig. 5). Remarkably, the association was independently replicated in other elite athletes (14 studies) (96) and the general population (1, 21, 56, 68, 86, 91, 99), suggesting that loss of α-actinin-3 is detrimental to elite sprint and muscle power performance.
Fig. 5.

A: the frequency of R577X ACTN3 genotypes (%RR, RX, and XX) differs in sprint populations (lower frequency of the XX genotype) compared with controls and endurance populations (higher frequency of the XX genotype in females) [data adapted from MacArthur et al. 2008 (51)]. B: WT and Actn3 knockout (KO) mice (pictured respectively) have been indispensable in modelling the human phenotype. Actn3 KO mice do not express α-actinin-3 on Western blot and have an upregulation of α-actinin-2 in skeletal muscle [MacArthur et al. 2008 (51)]. C: the highly conserved domain structures of α-actinin, with an in silico surface representation of the rod domain. The colors represent identical, conservative, and nonconservative residue substitutions when α-actinin-2 and -3 vertebrate sequences are aligned [figure from Lek et al. 2010 (46) used with permission from Elsevier]. D: in vitro and in vivo evidence demonstrates that the loss of α-actinin-3 alters calcineurin signaling, mitochondrial enzyme activity, and calcium handling (increased pump and release rates) to explain their improved fatigue resistance and also may result in enhanced heat production [figure from Head et al. 2015 (33)].
Actn3 KO mouse model mimics human α-actinin-3 deficiency.
To confirm the early associations and explore the molecular mechanisms by which α-actinin-3 deficiency affects muscle function, Actn3 KO mice were generated via targeted deletion of exons 2–7 of Actn3 (52). While this is not an exact “Actn3 R577X” KO mouse, they have complete deficiency of α-actinin-3 protein, by both immunohistochemistry and Western blot, which mimics complete loss in 577XX humans (Fig. 5). KO mice are morphologically indistinguishable from WT littermates, with normal sarcomere formation (52). Consistent with human performance studies, Actn3 KO mice display reduced grip strength but high capacity for endurance running. This reduction in strength is attributable to a reduction in muscle size, specifically due to decreased fast glycolytic 2B fibers (which express α-actinin-3 in WT mice) (51). Given the alterations in strength and performance we employed a systems biology approach over a number of years to investigate the molecular mechanisms influencing muscle metabolism, structure, and signaling.
In the Actn3 KO mouse, there is a compensatory upregulation of the closely related protein α-actinin-2 (52). α-Actinin-2 is the only sarcomeric isoform present in Actn3 KO and 577XX muscle, and this has a secondary effect on downstream pathways and binding partners (reviewed in Ref. 46). We have identified that this includes increases in myofibrillar (desmin, myotilin, y-filamin, ZASP, ALP) and metabolic (COX IV, porin, SERCA1) proteins accompanied with enhanced calcineurin signaling (51, 83, 84). Key metabolic enzymes demonstrate higher capacity for oxidative and fatty-acid oxidation in KO compared with WT mice (including increased activity of citrate synthase and reduced activity glycogen phosphorylase) (52, 76). More recently, we have identified that Actn3 KO mice have altered calcium handling with decreased peak twitch Ca2+ release but improved Ca2+ turnover (increased pump and release rates) to explain their fatigue resistance (33) (Fig. 5). The changes in Ca2+ handling provides a plausible reason for the positive selection of the ACTN3 577X null allele as it is consistent with enhanced cold acclimatization. This may have been beneficial for populations living in cold environments during recent evolution (33), which is a hypothesis we are further investigating in our Actn3 KO mouse model.
These studies demonstrate that α-actinin-3 deficiency leads to a shift toward oxidative metabolism in fast fibers through a combination of 1) decreased activity of glycogen phosphorylase (76), 2) enhanced calcineurin activity (84), and 3) changes in Ca2+ handling properties (33). These mechanistic investigations were influenced by in vitro studies of glycogen phosphorylase (20), mechanistic findings of the calsarcin-2 KO mouse (26), and environmental adaptation of mice to cold exposure (16).
Relevance for mechanism biology and human health.
The ACTN3 R577X polymorphism is one of the most replicated genes in performance and one of the few examples in the literature of demonstrating evidence for positive selection of a loss-of-function allele. Independent studies across different populations have reported that ACTN3 genotype is estimated to contribute ∼1–2.5% of the variance in human muscle strength, performance, and exercise training response (21, 39, 56). While the Actn3 KO mouse model provides an excellent model in which to explore the molecular mechanisms underlying the effects of α-actinin-3 deficiency on skeletal muscle performance, compared with mice, humans possess a much lower relative level of fast fibers and α-actinin-3 in skeletal muscle (reviewed in Ref. 28), which may enhance functional differences of loss of α-actinin-3. We have identified that Actn3 KO mice have enhanced endurance performance in young but not older Actn3 KO mice (82). This may explain some of the variability seen in the ACTN3 genotype and human endurance performance results that show an effect in some (72, 85, 97) but not all human studies (3, 24). Despite this limitation, the reductions in muscle strength/speed (14, 21, 56, 70), fast fiber size (90), and bone mineral density (98) and the alterations in oxidative metabolism (78), glycogen levels (76), and calcineurin activity (84) seen in the Actn3 KO mouse have all been replicated in healthy human cohorts. More recent evidence shows α-actinin-3 deficiency alters muscle adaptation and shift in fiber type (28), which may help explain the association with aging, disuse, and athletic performance (13, 23, 37, 97). The Actn3 mouse model in combination with human association studies has been crucial for modeling and understanding mechanisms of human genetic diversity, health/disease, migration, and environmental adaptation. Importantly, it has also been able to demonstrate roles of diverse structural, metabolic, and signaling function of the skeletal muscle Z-line in fast fibers.
Conclusion
Current research involving consortiums of large characterized public datasets are providing substantial resources to sufficiently power complex genotype-phenotypes of performance, health, and disease. The estimation of the cause and type of gene action at loci underlying quantitative traits, however, remains a major challenge in the field of quantitative genetics.
The caveat for research into common variants associated with health and disease is that each polymorphism only has small effects on health, performance, and anthropometry phenotypes (34), and the additive effect of these variants does not reach anywhere near the twin-study genetic heritability estimates (9, 74, 95). With the genome highly depleted for variants with large functional effects, the continued use of single-gene (followed by multigene) models will be required for accurate biological assessments of the individual candidate variants.
Here we describe in detail three uses of rodent models that have been applied successfully to advance knowledge that bridges our understanding of the connection between exercise capacity and health status. These models have demonstrated 1) a strong association between exercise and all-cause morbidity via the energy transfer hypothesis, 2) mechanisms and treatment for metabolic disease, and 3) human genetic diversity and molecular functions of the muscle Z-line. Unbiased (artificial selection) and targeted rodent (transgenic and KO) gene models, as demonstrated here, can help provide the molecular basis to understand the complexity of health and disease. Use of these models in combination with human studies will enhance predictive power of genome-phenome information.
GRANTS
The LCR/HCR rat model system was funded by the National Institutes of Health's Office of Research Infrastructure Programs/OD Grant R24OD-010950 and by Grant R01DK-099034 (to L. G. Koch and S. L. Britton). Contact L. G. Koch (lgkoch@umich.edu) or S. L. Britton (brittons@umich.edu) for information on the LCR and HCR rats; these rat models are maintained as an international resource with support from the Department of Anesthesiology at the University of Michigan, Ann Arbor, MI. Research by A. Lucia and G. Nogales-Gadea is funded by Fondo de Investigaciones Sanitarias Grants PI12/00914, PI15/01756, and CD14/00032 and Fondos FEDER. F. C. Garton and K. N. North are funded by National Health and Medical Research Council of Australia Grant NHMRC 1002033.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.C.G., K.N.N., L.G.K., S.L.B., G.N.-G., and A.L. conception and design of research; F.C.G., K.N.N., L.G.K., S.L.B., G.N.-G., and A.L. interpreted results of experiments; F.C.G., K.N.N., L.G.K., S.L.B., G.N.-G., and A.L. prepared figures; F.C.G., K.N.N., L.G.K., S.L.B., G.N.-G., and A.L. drafted manuscript; F.C.G., K.N.N., L.G.K., S.L.B., G.N.-G., and A.L. edited and revised manuscript; F.C.G., K.N.N., L.G.K., S.L.B., G.N.-G., and A.L. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Yannis Pitsiladis for input in this manuscript.
REFERENCES
- 1.Ahmetov II, Gavrilov DN, Astratenkova IV, Druzhevskaya AM, Malinin AV, Romanova EE, Rogozkin VA. The association of ACE, ACTN3 and PPARA gene variants with strength phenotypes in middle school-age children. J Physiol Sci 63: 79–85, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akar FG, Aon MA, Tomaselli GF, O'Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest 115: 3527–3535, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alfred T, Ben-Shlomo Y, Cooper R, Hardy R, Cooper C, Deary IJ, Gunnell D, Harris SE, Kumari M, Martin RM, Moran CN, Pitsiladis YP, Ring SM, Sayer AA, Smith GD, Starr JM, Kuh D, Day IN. ACTN3 genotype, athletic status, and life course physical capability: meta-analysis of the published literature and findings from nine studies. Hum Mutat 32: 1008–1018, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62: 1198–1211, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen ST, Duno M, Schwartz M, Vissing J. Do carriers of PYGM mutations have symptoms of McArdle disease? Neurology 67: 716–718, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Andreev-Andrievskiy AA, Popova AS, Borovik AS, Dolgov ON, Tsvirkun DV, Custaud M, Vinogradova OL. Stress-associated cardiovascular reaction masks heart rate dependence on physical load in mice. Physiol Behav 132: 1–9, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Angelos S, Valberg SJ, Smith BP, McQuarrie PS, Shanske S, Tsujino S, DiMauro S, Cardinet GH 3rd. Myophosphorylase deficiency associated with rhabdomyolysis and exercise intolerance in 6 related Charolais cattle. Muscle Nerve 18: 736–740, 1995. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin JE, Krebs H. The evolution of metabolic cycles. Nature 291: 381–382, 1981. [DOI] [PubMed] [Google Scholar]
- 9.Berndt SI, Gustafsson S, Magi R, Ganna A, Wheeler E, Feitosa MF, Justice AE, Monda KL, Croteau-Chonka DC, Day FR, Esko T, Fall T, Ferreira T, Gentilini D, Jackson AU, Luan J, Randall JC, Vedantam S, Willer CJ, Winkler TW, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet 45: 501–512, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhengu L, Davidson A, du Toit P, Els C, Gerntholtz T, Govendrageloo K, Henderson B, Mubaiwa L, Varughese S. Diagnosis and management of Pompe disease. S Afr Med J 104: 273–274, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Bouchard C, An P, Rice T, Skinner JS, Wilmore JH, Gagnon J, Perusse L, Leon AS, Rao DC. Familial aggregation of VO2 max response to exercise training: results from the Heritage family study. J Appl Physiol 87: 1003–1008, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Bowman TA, Ramakrishnan SK, Kaw M, Lee SJ, Patel PR, Golla VK, Bourey RE, Haram PM, Koch LG, Britton SL, Wisløff U, Lee AD, Najjar SM. Caloric restriction reverses hepatic insulin resistance and steatosis in rats with low aerobic capacity. Endocrinology 151: 5157–5164, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broos S, Malisoux L, Theisen D, Francaux M, Deldicque L, Thomis MA. Role of alpha-actinin-3 in contractile properties of human single muscle fibers: a case series study in paraplegics. PLoS One 7: e49281, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broos S, Van Leemputte M, Deldicque L, Thomis MA. History-dependent force, angular velocity and muscular endurance in ACTN3 genotypes. Eur J Appl Physiol 115: 1637–1643, 2015. [DOI] [PubMed] [Google Scholar]
- 15.Brull A, de Luna N, Blanco-Grau A, Lucia A, Martin MA, Arenas J, Marti R, Andreu AL, Pinos T. Phenotype consequences of myophosphorylase dysfunction: insights from the McArdle mouse model. J Physiol 593: 2693–2706, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruton JD, Aydin J, Yamada T, Shabalina IG, Ivarsson N, Zhang SJ, Wada M, Tavi P, Nedergaard J, Katz A. Increased fatigue resistance linked to Ca2+-stimulated mitochondrial biogenesis in muscle fibers of cold-acclimated mice. J Physiol 588: 4275–4288, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan Y, Tong H, Beggs AH, Kunkel LM. Human skeletal muscle-specific α-actinin-2 and-3 isoforms form homodimers and heterodimers in vitro and in vivo. Biochem Biophys Res Commun 248: 134–139, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, Minson CT, Nigg CR, Salem GJ, Skinner JS. American College of Sports Medicine position stand. Exercise and physical activity for older adults. Med Sci Sports Exerc 41: 1510–1530, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Choi J, Chandrasekaran K, Demarest TG, Kristian T, Xu S, Vijaykumar K, Dsouza KG, Qi NR, Yarowsky PJ, Gallipoli R, Koch LG, Fiskum GM, Britton SL, Russell JW. Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann Clin Transl Neurol 1: 589–604, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chowrashi P, Mittal B, Sanger JM, Sanger JW. Amorphin is phosphorylase; Phosphorylase is an alpha-actinin-binding protein. Cytoskeleton 53: 125–135, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Clarkson PM, Devaney JM, Gordish-Dressman H, Thompson PD, Hubal MJ, Urso M, Price TB, Angelopoulos TJ, Gordon PM, Moyna NM, Pescatello LS, Visich PS, Zoeller RF, Seip RL, Hoffman EP. ACTN3 genotype is associated with increases in muscle strength in response to resistance training in women. J Appl Physiol 99: 154–163, 2005. [DOI] [PubMed] [Google Scholar]
- 22.de Luna N, Brull A, Guiu JM, Lucia A, Martin MA, Arenas J, Marti R, Andreu AL, Pinos T. Sodium valproate increases the brain isoform of glycogen phosphorylase: looking for a compensation mechanism in McArdle disease using a mouse primary skeletal-muscle culture in vitro. Dis Model Mech 8: 467–472, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delmonico MJ, Zmuda JM, Taylor BC, Cauley JA, Harris TB, Manini TM, Schwartz A, Li R, Roth SM, Hurley BF. Association of the ACTN3 genotype and physical functioning with age in older adults. J Gerontol Ser A Biol Sci Med Sci 63: 1227–1234, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doring FE, Onur S, Geisen U, Boulay MR, Perusse L, Rankinen T, Rauramaa R, Wolfahrt B, Bouchard C. ACTN3 R577X and other polymorphisms are not associated with elite endurance athlete status in the Genathlete study. J Sports Sci 28: 1355–1359, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Fiuza-Luces C, Soares-Miranda L, Gonzalez-Murillo A, Palacio JM, Colmenero I, Casco F, Melen GJ, Delmiro A, Moran M, Ramirez M, Lucia A. Exercise benefits in chronic graft versus host disease: a murine model study. Med Sci Sports Exerc 45: 1703–1711, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Frey N, Frank D, Lippl S, Kuhn C, Kögler H, Barrientos T, Rohr C, Will R, Müller OJ, Weiler H. Calsarcin-2 deficiency increases exercise capacity in mice through calcineurin/NFAT activation. J Clin Invest 118: 3598, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garland T Jr, Bennett A, Daniels C. Heritability of locomotor performance and its correlates in a natural population. Experientia 46: 530–533, 1990. [Google Scholar]
- 28.Garton FC, Seto JT, Quinlan KG, Yang N, Houweling PJ, North KN. alpha-Actinin-3 deficiency alters muscle adaptation in response to denervation and immobilization. Hum Mol Genet 23: 1879–1893, 2014. [DOI] [PubMed] [Google Scholar]
- 29.Gavini CK, Mukherjee S, Shukla C, Britton SL, Koch LG, Shi H, Novak CM. Leanness and heightened nonresting energy expenditure: role of skeletal muscle activity thermogenesis. Am J Physiol Endocrinol Metab 306: E635–E647, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geel TM, McLaughlin PM, de Leij LF, Ruiters MH, Niezen-Koning KE. Pompe disease: current state of treatment modalities and animal models. Mol Genet Metab 92: 299–307, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Haller RG, Wyrick P, Taivassalo T, Vissing J. Aerobic conditioning: an effective therapy in McArdle's disease. Ann Neurol 59: 922–928, 2006. [DOI] [PubMed] [Google Scholar]
- 32.He Y, Liu W, Koch LG, Britton SL, Keep RF, Xi G, Hua Y. Susceptibility to intracerebral hemorrhage-induced brain injury segregates with low aerobic capacity in rats. Neurobiol Dis 49: 22–28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Head SI, Chan S, Houweling PJ, Quinlan KG, Murphy R, Wagner S, Friedrich O, North KN. Altered Ca2+ kinetics associated with alpha-actinin-3 deficiency may explain positive selection for ACTN3 null allele in human evolution. PLoS Genet 11: e1004862, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong H, Xu L, Liu J, Jones WD, Su Z, Ning B, Perkins R, Ge W, Miclaus K, Zhang L, Park K, Green B, Han T, Fang H, Lambert CG, Vega SC, Lin SM, Jafari N, Czika W, Wolfinger RD, Goodsaid F, Tong W, Shi L. Technical reproducibility of genotyping SNP arrays used in genome-wide association studies. PLoS One 7: e44483, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoydal MA, Stolen TO, Johnsen AB, Alvez M, Catalucci D, Condorelli G, Koch LG, Britton SL, Smith GL, Wisløff U. Reduced aerobic capacity causes leaky ryanodine receptors that trigger arrhythmia in a rat strain artificially selected and bred for low aerobic running capacity. Acta Physiol (Oxf) 210: 854–864, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hudson JW, Hefferon KL, Crerar MM. Comparative analysis of species-independent, isozyme-specific amino-acid substitutions in mammalian muscle, brain and liver glycogen phosphorylases. Biochim Biophys Acta 1164: 197–208, 1993. [DOI] [PubMed] [Google Scholar]
- 37.Judson RN, Wackerhage H, Hughes A, Mavroeidi A, Barr RJ, Macdonald HM, Ratkevicius A, Reid DM, Hocking LJ. The functional ACTN3 577X variant increases the risk of falling in older females: results from two large independent cohort studies. J Gerontol A Biol Sci Med Sci 66: 130–135, 2011. [DOI] [PubMed] [Google Scholar]
- 38.Kavanagh T, Mertens DJ, Hamm LF, Beyene J, Kennedy J, Corey P, Shephard RJ. Peak oxygen intake and cardiac mortality in women referred for cardiac rehabilitation. J Am Coll Cardiol 42: 2139–2143, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Kikuchi N, Yoshida S, Min Sk Lee K, Sakamaki-Sunaga M, Okamoto T, Nakazato K. ACTN3 R577X genotype is associated with muscle function in a Japanese population. App Phys Nutr Metab 4: 316–322, 2015. [DOI] [PubMed] [Google Scholar]
- 40.Kimura M, Crow JF. On the maximum avoidance of inbreeding. Genet Res 4: 399–415, 1963. [Google Scholar]
- 41.Koch LG, Britton SL. Aerobic metabolism underlies complexity and capacity. J Physiol 586: 83–95, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koch LG, Britton SL. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiol Genomics 5: 45–52, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Koch LG, Kemi OJ, Qi N, Leng SX, Bijma P, Gilligan LJ, Wilkinson JE, Wisløff H, Høydal MA, Rolim N, Abadir PM, van Grevenhof EM, Smith GL, Burant CF, Ellingsen O, Britton SL, Wisløff U. Intrinsic aerobic capacity sets a divide for aging and longevity. Circ Res 109: 1162–1172, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch LG, Pollott GE, Britton SL. Selectively bred rat model system for low and high response to exercise training. Physiol Genomics 45: 606–614, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kokkinos P, Myers J, Kokkinos JP, Pittaras A, Narayan P, Manolis A, Karasik P, Greenberg M, Papademetriou V, Singh S. Exercise capacity and mortality in black and white men. Circulation 117: 614–622, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Lek M, North KN. Are biological sensors modulated by their structural scaffolds? The role of the structural muscle proteins α-actinin-2 and α-actinin-3 as modulators of biological sensors. FEBS Lett 584: 2974–2980, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Lessard SJ, Rivas DA, Alves-Wagner AB, Hirshman MF, Gallagher IJ, Constantin-Teodosiu D, Atkins R, Greenhaff PL, Qi NR, Gustafsson T, Fielding RA, Timmons JA, Britton SL, Koch LG, Goodyear LJ. Resistance to aerobic exercise training causes metabolic dysfunction and reveals novel exercise-regulated signaling networks. Diabetes 62: 2717–2727, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci 6: 177, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, Croteau-Chonka DC, Esko T, Fall T, Ferreira T, Gustafsson S, Kutalik Z, Luan Ja Magi R, Randall JC, Winkler TW, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518: 197–206, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lucia A, Nogales-Gadea G, Perez M, Martin MA, Andreu AL, Arenas J. McArdle disease: what do neurologists need to know? Nat Clin Pract Neurol 4: 568–577, 2008. [DOI] [PubMed] [Google Scholar]
- 51.MacArthur DG, Seto JT, Chan S, Quinlan KG, Raftery JM, Turner N, Nicholson MD, Kee AJ, Hardeman EC, Gunning PW, Cooney GJ, Head SI, Yang N, North KN. An Actn3 knockout mouse provides mechanistic insights into the association between alpha-actinin-3 deficiency and human athletic performance. Hum Mol Genet 17: 1076–1086, 2008. [DOI] [PubMed] [Google Scholar]
- 52.MacArthur DG, Seto JT, Raftery JM, Quinlan KG, Huttley GA, Hook JW, Lemckert FA, Kee AJ, Edwards MR, Berman Y, Hardeman EC, Gunning PW, Easteal S, Yang N, North KN. Loss of ACTN3 gene function alters mouse muscle metabolism and shows evidence of positive selection in humans. Nat Genet 39: 1261–1265, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Mate-Munoz JL, Moran M, Perez M, Chamorro-Vina C, Gomez-Gallego F, Santiago C, Chicharro L, Foster C, Nogales-Gadea G, Rubio JC, Andreu AL, Martin MA, Arenas J, Lucia A. Favorable responses to acute and chronic exercise in McArdle patients. Clin J Sport Med 17: 297–303, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Mills M, Yang N, Weinberger R, Vander Woude DL, Beggs AH, Easteal S, North K. Differential expression of the actin-binding proteins, alpha-actinin-2 and -3, in different species: implications for the evolution of functional redundancy. Hum Mol Genet 10: 1335–1346, 2001. [DOI] [PubMed] [Google Scholar]
- 55.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191: 144–148, 1961. [DOI] [PubMed] [Google Scholar]
- 56.Moran CN, Yang N, Bailey ME, Tsiokanos A, Jamurtas A, MacArthur DG, North K, Pitsiladis YP, Wilson RH. Association analysis of the ACTN3 R577X polymorphism and complex quantitative body composition and performance phenotypes in adolescent Greeks. Eur J Hum Genet 15: 88–93, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Morris EM, Jackman MR, Johnson GC, Liu TW, Lopez JL, Kearney ML, Fletcher JA, Meers GM, Koch LG, Britton SL, Rector RS, Ibdah JA, MacLean PS, Thyfault JP. Intrinsic aerobic capacity impacts susceptibility to acute high-fat diet-induced hepatic steatosis. Am J Physiol Endocrinol Metab 307: E355–E364, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muncey A, Saulles A, Baghdoyan HA, Koch LG, Britton SL, Lydic R. Rats bred for low intrinsic aerobic running capacity exhibit decreased and more disrupted sleep compared with those bred for high intrinsic aerobic running capacity. Sleep 31: A27–A28, 2008. [Google Scholar]
- 59.Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med 346: 793–801, 2002. [DOI] [PubMed] [Google Scholar]
- 60.Nilsson MI, MacNeil LG, Kitaoka Y, Suri R, Young SP, Kaczor JJ, Nates NJ, Ansari MU, Wong T, Ahktar M, Brandt L, Hettinga BP, Tarnopolsky MA. Combined aerobic exercise and enzyme replacement therapy rejuvenates the mitochondrial-lysosomal axis and alleviates autophagic blockage in Pompe disease. Free Radic Biol Med 87: 98–112, 2015. [DOI] [PubMed] [Google Scholar]
- 61.Nilsson MI, Samjoo IA, Hettinga BP, Koeberl DD, Zhang H, Hawke TJ, Nissar AA, Ali T, Brandt L, Ansari MU, Hazari H, Patel N, Amon J, Tarnopolsky MA. Aerobic training as an adjunctive therapy to enzyme replacement in Pompe disease. Mol Genet Metab 107: 469–479, 2012. [DOI] [PubMed] [Google Scholar]
- 62.Nogales-Gadea G, Brull A, Santalla A, Andreu AL, Arenas J, Martin MA, Lucia A, de Luna N, Pinos T. McArdle disease: update of reported mutations and polymorphisms in the PYGM gene. Hum Mutat 36: 669–678, 2015. [DOI] [PubMed] [Google Scholar]
- 63.Nogales-Gadea G, Consuegra-Garcia I, Rubio JC, Arenas J, Cuadros M, Camara Y, Torres-Torronteras J, Fiuza-Luces C, Lucia A, Martin MA, Garcia-Arumi E, Andreu AL. A transcriptomic approach to search for novel phenotypic regulators in McArdle disease. PLoS One 7: e31718, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nogales-Gadea G, Pinos T, Lucia A, Arenas J, Camara Y, Brull A, de Luna N, Martin MA, Garcia-Arumi E, Marti R, Andreu AL. Knock-in mice for the R50X mutation in the PYGM gene present with McArdle disease. Brain 135: 2048–2057, 2012. [DOI] [PubMed] [Google Scholar]
- 65.Nogales-Gadea G, Rubio JC, Fernandez-Cadenas I, Garcia-Consuegra I, Lucia A, Cabello A, Garcia-Arumi E, Arenas J, Andreu AL, Martin MA. Expression of the muscle glycogen phosphorylase gene in patients with McArdle disease: the role of nonsense-mediated mRNA decay. Hum Mutat 29: 277–283, 2008. [DOI] [PubMed] [Google Scholar]
- 66.Nogales-Gadea G, Santalla A, Brull A, de Luna N, Lucia A, Pinos T. The pathogenomics of McArdle disease–genes, enzymes, models, and therapeutic implications. J Inherit Metab Dis 38: 221–230, 2015. [DOI] [PubMed] [Google Scholar]
- 67.Noland RC, Thyfault JP, Henes ST, Whitfield BR, Woodlief TL, Evans JR, Lust JA, Britton SL, Koch LG, Dudek RW, Dohm GL, Cortright RN, Lust RM. Artificial selection for high-capacity endurance running is protective against high-fat diet-induced insulin resistance. Am J Physiol Endocrinol Metab 293: E31–E41, 2007. [DOI] [PubMed] [Google Scholar]
- 68.Norman B, Esbjörnsson M, Rundqvist H, Österlund T, Glenmark B, Jansson E. ACTN3 genotype and modulation of skeletal muscle response to exercise in human subjects. J Appl Physiol 116: 1197–1203, 2014. [DOI] [PubMed] [Google Scholar]
- 69.North KN, Yang N, Wattanasirichaigoon D, Mills M, Easteal S, Beggs AH. A common nonsense mutation results in alpha-actinin-3 deficiency in the general population. Nat Genet 21: 353–354, 1999. [DOI] [PubMed] [Google Scholar]
- 70.Orysiak J, Sitkowski D, Zmijewski P, Malczewska-Lenczowska J, Cieszczyk P, Zembron-Lacny A, Pokrywka A. Overrepresentation of the ACTN3 XX genotype in elite canoe and kayak paddlers. J Strength Cond Res 29: 1107–1112, 2015. [DOI] [PubMed] [Google Scholar]
- 71.Overmyer KA, Evans CR, Qi NR, Minogue CE, Carson JJ, Chermside-Scabbo CJ, Koch LG, Britton SL, Pagliarini DJ, Coon JJ, Burant CF. Maximal oxidative capacity during exercise is associated with skeletal muscle fuel selection and dynamic changes in mitochondrial protein acetylation. Cell Metab 21: 468–478, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pasqua LA, Bueno S, Artioli GG, Lancha AH Jr, Matsuda M, Marquezini MV, Lima-Silva AE, Saldiva PH, Bertuzzi R. Influence of ACTN3 R577X polymorphism on ventilatory thresholds related to endurance performance. J Sports Sci: 1–8, 2015. [DOI] [PubMed] [Google Scholar]
- 73.Pedersen BK, Saltin B. Evidence for prescribing exercise as therapy in chronic disease. Scand J Med Sci Sports 16, Suppl 1: 3–63, 2006. [DOI] [PubMed] [Google Scholar]
- 74.Polderman TJC, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, Posthuma D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet 47: 702–709, 2015. [DOI] [PubMed] [Google Scholar]
- 75.Prigogine I. Time, structure, fluctuations. Science 201: 777–785, 1978. [DOI] [PubMed] [Google Scholar]
- 76.Quinlan KG, Seto JT, Turner N, Vandebrouck A, Floetenmeyer M, Macarthur DG, Raftery JM, Lek M, Yang N, Parton RG. α-Actinin-3 deficiency results in reduced glycogen phosphorylase activity and altered calcium handling in skeletal muscle. Hum Mol Genet 19: 1335–1346, 2010. [DOI] [PubMed] [Google Scholar]
- 77.Ren YY, Overmyer KA, Qi NR, Treutelaar MK, Heckenkamp L, Kalahar M, Koch LG, Britton SL, Burant CF, Li JZ. Genetic analysis of a rat model of aerobic capacity and metabolic fitness. PLoS One 8: e77588, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Riedl I, Osler ME, Benziane B, Chibalin AV, Zierath JR. Association of the ACTN3 R577X polymorphism with glucose tolerance and gene expression of sarcomeric proteins in human skeletal muscle. Physiol Rep 3: e12314, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Santalla A, Munguia-Izquierdo D, Brea-Alejo L, Pagola-Aldazabal I, Diez-Bermejo J, Fleck SJ, Ara I, Lucia A. Feasibility of resistance training in adult McArdle patients: clinical outcomes and muscle strength and mass benefits. Front Aging Neurosci 6: 334, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Santalla A, Nogales-Gadea G, Ortenblad N, Brull A, de Luna N, Pinos T, Lucia A. McArdle disease: a unique study model in sports medicine. Sports Med 44: 1531–1544, 2014. [DOI] [PubMed] [Google Scholar]
- 81.Seifert EL, Bastianelli M, Aguer C, Moffat C, Estey C, Koch LG, Britton SL, Harper ME. Intrinsic aerobic capacity correlates with greater inherent mitochondrial oxidative and H2O2 emission capacities without major shifts in myosin heavy chain isoform. J Appl Physiol (1985) 113: 1624–1634, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seto JT, Chan S, Turner N, MacArthur DG, Raftery JM, Berman YD, Quinlan KG, Cooney GJ, Head S, Yang N, North KN. The effect of alpha-actinin-3 deficiency on muscle aging. Exp Gerontol 46: 292–302, 2011. [DOI] [PubMed] [Google Scholar]
- 83.Seto JT, Lek M, Quinlan KG, Houweling PJ, Zheng XF, Garton F, MacArthur DG, Raftery JM, Garvey SM, Hauser MA. Deficiency of α-actinin-3 is associated with increased susceptibility to contraction-induced damage and skeletal muscle remodeling. Hum Mol Genet 20: 2914–2927, 2011. [DOI] [PubMed] [Google Scholar]
- 84.Seto JT, Quinlan KG, Lek M, Zheng XF, Garton F, MacArthur DG, Hogarth MW, Houweling PJ, Gregorevic P, Turner N, Cooney GJ, Yang N, North KN. ACTN3 genotype influences muscle performance through the regulation of calcineurin signaling. J Clin Invest 123: 4255–4263, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shang X, Huang C, Chang Q, Zhang L, Huang T. Association between the ACTN3 R577X polymorphism and female endurance athletes in China. Int J Sports Med 31: 913–916, 2010. [DOI] [PubMed] [Google Scholar]
- 86.Shang X, Zhang F, Zhang L, Huang C. ACTN3 R577X polymorphism and performance phenotypes in young Chinese male soldiers. J Sports Sci 30: 255–260, 2012. [DOI] [PubMed] [Google Scholar]
- 87.Tan P, Allen JG, Wilton SD, Akkari PA, Huxtable CR, Laing NG. A splice-site mutation causing ovine McArdle's disease. Neuromuscul Disord 7: 336–342, 1997. [DOI] [PubMed] [Google Scholar]
- 88.Thyfault JP, Rector RS, Uptergrove GM, Borengasser SJ, Morris EM, Wei Y, Laye MJ, Burant CF, Qi NR, Ridenhour SE, Koch LG, Britton SL, Ibdah JA. Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury. J Physiol 587: 1805–1816, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Damme R, Wilson RS, Vanhooydonck B, Aerts P. Evolutionary biology: performance constraints in decathletes. Nature 415: 755–756, 2002. [DOI] [PubMed] [Google Scholar]
- 90.Vincent B, De Bock K, Ramaekers M, Van den Eede E, Van Leemputte M, Hespel P, Thomis MA. ACTN3 (R577X) genotype is associated with fiber type distribution. Physiol Genomics 32: 58–63, 2007. [DOI] [PubMed] [Google Scholar]
- 91.Walsh S, Liu D, Metter EJ, Ferrucci L, Roth SM. ACTN3 genotype is associated with muscle phenotypes in women across the adult age span. J Appl Physiol 105: 1486–1491, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wikgren J, Mertikas GG, Raussi P, Tirkkonen R, Ayravainen L, Pelto-Huikko M, Koch LG, Britton SL, Kainulainen H. Selective breeding for endurance running capacity affects cognitive but not motor learning in rats. Physiol Behav 106: 95–100, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wisløff U, Bye A, Stolen T, Kemi OJ, Pollott GE, Pande M, McEachin RC, Britton SL, Koch LG. Blunted cardiomyocyte remodeling response in exercise-resistant rats. J Am Coll Cardiol 65: 1378–1380, 2015. [DOI] [PubMed] [Google Scholar]
- 94.Wisløff U, Najjar SM, Ellingsen Ø, Haram PM, Swoap S, Al-Share Q, Fernström M, Rezaei K, Lee SJ, Koch LG, Britton SL. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 307: 418–420, 2005. [DOI] [PubMed] [Google Scholar]
- 95.Wolfarth B, Rankinen T, Hagberg JM, Loos RJ, Perusse L, Roth SM, Sarzynski MA, Bouchard C. Advances in exercise, fitness, and performance genomics in 2013. Med Sci Sports Exerc 46: 851–859, 2014. [DOI] [PubMed] [Google Scholar]
- 96.Yang N, Garton F, North K. Alpha-actinin-3 performance. Med Sport Sci 54: 88–101, 2009. [DOI] [PubMed] [Google Scholar]
- 97.Yang N, MacArthur DG, Gulbin JP, Hahn AG, Beggs AH, Easteal S, North K. ACTN3 genotype is associated with human elite athletic performance. Am J Hum Genet 73: 627–631, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang N, Schindeler A, McDonald MM, Seto JT, Houweling PJ, Lek M, Hogarth M, Morse AR, Raftery JM, Balasuriya D, MacArthur DG, Berman Y, Quinlan KG, Eisman JA, Nguyen TV, Center JR, Prince RL, Wilson SG, Zhu K, Little DG, North KN. alpha-Actinin-3 deficiency is associated with reduced bone mass in human and mouse. Bone 49: 790–798, 2011. [DOI] [PubMed] [Google Scholar]
- 99.Zempo H, Tanabe K, Murakami H, Iemitsu M, Maeda S, Kuno S. Age differences in the relation between ACTN3 R577X polymorphism and thigh-muscle cross-sectional area in women. Genet Test Mol Biomarkers 15: 639–643, 2011. [DOI] [PubMed] [Google Scholar]