

Abstract
We report here the efficient, intramolecular trapping in a Diels–Alder (DA) sense of thermally generated benzynes by one of two pendant arene rings. A more electron rich ring (p-methoxyphenyl) reacted substantially faster than a simple phenyl ring, which was in turn, slightly more reactive vs. a 4-carbomethoxyphenyl ring. Photoinduced di-π-methane rearrangement of the initial DA adducts gave rise to unusual isomeric polycyclic adducts.
Benzene (and its substituted derivatives) are generally unwilling partners in typical Diels-Alder (DA) [4+2] cycloaddition reactions. In fact, a solvent like benzene, toluene, xylene, or 1,2-dichlorobenzene is often used as an effectively inert solvent medium for DA reactions between other pairs of diene/dienophile reactants. The energetic penalty of dearomatization of the benzenoid ring is typically too costly to permit it to participate in cycloaddition. o-Benzyne (1) and its substituted derivatives comprise one of the very few classes of dienophiles with sufficient reactivity to engage a benzenoid as a diene. The first example of the reaction of 1,2-dehydrobenzene (1) with benzene produced benzobar-relene (3a or 3b with X = H) in ≤12% yield (along with biphenyl and benzocycloctatetraene).1 This accentuates the inherent difficulty in DA reactions of arenes, even when a highly energetic benzyne is conscripted as the dienophile.
In 1977 Yoshida, Oda, and coworkers reported2 the relative reactivity of 1 with several monosubstituted benzenoid derivatives (i.e., 2) through sets of bimolecular competition experiments (Scheme 1). The reactivity pattern was that more electron-rich arenes reacted faster; however, the range of relative rates varied only from ca. 15 for the fastest (anisole) to 1 for the slowest (trifluoromethylbenzene). Anisole was also the only trapping agent that showed a propensity to favor addition across the C1/C4 positions of the arene ring (to give 3a); the other derivatives favored formation of the C2/C5 adduct 3b. In all of these experiments the aromatic trapping agents were used as the solvent; the sole isolated yield reported (for the anisole case) were 32% and 8% for 3a (X = OMe) and 3b (X = Me), respectively.
Scheme 1.
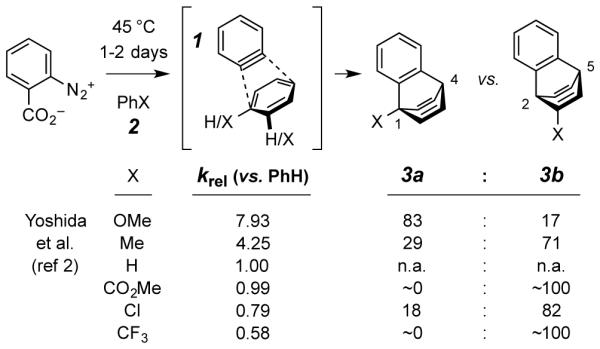
Reported bimolecular Diels–Alder reactions between o-benzyne (1) as the dienophile and various monosubstituted benzene derivatives 2 as the diene partner to give [4+2] adducts 3a-b
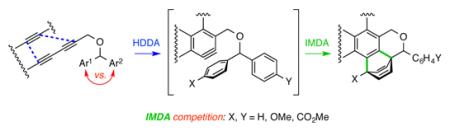
The cycloisomerization of triynes like 4 (and 7) to give benzynes like 5 (and 8) by a simple thermal process3 has considerable generality (Scheme 2).4 We have termed this cyclization a hexadehydro-Diels–Alder (HDDA) reaction. The reactive benzyne intermediates can be trapped in many useful ways, including by reaction in a [4+2] Diels–Alder sense with an aromatic benzenoid ring in either a bimolecular (e.g., 5 to 6)4 or unimolecular (e.g., 8 to 9)4,5 fashion. To understand in more detail the influence on reactivity of substituents on the aromatic ring acting in the role of the diene, we designed a substrate that would pit two aromatic rings in competition for the benzyne dienophile. Specifically, we have prepared and studied the reactivity of a number of substrates of general structure 10, having two aryl groups that bear substituents of differing electronic character, yet otherwise were equally well-poised to engage in intramolecular [4+2] trapping of the HDDA-generated benzyne. The ratio of the constitutionally isomeric products 11 (where X ≠ Y) formed upon heating 10 in an inert solvent would then provide a direct indication of the impact of substituents X vs. Y on the rate of the intramolecular Diels–Alder (IMDA) trapping reaction.
Scheme 2.

Examples of HDDA-generated benzynes reacting with arenes either (a) bimolecularly or (b) unimolecularly and (c) the substrates designed for the intramolecular competition reactions reported here
The substrates examined in this study were triynes 10a-f. Their preparation by cross coupling of the diyne 126 and bromoalkynes 13 (derived from the known benzhydrol derivatives 14) is outlined in Scheme 3 [see the Supporting Information (SI) for details].
Scheme 3.
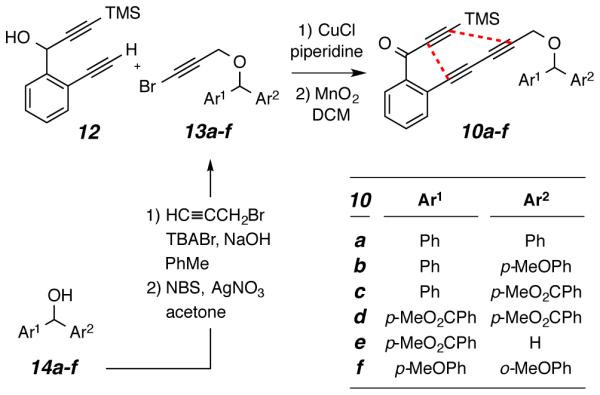
General route used for the synthesis of HDDA triyne substrates 10
The rate-limiting step in the conversion of 10 to 11 (Scheme 2c) is the initial HDDA cycloisomerization reaction; the subsequent Diels–Alder trapping event by the pendant arene is relatively rapid. We have reported that the half-life for the HDDA reaction of a triynone similar to that of 10 is ca. 4 h at 85 °C.4 Accordingly, we heated each of 10a-f in an 80–85 °C bath in a sealed vial containing a solution of the triyne for ca. 48 hours in CDCl3. This solvent has proven repeatedly to be inert toward HDDA-generated benzynes, underscoring the fact that these benzynes should not be considered to engage in reaction processes that are radical in character. Analysis of the 1H NMR spectra of the reaction mixture itself was used to judge the product ratios in the cases where competitive reaction pathways were detected.
The diphenyl-containing triyne 10a gave the trapped adduct 11a (Figure 1.) in 70% yield following chromatographic purification.7 Hence, a simple phenyl ring is an efficient and competent trap for the benzyne intermediate (cf. 5, R = CH2OCHAr1Ar2). The first substrate we studied with two different pendant arenes was the mono-p-methoxy benzhydryl ether 10b. It produced a single isolable IMDA adduct—namely, 11b-maj in 88% yield. We did not definitively detect the isomeric, phenyl-trapped adduct 11b-min (1H NMR). Thus, the more electron-rich arene is considerably more reactive (cf. kanisole/kbenzene = 8, Scheme 1).
Figure 1.

Products from competitive IMDA trapping within benzyne intermediates 15, derived from 10a-f
We then studied the mono-carbomethoxy mono-phenyl ether 10c. In this substrate an electron-poor arene is pitted against the parent phenyl ring. The major product in this case was the phenyl-trapped adduct 11c-maj, isolated in 64% yield. The minor component 11c-min was also isolated following careful separation by HPLC (product ratio = 3.2:1 by 1H NMR analysis of the reaction mixture). An interesting behavior was observed for 11c-min (as well as for the analogous 11d and 11e, which also contain a bridgehead carbomethoxy group). Several resonances in both the 1H and 13C NMR spectra were broadened when recorded at ambient temperature. We attributed this to slow rotation about the bridgehead to carbonyl carbons in these hindered esters. These resonances sharpened considerably when the 1H spectrum was recorded at 100 °C (see SI), consistent with this hindered rotation hypothesis. Similarly, when the bisester-containing substrate 10d (or the mono-ester 10e) was heated (in the dark, see below), only the primary Diels-Alder adduct 11d (or 11e) was observed.
We also examined, in one instance, the effect of substitution pattern. Substrate 10f contains both a para- and an ortho-methoxyphenyl substituent. When heated in 1,2-dichloroethane at 85 °C for 2 days, 10f produced both 11f-maj and the ketone 11f-min8 (8.8:1 ratio from 1H NMR analysis) in 89% and 5% isolated yield (MPLC separation). Oda et al. reported facile in situ hydrolysis of a primary enol ether product arising from anisole trapping to its ketone, the process we also presume gives rise to 11f-min. They also observed a similar preference for formation of the bridgehead rather than the alkenyl methoxylated isomer (cf. Scheme 1).
We examined whether DFT calculations are effective at rationalizing the observed selectivities. Specifically, we have computed the transition state (TS) structures for the competing IMDA trapping reactions of the benzynes derived from 10b, 10c, and 10f. These lead via TS15 (Figure 2a) to the major and minor products of 11b, 11c, and 11f. Also given in Figure 2a are the differences in computed free energy of activation (ΔΔG‡) along with the associated computed and experimentally observed product ratios. DFT does a good job of accounting for the relative energies of the competing TS structures for the trapping of the benzyne. The geometries of those TSs all indicated a high degree of asynchroneity in these concerted IMDA reactions (Figure 2b).
Figure 2.

(a) DFT calculation [M06-2X/6-31G(d) with SMD (chloroform)] of the relative TS energies for the competitive IMDA reactions proceeding via TS15 to 11-maj vs. 11-min. (b) DFT geometries and relative energies for the TSs accounting for the preferential formation of 11f-maj as well as the observed anti relative configuration in 11f-min from 10f.
We were also successful in using DFT to account for the sense of diastereoselectivity (i.e., anti) observed in the formation of 11f-min8 (cf. Figure 1). The TS TS15f- min-anti was favored by 1.8 kcal•mol−1 over the alternative TS15f-min-syn (Figure 2b), presumably reflecting the greater steric congestion in the latter, which possesses cis-(and endo-)oriented methoxy and aryl groups.
We observed that some of the IMDA products containing bridgehead substituents on their benzobicy-clo[2.2.2]octatriene units underwent facile photo-induced rearrangement reactions (Figure 3.). Either ambient laboratory light or exposure of a chloroform solution to a 75 W incandescent light source was sufficient to induce these changes in these typically yellow fluorenone compounds. Specifically, the primary HDDA-IMDA products 11c-min, 11d, and 11e gave rise via di-π-methane rearrangement9 to the cyclopropane-containing products 16c, 16d, and 16e, respectively. Irradiation of 11f in CDCl3 produced a structure whose 1H NMR spectrum was consistent with the cyclopropane 16f, which, upon chromatographic purification on silica gel, cleanly gave rise to the ketone 17f. The structure assignments for 16c and 17f were confirmed by single crystal X-ray analysis. The hydrogen locations in the renderings in Figure 3 are idealized and some have been omitted for clarity. Interestingly, products of trapping by a parents phenyl ring, i.e. 11a and 11c-maj, did not show evidence of the di-π-methane rearrangement, likely reflecting the greater steric strain in those dibenzobar-relenes having a non-hydrogen bridgehead substituent.
Figure 3.
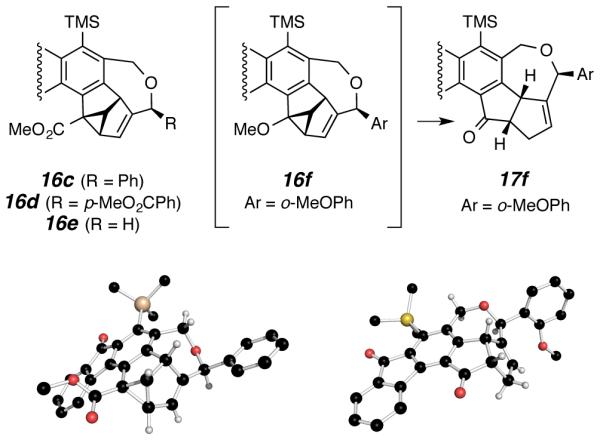
Di-π-methane rearrangement products from light-induced transformations of the primary HDDA-IMDA products 11c-min, 11d, 11e, and 11f-maj.
Finally, a pendant naphthalene substituent was shown to efficiently capture the HDDA benzyne. Triyne 18 gave the Diels–Alder adduct 19 in high yield following chromatographic purification (Figure 4). The in situ 1H NMR spectrum of this reaction mixture is indicative of the cleanliness of this transformation.
Figure 4.

1H NMR spectrum of the product mixture produced directly upon heating 18 in CDCl3 at 85 °C.
In conclusion, we have studied intramolecular Diels-Alder reactions between various pendent aromatic groups to trap thermally generated benzynes. The arene functions as the 4π dienic component and the HDDA benzyne the 2π dienophile. More electron rich arenes are more reactive dienes, consistent with the view of the benzyne as an electrophilic partner. DFT calculations were effective in accounting for the observed product ratios for three substrates containing two different and competing arenes. The transition state structures for these reactions show a substantial degree of asynchroneity (Figure 2b). Adducts containing a bridgehead carbomethoxy substituent show hindered rotation about the ester carbonyl to barrelene bridgehead C–C bond. Some of the initial Diels-Alder adducts—yellow, dibenzobarrelene, fluorenone-containing derivatives—underwent smooth di-π-methane rearrangement when exposed to ambient light. Finally, a pendant naphthalene substituent is a particularly effective diene. Collectively these results provide guidance about what types of substituents can be selected to enhance bimolecular Diels-Alder trapping of arynes by structurally more complex arenes, a topic that is under further study here.
Supplementary Material
ACKNOWLEDGMENT
This investigation was supported by a grant awarded by the U.S. Department of Health and Human Services (National Institute of General Medical Sciences, GM-65597). Portions of this work were performed with hardware and software resources available through the University of Minnesota Supercomputing Institute (MSI). T. W. acknowledges the support of a Wayland E. Noland Fellowship and a University of Minnesota Graduate School Doctoral Dissertation Fellowship. We thank Victor G. Young, Jr. of the University of Minnesota for assistance with the X-ray diffraction analysis.
Footnotes
Supporting Information
Experimental procedures, characterization data, copies of 1H- and 13C NMR spectra for all isolated compounds, ORTEP renderings, and DFT computational methods (146 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
V.P. performed all of the experiments; T.W. performed all of the computations. / The manuscript was written through contributions from all authors. / All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interests.
REFERENCES
- 1.Miller RG, Stiles M. J. Am Chem. Soc. 1963;85:1798–1800. [Google Scholar]
- 2.Tabushi I, Yamada H, Yoshida Z, Oda R. Bull. Chem. Soc. Jpn. 1977;50:285–290. [Google Scholar]
- 3.a Miyawaki K, Suzuki R, Kawano T, Ueda I. Tetrahedron Lett. 1997;38:3943–3946. [Google Scholar]; b Bradley AZ, Johnson RP. J. Am. Chem. Soc. 1997;119:9917–9918. [Google Scholar]
- 4.Hoye TR, Baire B, Niu D, Willoughby PH, Woods BP. Nature. 2012;490:208–212. doi: 10.1038/nature11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niu D, Wang T, Woods BP, Hoye TR. Org. Lett. 2014;16:254–257. doi: 10.1021/ol403258c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suffert J, Abraham E, Raeppel S, Brückner R. Liebigs Ann. 1996:447–456. [Google Scholar]
- 7. We typically observed some amount of competitive cleavage of the benzhydryl ether to its corresponding ketone (or benzylic ether to aldehyde in the case of 10e). For 10a benzophenone itself was ob- served. Although this byproduct formation reduced the overall yield of IMDA products isolated from these experiments, it did not compromise our ability to assess the relative propensity of two different arenes to participate in the Diels–Alder trapping reactions. The IMDA products do not further fragment to give these benzophenone derivatives.
- 8. The relative configuration of the two stereocenters in 11f-min was assigned on the basis of NOE experiments (see Supporting Information)
- 9.Liu RSH, Krespan CG. J. Org. Chem. 1969;34:1271–1278. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.