

Abstract
Glucocorticoids (GCs) have been widely applied to treat patients with chronic obstructive pulmonary disease (COPD). But the effect of GCs was not ideal. This study was to observe whether erythromycin could enhance the anti-inflammatory activity of budesonide in COPD model rats and to explore the mechanism involved. In this study, male Sprague-Dawley rats were divided into five groups: healthy control group (H group), COPD model group (C group), erythromycin group (E group), budesonide group (B group) and erythromycin + budesonide group (E+B group). The rats in groups of C, E, B and E+B were developed into COPD models. Different groups were given different drug interventions. The levels of 8-iso-PGF2α, IL-8, and TNF-α in BALF and serum were measured with ELISA. The protein expression levels of HDAC2, PI3K, and p-AKT in lung tissue were measured with Western-blot and immunohistochemistry. The levels of 8-iso-PGF2α, IL-8, and TNF-α in BALF and serum were lower in E+B group than those in B group and C group (all P<0.001).The protein expression level of HDAC2 was higher and PI3K and p-AKT were lower in E+B group than those in B group and C group (all P<0.001). Moreover, the expression levels of HDAC2 were negatively correlated with the levels of 8-iso-PGF2α, IL-8 and TNF-α both in serum and BALF and the expression levels of PI3K and p-AKT among the five groups, with all P<0.001. We conclude that erythromycin can enhance the anti-inflammatory activity of budesonide in COPD model rats, possibly through inhibiting the PI3K/AKT pathway and enhancing the activity of HDAC2.
Keywords: Budesonide, chronic obstructive pulmonary disease, erythromycin, histone deacetylase 2, PI3K/AKT
Introduction
Chronic obstructive pulmonary disease (COPD) is an airway inflammatory disease. Glucocorticoids (GCs) are the most important and the most effective anti-inflammatory agent and had been widespread applied in patients with COPD, however, the effect was not ideal. Studies have suggested many possible mechanisms reducing the anti-inflammatory effect of glucocorticoid, namely the glucocorticoid resistance, including a decrease in histone deacetylase 2 (HDAC2) activity, abnormality (dysfunction) of the GC receptor, and the lack of a nuclear factor-κB (NF-κB) activation pathway. Among these mechanisms, the decrease in HDAC2 is the main cause of glucocorticoid resistance [1] Erythromycin is a macrolide antibiotic with significant anti-inflammatory and antioxidant activity [2-5]. It can decrease the release of interleukin-8 (IL-8) and the activation of neutrophil [6]. It can also reduce the levels of reactive oxygen species, which are produced by macrophages that have been treated with cigarette smoke extract (CSE) in vitro. In addition, some other macrolide antibiotics were proved that they can inhibit the activation of the PI3K/AKT signalling pathway under oxidative stress (OS) to reduce HDAC2 activity [7] or enhances steroid effects in cells from smoke exposed mice [8]. However, studies about erythromycin whether and how to inhibit OS, inhibit the activation of the PI3K/AKT signaling pathway, enhance HDAC2 activity or reverse GC resistance were rare. Budesonide is the most commonly used hormone in clinical treatments. It can be administered by inhalation and can reduce airway mucosal inflammation. We hypothesis that erythromycin can enhance the sensitivity of budesonide locally or systematically. In this study, COPD model rats were developed by airway instillation of lipopolysaccharide combined with exposure to passive smoking. Then, erythromycin and budesonide were administered to COPD model rats to observe the effect of erythromycin on HDAC2 activity and the anti-inflammatory activity of budesonide and to explore the role of the PI3K/AKT signaling pathway.
Materials and methods
Ethics statement
The experimental protocol was approved by the Experimental Animal Care and Ethics committees of the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China.
Preparation of COPD model
Fifty-three ten-week-old male Sprague-Dawley (SD) rats were purchased from the Experimental Animal Center of Henan province (Zhengzhou, China), weighing 200 ± 20 g (Animal qualified number: Henan 2010-0002). These animals were adapted in a temperature- and humidity-controlled condition and kept on a 12 h light/dark cycle, with free access to food and water. The rats were randomly divided (according to the computer-generated randomisation list) into the healthy control group (H group, n=11) and the LPS + smoke group (n=42). On the 1st and 14th day, after being anaesthetized with 10% chloral hydrate (3 mg/kg, yulong seaweed company, Qingdao, China) by intraperitoneal injection, the rats in the H group and LPS + smoke group were given saline (0.2 ml/rat) and lipopolysaccharide (LPS, Sigma Company) (0.2 ml/rat, 1 g/L) respectively through tracheal instillation. During the 2nd-13th days and 15th-30th days, the rats in the H group and LPS + smoke group were exposed to clean air and cigarette smoke in two same custom-built fumigation chamber, which has a volume of 72 L and a side wall that has a ventilation hole diameter of 1 cm to maintain a stable pressure, to inhale clean air and for the passive smoke exposure (1 cigarette/rat) (lasting 0.5 h for every treatment, 2 times a day, at an interval of at least 6 h), respectively. The cigarette (produced by Henan Tobacco Industry Corporation) tar content was 13 mg, and the nicotine content was 0.8 mg.
Administration
On the 31st day, one rat from H group and two rats from LPS + smoke group were sacrificed to validate the success of the model by measure the lung function and lung tissue morphology in light microcopy of the rats. Then, the rest of the rats in LPS + smoke group were randomly divided (according to the computer-generated randomisation list) into four groups: COPD model group (C group), erythromycin group (E group), budesonide group (B group) and erythromycin + budesonide group (E+B group), with 10 rats in each group. The rats in the four groups were given the following treatment for 2 weeks: C group, given gavages of normal saline (1 time/day, 2 ml/rat); E group, given gavages of erythromycin (1.25% erythromycin liquid, prepared when used, produced by Shanghai Sangon Company) (1 time/day, 100 mg/kg); B group, given aerosol inhalations of a medical budesonide suspension (Pulmicort) (1 time/day, 2 ml/day); E+B group, given gavages of erythromycin (the dose and time as above) and aerosol inhalations of the budesonide suspension (the dose and time as above).
On the 45th day, we tested the pulmonary function of the rats using a small animal spirometer YL-S-001 (Buxco Electronics, Inc. USA), and collected specimens of bronchoalveolar lavage fluid (BALF) from the left lung and blood from the abdominal aorta after being anesthetized.Lung tissues were sampled along the maximum diameter of the right lower lobe.
The level of 8-iso-PGF2α, IL-8, TNF-α measurement by ELISA
The levels of 8-isoPGF2α, IL-8, and TNF-α were quantified using sandwich ELISA (R&D Systems Europe, Abingdon, UK) according to the manufacturer’s instructions.
Morphology
Lung tissues were cut into 3 mm-thick slices and fixed in a 4% paraformaldehyde solution for 72 h. The samples were embedded in paraffin, and 4 μm thick sections were cut and stained with hematoxylin and eosin (HE). Then Olympus PM-10 AD optical microscope and photographic system (Olympus, Tokyo, Japan) were used to observe the morphology.
Immunohistochemistry analysis for the protein expression of HDAC2, PI3K, p-AKT
Immunostaining in formalin fixed, paraffin embedded 4 μm thick sections was performed according to the SP kit instructions. Rabbit anti-HDAC2, PI3K or p-AKT antibody (BioVision Inc. USA; 1:500 dilution) were used as primary antibody. Results were quantified by measuring the integrated optical density (IOD) of the positive staining area.
Western blot analysis for the protein expression of HDAC2, PI3K, p-AKT
Lung tissues were homogenised and total protein was extracted. Fifty micrograms of isolated soluble protein was separated by polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride (PVDF) membranes (PALL, USA), then incubated with polyclonal rabbit anti-HDAC2, PI3K, p-AKT antubody or β-actin (BioVision Inc. USA) according to the instructions. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies (BioVision Inc. USA) were also added.
Data analysis
All statistical analyses were performed with SPSS software version 17.0 (SPSS, Inc., Chicago, IL, USA). The differences were compared with one way analysis of variance (one way-ANOVA) among all the groups. LSD-t test was used for further analysis between two groups. The inspection standard was α =0.05. The correlation coefficients were calculated with the Spearman’s rank method.
Results
Lung function changes in rats
The values of tidal volume (TV), peak expiratory flow (PEF), 50% of tidal volume and peak expiratory flow rate (EF50) of five groups were showed in Table 1.
Table 1.
Lung function changes in rats (x̅±s)
| Group | n | TV (ml) | PEF (ml/s) | EF50 (ml/s) |
|---|---|---|---|---|
| Healthy control group | 10 | 1.87±0.76 | 24.96±11.38 | 1.73±1.02 |
| COPD model group | 10 | 1.25±0.73* | 18.49±3.24* | 1.38±0.21* |
| Budesonide group | 10 | 1.52±0.88† | 19.62±10.25† | 1.47±0.97† |
| Erythromycin group | 10 | 1.68±0.54† | 23.30±10.34† | 1.61±0.54† |
| Erythromycin + budesonide group | 10 | 1.72±0.97† | 24.87±10.58† | 1.68±0.40† |
Compared with the healthy control group, P<0.05;
Compared with the COPD model group, P<0.05.
Light microscopy of lung tissue morphology
Compared with the H group, the tracheal and bronchial epithelial cells were changed in the C group, along with evidence of bronchial smooth muscle thickening, inflammatory cell infiltration, collagen deposition, alveolar wall thinning, or, in some cases, rupture fused to the bulla. There was less changes to the lung tissue in the drug intervention groups than to those in the C group. The changes in the airways of the E+B group rats were the least significant (Figure 1).
Figure 1.
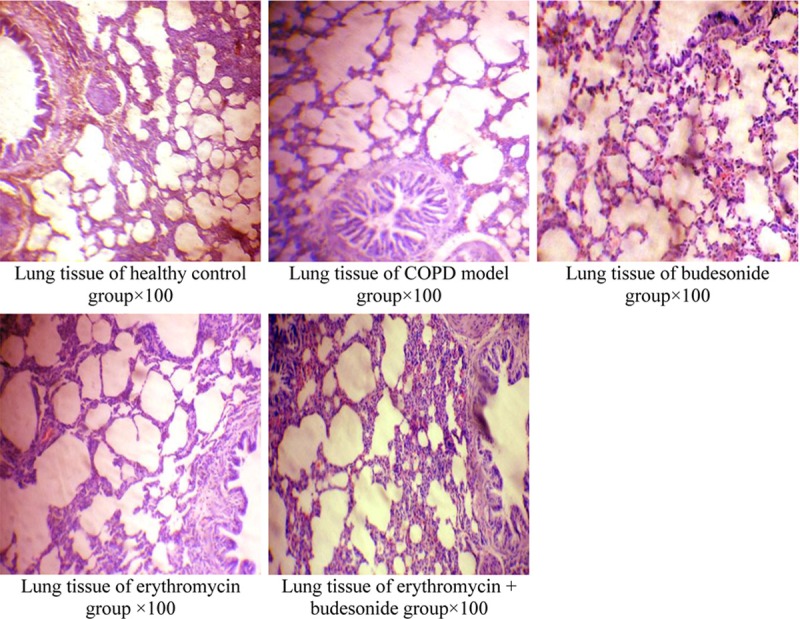
Light microscopy of lung tissue morphology.
Levels of 8-iso-PGF2α, IL-8 and TNF-α in BALF and serum
In the C group, the levels of 8-iso-PGF2α, IL-8, and TNF-α were higher than those of H group, E group, and E+B group in BALF and serum. The levels in C group were higher than those of B group in BALF (all P<0.001), but the difference was not statistically significant in the serum of B group (P=0.435, 0.693, 0.505, respectively). In B group, the BALF and serum levels were higher than those in and E+B group (all P<0.001). The levels of 8-iso-PGF2α in BALF and serum and IL-8 and TNF-α in BALF was lower in E+B group than those in E group, but the difference was no statistically significant (P=0.621, 0.797 and P=0.787, 0.902) (Tables 2 and 3).
Table 2.
The levels of 8-iso-PGF2α in BALF and serum (x̅ ±s)
| Group | n | BALF (ng/ml) | Serum (ng/ml) |
|---|---|---|---|
| Healthy control group | 10 | 19.10±0.91 | 31.65±0.45 |
| COPD model group | 10 | 37.14±0.21* | 40.74±6.86* |
| Budesonide group | 10 | 23.50±1.01† | 38.14±3.37‡ |
| Erythromycin group | 10 | 19.62±0.76† | 28.96±0.55† |
| Erythromycin + budesonide group | 10 | 19.38±0.74†,§,** | 29.73±0.69†,§,** |
Compared with healthy control group, P<0.05;
Compared with COPD model group, P<0.05;
Compared with budesonide group, P<0.05;
Compared with COPD model group, P=0.435;
Compared with erythromycin group, P=0.621 and 0.797.
Table 3.
The levels of IL-8 and TNF-α in BALF and serum (ng/L) (x̅±s)
| Group | n | BALF (ng/L) | Serum (ng/L) | ||
|---|---|---|---|---|---|
|
|
|
||||
| IL-8 | TNF-α | IL-8 | TNF-α | ||
| Healthy control group | 10 | 63.79±3.42 | 110.39±9.38 | 99.16±2.23 | 240.70±6.68 |
| COPD model group | 10 | 127.70±9.32* | 280.81±4.48* | 153.82±2.21* | 436.76±3.33* |
| Budesonide group | 10 | 80.31±3.71† | 170.78±12.51† | 134.97±2.67‡ | 398.68±7.71‡ |
| Erythromycin group | 10 | 64.97±3.05† | 117.59±10.71† | 110.60±4.33† | 275.00±8.28† |
| Erythromycin + budesonide group | 10 | 65.47±2.29†,§,** | 116.86±6.64†,§,** | 102.11±2.35†,§,*** | 252.15±7.95†,§,*** |
Compared with healthy control group, P<0.05;
Compared with COPD model group, P<0.05;
Compared with budesonide group, P<0.05;
Compared with COPD model group, P=0.693 and 0.505;
Compared with erythromycin group, P=0.787, 0.902;
Compared with erythromycin group, P<0.05.
Expression levels of HDAC2, PI3K, and p-AKT in lung tissue by immunohistochemistry and Western blot
The results of immunohistochemistry showed HDAC2 was widely expressed in bronchial mucosal epithelial cells, vascular endothelial cells, alveolar epithelial cells, and alveolar macrophage nuclei. PI3K and P-AKT were mostly expressed in bronchial epithelial cells, airway smooth muscle cells, and inflammatory cells (Figures 2 and 3).
Figure 2.
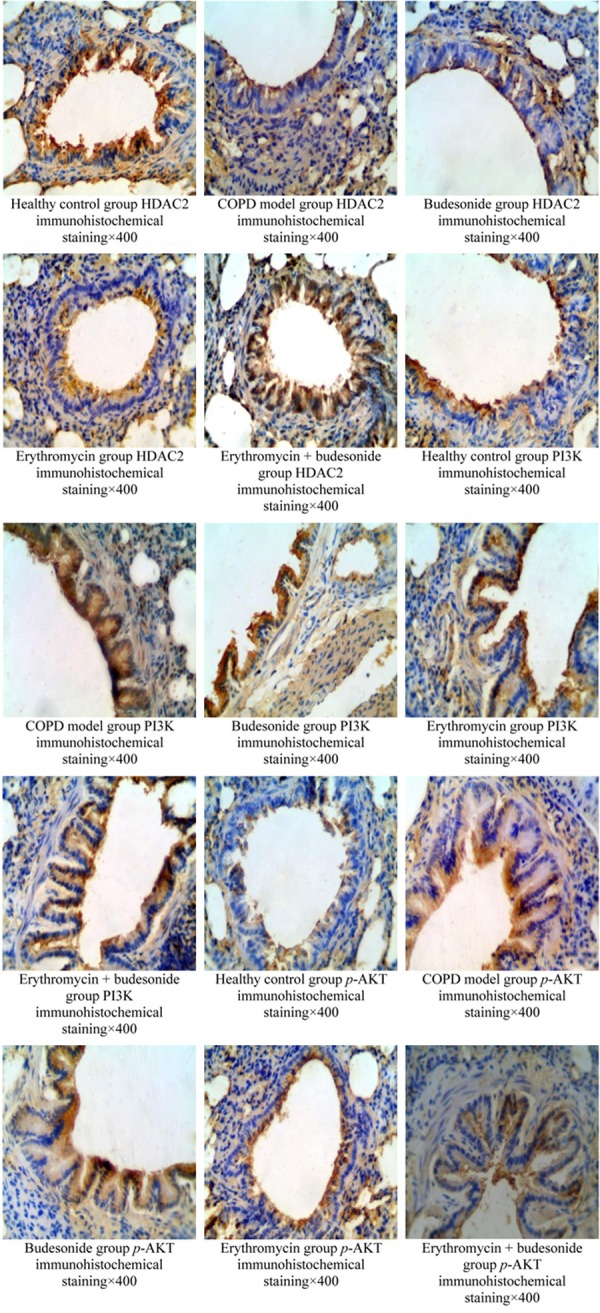
The results of HDAC2, PI3K, p-AKT expression in lung tissue by immunohistochemistry.
Figure 3.
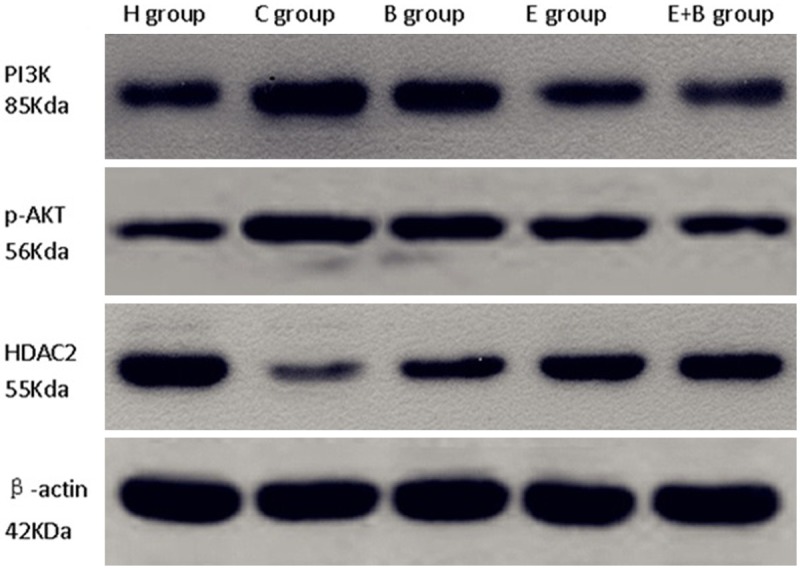
PI3K, p-AKT, HDAC2 protein expression by Western blot.
Immunohistochemistry and Western blot showed that the expression levels of HDAC2 were lower and the PI3K and p-AKT were higher in the C group than in the H group, E group and E+B group (all P<0.001), but the differences in the values between C and B groups were not statistically significant (P=0.145, 0.203, 0.178 and P=0.423, 0.706, 0.693, with immunohistochemistry and Western blot respectively). In the B group, the expression level of HDAC2 was lower, and PI3K and p-AKT were higher than in the E+B group (all P<0.001) (Table 4).
Table 4.
The protein of HDAC2, PI3K, p-AKT expression in lung tissue by Western-blot and the integral optical density (IOD) by immunohistochemistry (x̅ ±s)
| Group | n | Immunohistochemistry | Western-blot | ||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| HDAC2 | PI3K | p-AKT | HDAC2 | PI3K | p-AKT | ||
| Healthy control group | 10 | 90.05±2.38 | 21.22±2.19 | 23.76±1.26 | 0.636±0.032 | 0.193±0.013 | 0.225±0.085 |
| COPD model group | 10 | 18.06±1.27* | 99.15±6.85* | 104.43±5.69* | 0.115±0.014* | 0.745±0.029* | 0.786±0.067* |
| Budesonide group | 10 | 32.49±5.49‡ | 76.43±3.32‡ | 87.64±4.23‡ | 0.201±0.098** | 0.632±0.035** | 0.699±0.049** |
| Erythromycin group | 10 | 53.33±3.24†,§ | 57.72±4.51†,§ | 64.38±3.79†,§ | 0.479±0.046†,§ | 0.458±0.038†,§ | 0.583±0.025†,§ |
| Erythromycin + budesonide group | 10 | 67.02±2.28†,§ | 44.83±2.67†,§ | 57.21±5.65†,§ | 0.501±0.037†,§ | 0.332±0.043†,§ | 0.417±0.087†,§ |
Compared with healthy control group, P<0.05;
Compared with COPD model group, P<0.05;
Compared with budesonide group, P<0.05;
Compared with COPD model group, P=0.145, 0.203, 0.178;
Compared with COPD model group, P=0.423, 0.706, 0.693.
Correlation analysis between the expression levels of HDAC2 in the lung tissue and 8-iso-PGF2α, IL-8, TNF-α in the serum (B) and BALF (F), and the expression levels of PI3K, P-AKT in the lung tissue
HDAC2 protein expression levels in lung tissue were negatively correlated with the levels of 8-iso-PGF2α, IL-8 and TNF-α in serum or BALF among the five groups, Also, the expression levels of HDAC2, PI3K and p-AKT were negatively correlated (Figure 4). The Spearman correlation coefficient were showed in Table 5 (all P<0.001).
Figure 4.
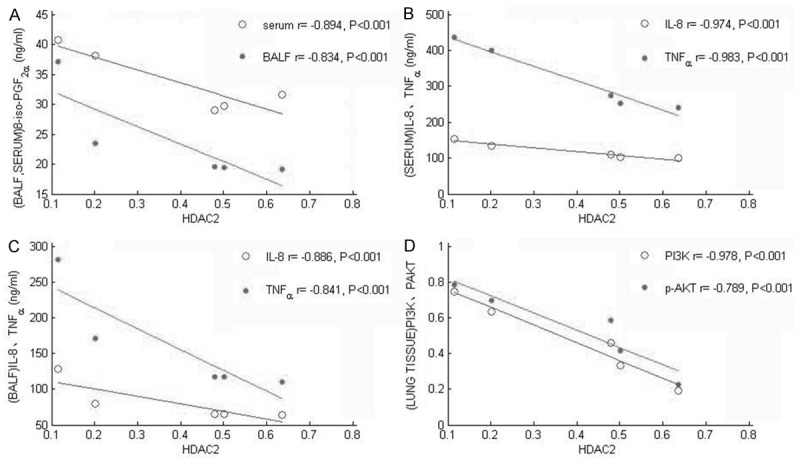
Correlation analysis between HDAC2 in the lung tissue and 8-iso-PGF2α, IL-8, TNF-α in the serum and BALF, and the expression levels of PI3K, P-AKT in the lung tissue.
Table 5.
Correlation analysis between HDAC2 in the lung tissue and 8-iso-PGF2α, IL-8, TNF-α in the serum and BALF, and the expression levels of PI3K, P-AKT in the lung tissue
| LUNG TISSUE (r) | SERUM (r) | BALF (r) | LUNG TISSUE (r) | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| 8-iso-PGF2α* | IL-8* | TNF-α* | 8-iso-PGF2α* | IL-8* | TNF-α* | PI3K* | p-AKT* | |
| HDAC2 | -0.894 | -0.974 | -0.983 | -0.834 | -0.886 | -0.841 | -0.978 | -0.789 |
All P<0.001.
Discussion
The incidence and mortality rate of COPD has been increasing in recent years. It is the fourth leading cause of death globally [9] and more than 10% of adults over the age of 40 have airflow limitations [10-12]. Moreover, this disease is chronically progressive, and there is no effective drug that can slow the progression of and reduce the mortality from the disease [13]. So, better understanding the cellular and molecular mechanisms of COPD, looking for new therapeutic targets and developing effective treatment strategies Is an urgent matter. StudIes found that repeated injection of LPS can cause chronic airway inflammation, emphysema, pulmonary function changes [14]. Exposed to smoke for 36 weeks could established COPD model rats [15]. In this study, COPD model rats were developed by airway instillation of lipopolysaccharide combined with exposure to smoke.
Our study showed that TV, PEF, and EF50 in the C group were significantly lower than in the H group (P<0.05). This change is consistent with the lung function of COPD rat models established by Li Ya [16]. The pathological changes in lung tissue in the C group were similar to those in human patients with COPD. Inflammation and OS are important pathogenesis factors in COPD. 8-iso-PGF2α has been a good indicator of OS levels [17,18] because of its stability and its generation being unaffected by enzyme activity. Our study found that 8-iso-PGF2α, IL-8 and TNF-α in serum and BALF of the C group were significantly higher than in the H group, which demonstrated that there was increased OS and inflammation systemically and locally in the lung tissue of the COPD model rats. This is consistent with reports by Santus [19]. All these demonstrated that the COPD model rats were successfully established.
Anti-inflammatory treatment is an important part of the treatment of COPD. Thus far, GCs are the most important anti-inflammatory agents. This study showed that, compared with the C group, the level of 8-iso-PGF2α, IL-8 and TNF-α in BALF were significantly reduced in the B group, but this reduction was not statistically significant in serum. These results suggest that the effect of the anti-inflammatory and anti-oxidant activity of GC inhalation has some limitations that may be associated with GC resistance. Not only does erythromycin have antibacterial activity, but it also has anti-inflammatory and immunomodulatory activity [2-5]. Compared with the C group, the indicators listed above in both BALF and serum were significantly reduced in the E group, which proved once again that erythromycin had anti-inflammatory and antioxidant activity and could alleviate inflammation and OS in the COPD model rats. Comparing the E+B group with the B group, we found that the above indicators in BALF and serum were significantly reduced, which supports our hypothesis that erythromycin could enhance the anti-inflammatory activity of budesonide.
Histone deacetylase (HDACs) plays an important role in the regulation of cell growth [20] and inflammatory gene expression [21-23]. HDAC2 is an HDAC subtype and is the main subtype involved in the pathogenesis of COPD [24]. GCs can recruit HDAC2 to the region of inflammatory gene transcription, making DNA highly helical, and thereby inhibiting inflammatory gene transcription which produces the anti-inflammatory effect. Studies show that the activity of HDAC2 decreases in COPD patients [25-27]. This may explain the poor response to GC in COPD. This study showed that, compared with the H group, the expression level of HDAC2 protein in the C group was significantly lower. However, the B group showed no significant increase compared with the C group, suggesting that budesonide had a poor effect on increasing HDAC2 activity. Comparing the E+B group with the B group, we found that the HDAC2 activity was significantly elevated, and the levels of IL-8 and TNF-α in BALF and serum were significantly decreased. Moreover, the expression of HDAC2 was negatively correlated with IL-8 and TNF-α. This is consistent with a previous report [28] that erythromycin inhibits the expression of inflammatory cytokines such as IL-8 by enhancing the activity of HDAC2. Therefore, erythromycin could improve the anti-inflammatory effect of GCs by enhancing HDAC2 activity.
OS is thought to be the main reason for the decrease in HDAC2 activity. OS can activate the PI3K/AKT signaling pathway resulting in the phosphorylation and inactivation of HDAC2 [29,30]. Rats whose PI3Kδ genes were knocked out do not show a decrease in HDAC2 activity or GC resistance after exposure to cigarette smoke [31]. Mercado [32] also found that the expression levels of PI3K and p-AKT increased and HDAC2 correspondingly decreased when U937 human monocyte cells pre-treated by nortriptyline were exposed to smoke extract and hydrogen peroxide. Also the activation of PI3K-δ in the lung tissues and cells of COPD patients can lead to the increase in p-AKT levels [33]. In the current study, the expression levels of PI3K and P-AKT were negatively correlated with the level of HDAC2. This suggests that the PI3K/AKT signalling pathway was involved in the regulation of HDAC2 activity during OS in COPD. This is consistent with the studies conducted by Steffen [29] and Barnes [30]. Compared with the C group, the levels of 8-iso-PGF2α, PI3K, and p-AKT protein expression in lung tissue were significantly decreased in the E+B group and the HDAC2 protein expression was significantly increased. Those indicators in the B group showed no significant differences to those in the C group. This suggests that erythromycin can enhance the expression level of HDAC2, most likely by inhibiting OS and, therefore, inhibiting the PI3K/AKT signalling pathway, and that budesonide has no such effect.
Disclosure of conflict of interest
None.
References
- 1.Butler CA, McQuaid S, Taggart CC, Weldon S, Carter R, Skibinski G, Warke TJ, Choy DF, McGarvey LP, Bradding P, Arron JR, Heaney LG. Glucocorticoid receptor β and histone deacetylase 1 and 2 expression in the airways of severe asthma. Thorax. 2012;67:392–8. doi: 10.1136/thoraxjnl-2011-200760. [DOI] [PubMed] [Google Scholar]
- 2.Yamaya M, Azuma A, Takizawa H, Kadota J, Tamaoki J, Kudoh S. Macrolide effects on the prevention of COPD exacerbations. Eur Respir J. 2012;40:485–94. doi: 10.1183/09031936.00208011. [DOI] [PubMed] [Google Scholar]
- 3.Simoens S, Laekeman G, Decramer M. Preventing COPD exacerbations with macrolides: a review and budget impact analysis. Respir Med. 2013;12:506–9. doi: 10.1016/j.rmed.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 4.Hodge S, Reynolds PN. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophages in chronic obstructive pulmonary disease subjects. Respirology. 2012;17:802–7. doi: 10.1111/j.1440-1843.2012.02135.x. [DOI] [PubMed] [Google Scholar]
- 5.Zarogoulidis P, Papanas N, Kioumis I, Chatzaki E, Maltezos E, Zarogoulidis K. Macrolides: from in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur J Clin Pharmacol. 2012;68:479–503. doi: 10.1007/s00228-011-1161-x. [DOI] [PubMed] [Google Scholar]
- 6.Desaki M, Okazaki H, Sunazuka T, Omura S, Yamamoto K, Takizawa H. Molecular mechanisms of anti-inflammatory action of erythromycin in human bronchial epithelial cells: possible role in the signaling pathway that regulates nuclear factor-kappaB activation. Antimicrob Agents Chemother. 2004;48:1581–5. doi: 10.1128/AAC.48.5.1581-1585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu LQ, Dai YR, Xia XD, Jin LD, Lin J. Phosphatidylinositol 3-kinase pathway on proliferation of airway smooth muscle cells in asthmatic rats and the intervention effect of roxithromycin. Chin J Tuberc Respir Dis. 2009;32:304–6. [Google Scholar]
- 8.Kobayashi Y, Wada H, Rossios C, Takagi D, Charron C, Barnes PJ, Ito K. A novel macrolide/fluoroketolide, solithromycin (CEM-101), reverses corticosteroid insensitivity via phosphoinositide 3-kinase pathway inhibition. Br J Pharmacol. 2013;169:1024–1034. doi: 10.1111/bph.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.GOLD Science Committee. Updated: Global Initiative for chronic obstructive pulmonary disease, 2014. http://www.goldcopd.org/Guidelines/guidelines-resources.html. Accessed: 25 June 2014.
- 10.Soriano JB, Rodríguez-Roisin R. Chronic obstructive pulmonary disease overview: epidemiology, risk factors, and clinical presentation. Proc Am Thorac Soc. 2011;8:363–7. doi: 10.1513/pats.201102-017RM. [DOI] [PubMed] [Google Scholar]
- 11.Buist AS, McBurnie MA, Vollmer WM, Gillespie S, Burney P, Mannino DM, Menezes AM, Sullivan SD, Lee TA, Weiss KB, Jensen RL, Marks GB, Gulsvik A, Nizankowska-Mogilnicka E BOLD Collaborative Research Group. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet. 2007;370:741–50. doi: 10.1016/S0140-6736(07)61377-4. [DOI] [PubMed] [Google Scholar]
- 12.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–73. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 13.Takimoto T, Yoshida M, Hirata H, Kashiwa Y, Takeda Y, Goya S, Kijima T, Kumagai T, Tachibana I, Kawase I. 4-Hydroxy-2-nonenal induces chronic obstructive pulmonary diseaselike histopathologic changes in mice. Biochem Biophys Res Commun. 2012;420:84–90. doi: 10.1016/j.bbrc.2012.02.119. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura S, Yanagihara K, Araki N, Yamada K, Morinaga Y, Izumikawa K, Seki M, Kakeya H, Yamamoto Y, Kamihira S, Kohno S. High-dose tobramycin inhibits lipopolysaccharide-induced MUC5AC production in human lung epithelial cells. Eur J Pharmacol. 2011;659:67–71. doi: 10.1016/j.ejphar.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Zheng H, Liu Y, Huang T, Fang Z, Li G, He S. Development and characterization of a rat model of chronic obstructive pulmonary disease (COPD) induced by sidestream cigarette smoke. Toxicol Lett. 2009;189:225–234. doi: 10.1016/j.toxlet.2009.06.850. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Li SY, Li JS, Deng L, Tian YG, Jiang SL, Wang Y, Wang YY. A rat model for stable chronic obstructive pulmonary disease induced by cigarette smoke inhalation and repetitive bacterial infection. Biol Pharm Bull. 2012;35:1752–60. doi: 10.1248/bpb.b12-00407. [DOI] [PubMed] [Google Scholar]
- 17.Smith KA, Shepherd J, Wakil A, Kilpatrick ES. A comparison of methods for the measurement of 8-isoPGF2: a marker of oxidative stress. Ann Clin Biochem. 2011;48:147–54. doi: 10.1258/acb.2010.010151. [DOI] [PubMed] [Google Scholar]
- 18.Jonasson S, Hjoberg J, Hedenstierna G, Basu S. Allergen-induced formation of F2-isoprostanes in a murine asthma model identifies oxidative stress in acute airway inflammation in vivo. Prostaglandins Leukot Essent Fatty Acids. 2009;80:1–7. doi: 10.1016/j.plefa.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Santus P, Sola A, Carlucci P, Fumagalli F, Di Gennaro A, Mondoni M, Carnini C, Centanni S, Sala A. Lipid peroxidation and 5-lipoxygenase activity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171:838–43. doi: 10.1164/rccm.200404-558OC. [DOI] [PubMed] [Google Scholar]
- 20.Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol. 2004;31:633–42. doi: 10.1165/rcmb.2004-0006OC. [DOI] [PubMed] [Google Scholar]
- 21.Galasinski SC, Resing KA, Goodrich JA, Ahn NG. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem. 2002;277:19618–26. doi: 10.1074/jbc.M201174200. [DOI] [PubMed] [Google Scholar]
- 22.Mercado N, To Y, Ito K, Barnes PJ. Nortriptyline reverses corticosteroid insensitivity by inhibition of phosphoinositide-3-kinase-δ. J Pharmacol Exp Ther. 2011;337:465–70. doi: 10.1124/jpet.110.175950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Bao H, Wu J, Duan X, Liu B, Sun J, Gong W, Lv Y, Zhang H, Luo Q, Wu X, Dong J. Baicalin is anti-inflammatory in cigarette smoke-induced inflammatory models in vivo and in vitro: A possible role for HDAC2 activity. Int Immunopharmacol. 2012;13:15–22. doi: 10.1016/j.intimp.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Nakamaru Y, Vuppusetty C, Wada H, Milne JC, Ito M, Rossios C, Elliot M, Hogg J, Kharitonov S, Goto H, Bemis JE, Elliott P, Barnes PJ, Ito K. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 2009;23:2810–9. doi: 10.1096/fj.08-125468. [DOI] [PubMed] [Google Scholar]
- 25.Kim DR, Park MY, Lee CS, Shim SH, Yoon HI, Lee JH, Sung MW, Kim YS, Lee CT. Combination of vorinostat and adenovirus-TRAIL exhibits a synergistic antitumor effect by increasing transduction and transcription of TRAIL in lung cancer cells. Cancer Gene Ther. 2011;18:467–77. doi: 10.1038/cgt.2011.11. [DOI] [PubMed] [Google Scholar]
- 26.Tønnesen P. Smoking cessation and COPD. Eur Respir Rev. 2013;22:37–43. doi: 10.1183/09059180.00007212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–76. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 28.Charron C, Sunazuka T, Oomura S, Ito K, editors. American Thoracic Society International Meeting. San Francisco, USA: 2007. May 18-23, Em-703, a non-antibacterial erythromycin derivative, restores HDAC2 promoter activation diminished under hypoxia and oxidative stress; p. A640. [Google Scholar]
- 29.Wedel S, Hudak L, Seibel JM, Juengel E, Tsaur I, Wiesner C, Haferkamp A, Blaheta RA. Inhibitory effects of the HDAC inhibitor valproic acid on prostate cancer growth are enhanced by simultaneous application of the mTOR inhibitor RAD001. Life Sci. 2011;88:418–24. doi: 10.1016/j.lfs.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Barnes PJ. Development of new drugs for COPD. Curr Med Chem. 2013;20:1531–40. doi: 10.2174/0929867311320120005. [DOI] [PubMed] [Google Scholar]
- 31.Marwick JA, Caramori G, Stevenson CS, Casolari P, Jazrawi E, Barnes PJ, Ito K, Adcock IM, Kirkham PA, Papi A. Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med. 2009;179:542–8. doi: 10.1164/rccm.200810-1570OC. [DOI] [PubMed] [Google Scholar]
- 32.Mercado N, Thimmulappa R, Thomas CM, Fenwick PS, Chana KK, Donnelly LE, Biswal S, Ito K, Barnes PJ. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem Biophys Res Commun. 2011;406:292–8. doi: 10.1016/j.bbrc.2011.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.To Y, Ito K, Kizawa Y, Failla M, Ito M, Kusama T, Elliott WM, Hogg JC, Adcock IM, Barnes PJ. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:897–904. doi: 10.1164/rccm.200906-0937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]