

Abstract
Diazotrophs provide the only biological source of fixed atmospheric nitrogen in the biosphere. Although they are the key player for plant-available nitrogen, less is known about their diversity and potential importance in arid ecosystems. We investigated the nitrogenase gene diversity in native and agricultural desert soil as well as within root-associated microbiota of medicinal plants grown in Egypt through the combination of nifH-specific qPCR, fingerprints, amplicon pyrosequencing and fluorescence in situ hybridization–confocal laser scanning microscopy. Although the diazotrophic microbiota were characterized by generally high abundances and diversity, statistically significant differences were found between both soils, the different microhabitats, and between the investigated plants (Matricaria chamomilla L., Calendula officinalis L. and Solanum distichum Schumach. and Thonn.). We observed a considerable community shift from desert to agriculturally used soil that demonstrated a higher abundance and diversity in the agro-ecosystem. The endorhiza was characterized by lower abundances and only a subset of species when compared to the rhizosphere. While the microbiomes of the Asteraceae were similar and dominated by potential root-nodulating rhizobia acquired primarily from soil, the perennial S. distichum generally formed associations with free-living nitrogen fixers. These results underline the importance of diazotrophs in desert ecosystems and additionally identify plants as important drivers in functional gene pool diversity.
Keywords: desert farming, diazotrophs, medicinal plants, nitrogen-fixing communities, organic agriculture, Rhizobiales
The diazotrophic microbiome of desert ecosystems is characterized by a high diversity and abundance and specific for each plant rhizosphere.
INTRODUCTION
Nitrogen is one of the most yield-limiting factors in agricultural production systems throughout the world and an essential macronutrient for plants. Nitrogen-fixing microorganisms provide the only natural source of fixed atmospheric nitrogen in the biosphere (Gaby and Buckley 2012), and the capability for nitrogen fixation is widely dispersed among prokaryotic taxa including very divergent, distantly related bacteria and archaea (Zehr and Turner 2001; Zehr et al.2003). Biological nitrogen fixation by diazotrophic bacteria together with the input of recycled organic waste, such as manure or compost, is considered a sustainable alternative to chemical nitrogen fertilizers and a possibility to reduce rates of inorganic fertilizer application (Yang, Kloepper and Ryu 2009). The dispersal of inorganic nitrate into surface and groundwater often leads to eutrophication and severe environmental and health problems, yet can be largely avoided through biological inoculants (Orr et al.2011).
The nifH gene that encodes the nitrogenase reductase subunit is highly conserved over all nitrogenase types and has become the marker gene for studies of phylogeny, diversity and abundance of diazotrophic microorganisms (Zehr and Turner 2001). Phylogenetic analyses of nifH genes have revealed five primary clusters of homologous genes (Raymond et al.2004; Gaby and Buckley 2012). Although a wide range of environments has already been investigated for nifH gene diversity (López-Lozano et al.2012; Farnelid et al.2013; Yousuf et al.2014), the global census of diazotrophic diversity remains far from complete (Gaby and Buckley 2011, 2012). Next-generation sequencing techniques in combination with network analyses will allow a deeper insight into microbiome functioning involving nitrogen fixation.
Microbial viability and bioactivity play an important role in arid terrestrial ecosystems. Our previous research in desert ecosystems showed a higher indigenous antagonistic potential against soil-borne phytopathogens, a higher overall bacterial diversity, and better ecosystem function for plant health in soil used for desert agriculture in comparison to uncultivated native desert soil (Köberl et al.2011, 2013). Based on these results, we hypothesized (i) that the agricultural use of desert soil, especially crop rotation with leguminous cover crops, also enhances the diversity of the nitrogen-fixing underground communities. Plant specificity of associated bacteria as well as co-evolution of plant–microbe interactions are well-studied (Berg and Smalla 2009; Bulgarelli et al.2012; Oldroyd 2013), and root exudates are some of the most important drivers for this selecting effect from the soil microbiome (Bais et al.2006). Because we observed such a high specificity in the structural microbial diversity associated with medicinal plants (Köberl et al.2013), we hypothesized (ii) that this high specificity is visible in the functional nifH gene pool as well. All three selected plants for this study—German chamomile (Matricaria chamomilla L.), pot marigold (Calendula officinalis L.) and African nightshade (Solanum distichum Schumach. and Thonn.)—are well-known for their anti-microbial effects and bioactive ingredients (McKay and Blumberg 2006; Ukiya et al.2006; Bahgat et al.2008). Although the highly specific association between Rhizobiaceae and legumes is well investigated (Oldroyd 2013), less is known about the interactions of diazotrophic bacteria with non-legumes. In recent years, endophytic rhizobia were detected in some non-legumes as well, e.g. in rice, maize, barley, wheat, canola and lettuce, colonizing both the intercellular and intracellular spaces of epidermis, cortex and vascular system. They were associated with general plant growth promotion and improved grain yields in cereal crops (Yanni et al.2001; Chi et al.2010). Due to the extensive organic soil management, we hypothesized iii) that endophytic colonization with rhizobia occurs in non-leguminous plants cultivated on Sekem farms as well.
The objective of this research was to study the nitrogen-fixing communities in the endorhiza and rhizosphere of medicinal plants, and the bulk soil of long-term organically managed agricultural soil from the Sekem farms at Sharqia governorate in Egypt compared to unexploited native desert soil. We used a robust methodological approach combining nifH-specific qPCR, fingerprints and amplicon pyrosequencing. Complementary fluorescence in situ hybridization–confocal laser scanning microscopy (FISH–CLSM) analyses targeting the most dominant diazotrophic taxa were performed in order to reveal their habitat preferences and colonization type.
MATERIALS AND METHODS
Experimental design and sampling
Nitrogen-fixing communities were studied at the organically managed Sekem farm Adleya (www.sekem.com) located in the north-eastern desert region of Egypt near Bilbeis (30°13′44″N, 31°23′39″E) at Sharqia governorate. Physicochemical data of the soil is provided in Luske and van der Kamp (2009). The diazotrophic community of bulk agricultural soil was also investigated in comparison to the community of native desert soil from the Sinai Peninsula, Egypt (30°21′00″N, 32°15′18″E). At each site, four composite samples of soil in a horizon of 10–30 cm depth were collected. Profiles of the nifH gene in communities associated with the rhizosphere and endorhiza of three different species of medicinal plants (M. chamomilla L., C. officinalis L., and S. distichum Schumach. and Thonn.) cultivated on Adleya farm were studied and compared. From each plant species, four independent composite samples consisting of 5–10 plant roots with adhering soil were taken. The detailed sampling strategy is described by Köberl et al. (2011).
To isolate total community DNA from the soil and rhizosphere, 5 g of soil or roots with adhering soil were added to 45 mL of sterile 0.85% NaCl and vortexed. For isolation from the endorhiza, 5 g of roots were surface-sterilized with 4% NaOCl for 5 min. The roots were washed three times with sterile distilled water, then 10 mL sterile 0.85% NaCl were added and further homogenized using mortar and pestle. For isolation of total DNA from the rhizosphere, endorhiza and soil, 4 mL of the suspensions were centrifuged (16,000 × g, 4°C) for 20 min and the resulting microbial pellets were stored at –70°C. In the desert soil, a lower concentration of DNA was expected. Therefore, the pellets of 10 mL suspension were used for the isolation of total DNA. Total community DNA was extracted using the FastDNA SPIN Kit for Soil (MP Biomedicals, Solon, OH, USA).
Quantification of microbial nifH genes by qPCR
To determine nifH gene abundances, quantitative PCRs were performed according to Hai et al. (2009) with following modifications. Reactions were conducted in a total volume of 10 μL containing 1× KAPA SYBR FAST qPCR MasterMix Universal (PEQLAB, Polling, Austria), 0.6 mg mL−1 BSA, 0.125 μM of primers nifH-F and nifH-R (Rösch, Mergel and Bothe 2002), and 0.8 μL template DNA dilutions with a concentration of ∼1 ng μL−1 (95°C, 10 min; 39 cycles of 95°C, 45 s; 55°C, 45 s; 72°C, 45 s; and melt from 72 to 95°C). Rotor-Gene 6000 real-time rotary analyzer (Corbett Research, Sydney, Australia) was used for fluorescence quantification. For absolute quantification, the PCR amplified nifH gene fragment from Pectobacterium atrosepticum SCRI1043 was ligated into the pGEM-T Easy Vector (Promega, Mannheim, Germany) and transformed into Escherichia coli DH5α. Serial dilutions of PCR fragments generated with the vector-specific primers USP and RSP (Köberl et al.2011) were used as standards for calculation of nifH gene copy numbers. Concentrations determined by absolute quantification were calculated to copy number per g soil or fresh weight (fw) of root. Each replicate sample was analyzed in duplicates in three independent runs. Statistical analysis was performed with PASW Statistics 18 (SPSS Inc., Chicago, IL, USA) using the independent samples t-test for differences between desert and agricultural soil, and the Games–Howell post hoc test for plant samples.
Fingerprints from single-stranded conformational polymorphism analysis of the nifH gene
Fingerprinting of microbial communities using single-stranded conformational polymorphism was conducted as described by Schwieger and Tebbe (1998). Amplification of the nifH gene fragment was performed using a nested PCR approach with primer pairs nifH4/nifH3 (Zani et al.2000) and nifH11/nifH22P (Yeager et al.2004). The obtained amplicons were separated and analyzed according to Bragina et al. (2012). Comparisons of SSCP generated nifH community profiles were performed using GelCompar II 5.1 (Applied Maths, Kortrijk, Belgium). The cluster analysis was performed with the following settings: dendrogram type: unweighted pair group method with arithmetic mean (UPGMA); similarity coefficient: curve based: Pearson correlation; position tolerances: optimization: 0%, position tolerance: 1%. Based on the Pearson similarity matrix, a multidimensional scaling (MDS) ordination plot was constructed. Pearson correlation matrices were additionally subjected to significance tests of pair-wise similarities by applying permutation analyses (p ≤ 0.05) using the permtest package of R statistics 2.13.1 (The R Foundation for Statistical Computing, Vienna, Austria) with 105 random permutations of sample elements (Kropf et al.2004; R Development Core Team 2011).
nifH gene profiling using 454 pyrosequencing
The nitrogenase gene nifH was amplified according to a nested PCR protocol with primers designed by Zani et al. (2000). The first PCR was performed with the primer pair nifH4/nifH3 as described above for SSCP analysis. Generated amplicons served as templates for the second PCR (30 μL) with the primer pair nifH1 and nifH2 designed by Zehr and McReynolds (1989) that contained the 454 pyrosequencing adaptors, linkers, and sample-specific tags (Table S1). Accordingly, 3 μL template dilutions were added to 1× Taq&Go (MP Biomedicals, Solon, OH, USA), 1.5 mM MgCl2 and 0.2 μM of each primer (95°C, 5 min; 30 cycles of 95°C, 1 min; 65.5°C, 1 min; 72°C, 30 s; and elongation at 72°C, 5 min). PCR products were purified by employing the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). For rhizosphere samples, PCR products of two independent PCR reactions were pooled. For soil samples, respective PCR products from four independent replicate samples were pooled from each habitat in equal volumes. Pyrosequencing read libraries were sequenced by Eurofins Genomics (Ebersberg, Germany) using the Roche 454 GS-FLX+ Titanium sequencing platform. The nucleotide sequences are available in the European Nucleotide Archive (www.ebi.ac.uk/ena) under the BioProject accession number PRJEB10243.
Primer sequences were cropped, reads with low quality (minimum average base quality score 20) and a read length shorter than 200 nucleotides were removed, and remaining sequences were translated into their amino acid sequence using the tool FrameBot of RDP's FunGene pipeline (Fish et al.2013; Wang et al.2013). All subsequent analyses were carried out based on amino acid sequence datasets that were normalized to the same number of sequences within a habitat (5217 sequences per soil sample and 553 sequences per rhizosphere sample) using Subsetify 1.4 (Bragina et al.2013). Amino acid sequences were aligned and clipped at the same alignment reference position (∼108 amino acids) by using ClustalX 2.1 (Larkin et al.2007). OTUs were classified and rarefaction curves were constructed based on the distance matrices of amino acid sequences at 0%, 4% and 8% dissimilarity (Farnelid et al.2011) using mcClust and rarefaction of RDP's FunGene pipeline. Diversity indices were ascertained based on the clustering data (Shannon 1997; Chao and Bunge 2002). Significant differences in diversity indices were calculated with PASW Statistics 18 using Tukey and Games–Howell post hoc tests, depending on the homogeneity of variances. Representative sequences at 8% dissimilarity were selected for the following taxonomic and phylogenetic analysis (Farnelid et al.2011) where clusters with less than 1% of relative abundance were not designated. Nearest relatives were retrieved using the search tool tblastn of the NCBI database.
A neighbor-joining tree with 100 bootstrap replications was created with the tools seqboot, protdist, neighbor, consense and fitch of PHYLIP 3.69 (Felsenstein 1989). The phylogenetic tree was visualized and edited in MEGA4 (Tamura et al.2007). A heatmap showing the number of sequences for each OTU was added. As an outgroup root, a partial sequence of the light-independent photochlorophyllide reductase subunit L (BchL) from Chlorobaculum tepidum (accession number AAG12203) was selected. Chlorophyllide reductases share a small but significant degree of similarity with NifH (Zehr and Turner 2001).
A profile clustering network analysis was performed in order to highlight single OTUs (8% dissimilarity) with considerable differences between the rhizospheres of the medicinal plants. The network analysis was carried out with OTUs exhibiting a mean read change between plants of more than 1% of the normalized dataset. If the ratio of mean OTU read numbers exceeded two, the OTUs were regarded as altered and assigned to the respective profile. Visualization of the network was carried out using Cytoscape 2.8.2 (Smoot et al.2011). Significant differences between medicinal plants were calculated with Metastats (White et al.2009). P values were computed using a combination of the nonparametric t-test, exact Fisher's test and the false discovery rate with 103 permutations.
FISH and CLSM
In order to unravel habitat preferences and the colonization type of potential diazotrophs in situ, we carried out FISH–CLSM approaches using a robust set of specific fluorescent probes. The root system of M. chamomilla was used as representative subject. FISH was carried out as described by Cardinale et al. (2008). In brief, root samples were washed once with phosphate-buffered saline and then fixed in 4% paraformaldehyde. For the detection of Alphaproteobacteria, the Cy5-labeled ALF968 probe (Loy et al.2007) was used. Betaproteobacteria were detected with the Atto488-labeled BET42a probe applied together with an unlabeled competitor probe to avoid unspecific hybridizations (Manz et al.1992). An equimolar mixture of Cy3-labeled EUB338, EUB338II, and EUB338III probes (Amann et al.1990; Daims et al.1999) was used for the detection of all bacteria. In an additional ternary staining approach to visualize Rhizobiales with a high coverage, EUB338-MIX (Cy3) and ALF968 (Alexa488) were applied together with a combination of Cy5-labeled RHIZ1244 (Thayanukul et al.2010) and RHIZ3r (Erlacher et al.2015) probes. As negative controls, non-sense FISH probes labeled with all three fluorochromes (NONEUB) (Wallner, Amann and Beisker 1993) were applied. For better contrast, plant tissue sections were additionally stained by calcofluor-white staining (0.15% in H2O, 15 min incubation). Micrographs were acquired with a Leica TCS SPE confocal laser scanning microscope (Leica Microsystems GmbH, Mannheim, Germany) using the oil immersion objectives Leica ACS APO 40.0 × 1.15 (183.33 μm × 183.33 μm) and ACS APO 63 × 1.30 (116.40 μm × 116.40 μm). The Z-step was 0.7 μm. Solid state lasers were used with 405, 488, 532, 635 nm excitation. The colors red, green, and blue were assigned to the fluorochromes Cy3, Cy5 and Atto/Alexa488, respectively. Imaris 7.0 (Bitplane, Zurich, Switzerland) was used for micrograph post-processing and the assembly of iso-surface renderings.
RESULTS
Abundances of nifH genes in different underground communities
Analysis of nifH gene copy numbers resulted in a statistically significant higher abundance in the agriculturally used soil (6.0 ± 0.3 log10 g−1) in comparison to the native desert soil (4.4 ± 0.7 log10 g−1; Fig. 1). Among the medicinal plant-associated microenvironments, rhizospheres showed significantly higher nifH gene copy numbers than endorhiza samples. Calculated abundances in the rhizospheres ranged from 7.9 ± 0.1 to 8.3 ± 0.1 log10 g−1 root fresh weight (fw) and were not significantly different between medicinal plants. Conversely, the endorhiza of the perennial S. distichum was more significantly colonized by nitrogen-fixing microorganisms (5.9 ± 0.1 log10 g−1 fw) than the endorhizas of the annual Asteraceae M. chamomilla (4.7 ± 0.3 log10 g−1 fw) and C. officinalis (4.9 ± 0.2 log10 g−1 fw; Fig. 1).
Figure 1.

Abundances of nifH genes in bulk soils, rhizosphere, and endorhiza of medicinal plants detected by qPCR. Wb = desert soil, Sb = agricultural soil; Mc = M. chamomilla, Co = C. officinalis, Sd = S. distichum; Re = endorhiza, rhizosphere has no further designation. Averages of nifH gene copy numbers per gram soil or root fresh weight (fw) as log10 and confidences are shown. Significant differences between samples (p ≤ 0.05) are indicated by different letters.
Molecular fingerprinting of diazotrophic underground communities
SSCP fingerprints of nifH genes in microbial communities revealed the first insight into diversity (Fig. S1, Supporting Information). According to the statistical comparison analysis (Fig. S2, Supporting Information), the diazotrophic community composition of field soil differed significantly from the less diverse desert soil (p = 0.0286) by approximately 70%. Rhizospheres exhibited significantly different profiles from bulk agricultural soil (p = 0.0005) as well as from the inner tissue of the root (p < 0.0001). Also, statistically significant differences in nitrogen-fixing rhizosphere communities were detected between the three different medicinal plants (p values between 0.0285 and 0.0290). Approximately half of the nifH gene community was shared by all three plants; M. chamomilla and C. officinalis were more similar to each other, but still nearly 40% of their communities were determined by plant-specific nitrogen fixers (Fig. S2, Supporting Information). Dominant bands were identified as Rhizobium, Bradyrhizobium and Burkholderia. Furthermore, Paenibacillus spp. were assigned to dominant endorhiza bands. Throughout all plant-associated microenvironments, several cyanobacteria from the genera Anabaena and Nostoc were found. Additionally, the nifH gene sequence of the methanogenic archaeon Methanocella was identified (Fig. S1, Supporting Information).
Pyrosequencing-based nifH profiling
A pyrosequencing-based analysis of the nifH gene was employed to gain a deeper insight into the diazotrophic community composition and diversity in soils and medicinal plant rhizospheres. Normalized NifH sequence datasets were rarefied at three cut-off levels (0%, 4% and 8% amino acid dissimilarity; Fig. S3, Supporting Information). Sequences from both soil types (10 434 reads) were classified into 361 OTUs with a dissimilarity cut-off of 8% (desert soil 118 OTUs; agricultural soil 290 OTUs), and rhizosphere sequences (6636 reads) were clustered into 400 OTUs (86–124 OTUs per sample). At a genetic dissimilarity level of 8%, the coverage of Chao1 estimated richness reached 61.1% and 56.9% for desert and agricultural soil, respectively (Table S2, Supporting Information). The calculated Shannon diversity index (H′) was much lower for the desert soil (H′ at a dissimilarity cut-off of 8% = 1.87) than for the agricultural soil (H′ = 3.92) indicating a higher diazotrophic diversity due to the agricultural use of the desert. The coverage of the rhizosphere samples was between 78.9 and 42.0%, and diversity indices showed no statistically significant differences (p ≤ 0.05) between plants; Shannon values ranged from 3.26 to 3.92 at 8% dissimilarity (Table S2, Supporting Information).
Among quality amino acid sequences, 70.6% of soil reads and 68.1% of rhizosphere reads could be taxonomically assigned to at least class level (Fig. 2). The desert soil in particular exhibited an overwhelming dominance of NifH sequences related to Alphaproteobacteria (82.4%). Field soil revealed a high proportion of unclassified sequences, yet still contained 41.0% Alphaproteobacteria. In regards to phyla with greater than 1% of quality reads: 1.9% of field soil sequences could be affiliated to Deltaproteobacteria, 10.8% to Bacilli, 1.2% to Clostridia, 2.8% to Spirochaetes and 1.1% to Cyanobacteria. Rhizosphere samples also revealed a high abundance of Alphaproteobacteria (58.0%–15.4%). Additionally, Betaproteobacteria were found in all samples (55.7%–1.3%). Higher proportions of Alphaproteobacteria were found in the rhizospheres of the Asteraceae (M. chamomilla 55.2%–39.8% and C. officinalis 58.0%–42.1%) in comparison to Betaproteobacteria which were more dominant in the rhizosphere of S. distichum (55.7%–27.1%). Gammaproteobacteria were additionally found in the rhizosphere of S. distichum (19.7%–2.4%) and M. chamomilla (≤1.8%), yet Deltaproteobacteria were only identified in the S. distichum rhizosphere (≤2.2%). Among Firmicutes, Bacilli were found associated with M. chamomilla (≤17.2%), and Clostridia with M. chamomilla (≤1.6%) and C. officinalis (≤1.3%). Cyanobacteria were found in the rhizospheres of M. chamomilla (≤2.7%) and C. officinalis (≤1.8%).
Figure 2.

Taxonomic classification of NifH sequences in bulk soils and associated with the rhizosphere of medicinal plants obtained by 454 amplicon sequencing. Wb = desert soil, Sb = agricultural soil, Mc = M. chamomilla, Co = C. officinalis, Sd = S. distichum. NifH amino acid sequences were classified at class (a) and genus (b) level. From each medicinal plant, four independent replicate samples were investigated separately. PCR products of soil replicates were pooled to one composite sample per soil type. Clusters containing less than 1% of quality sequences were not designated. Multi-colored charts at the legend are shown for each soil or rhizosphere type correspondingly.
Representative sequences of the dominant alphaproteobacterial NifH clusters in native desert soil showed 99%–100% similarity to the NifH sequences of the genus Rhizobium (Table S3, Supporting Information). Further, Bradyrhizobium and Mesorhizobium were also found. The diversity within Alphaproteobacteria was higher in the agriculturally used soil with 40.9% identified as Rhizobium, 12.9% as Agrobacterium, 38.2% as Methylocystis, 5.1% as Bradyrhizobium and 2.9% as Methylocella. Classifiable Bacilli in unplanted field soil were identified as Paenibacillus, and all cyanobacterial reads revealed the genus Anabaena as the closest hit. Deltaproteobacteria, Clostridia and Spirochaetes could not be identified at the genus level (<95% of sequence similarity). In all three investigated medicinal plant rhizospheres, alphaproteobacterial sequences of the genera Rhizobium, Ensifer and Bradyrhizobium were found, and Agrobacterium was identified in the rhizospheres of the Asteraceae. Methylocystis was found associated with roots of C. officinalis and S. distichum, and Azospirillum with M. chamomilla and S. distichum. Clusters of Betaproteobacteria found in all rhizospheres were identified as Ideonella and Derxia, and Zoogloea was found associated with both Asteraceae. In the rhizospheres of C. officinalis and S. distichum, nitrogen-fixing Burkholderia and Azoarcus were additionally identified. Dechloromonas was found in rhizospheres of M. chamomilla and S. distichum, and Azospira and Azonexus were only associated with the S. distichum root. Classifiable Gammaproteobacteria in the rhizospheres of M. chamomilla and S. distichum were affiliated with the genus Azomonas. All deltaproteobacterial reads found associated with S. distichum were classified in the genus Geoalkalibacter, and all classifiable Bacilli reads in the rhizosphere of M. chamomilla were identified as Paenibacillus.
In a phylogenetic tree distinguishing between the major nifH gene types, all NifH sequences from the soil sample libraries were affiliated with the canonical nifH clusters I and III (Fig. 3). Conventional molybdenum nitrogenases (cluster I) were dominated by Alphaproteobacteria (closest related to Rhizobium, Methylocella, Bradyrhizobium, Methylocystis, Mesorhizobium, Skermanella and Agrobacterium), but also contained sequences from Cyanobacteria (Anabaena), Actinobacteridae (Frankia) and Bacilli (Paenibacillus). Reads affiliated with the conventional molybdenum nitrogenases from anaerobes (cluster III) were most closely related to Spirochaetes (Treponema), Clostridia (Clostridium, Acetobacterium and Desulfosporosinus) and Deltaproteobacteria (Desulfovibrio). No sequences of alternative nitrogenases (cluster II) and nifH paralogs (clusters IV and V) were found in the soil libraries.
Figure 3.
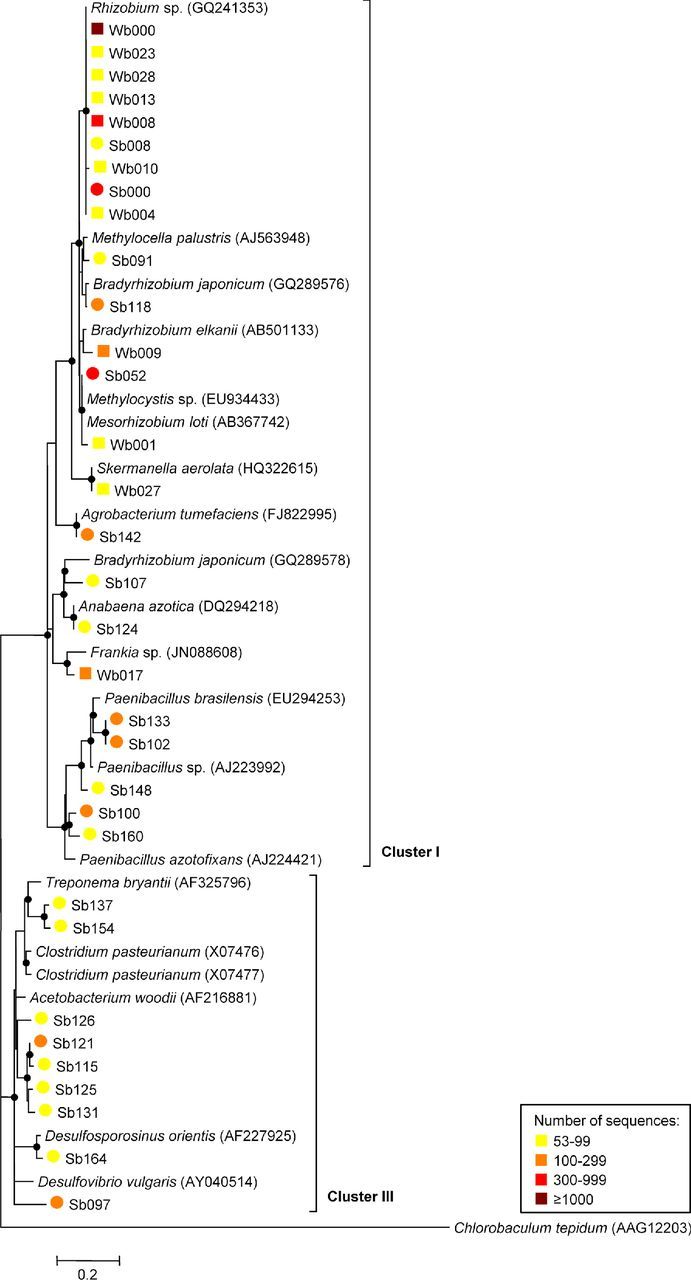
Phylogenetic composition of diazotrophic communities in desert (Wb, squares) and agricultural soil (Sb, circles). Each dataset comprised 5217 high-quality NifH sequences clustered at 8% dissimilarity. The neighbor-joining phylogenetic tree was constructed with both one representative sequence per each OTU and the closest database match (accession numbers in brackets). A partial sequence of the light-independent photochlorophyllide reductase subunit L (BchL) from C. tepidum (AAG12203) was used as an outgroup. Reliability of the tree topology was evaluated using bootstrap analysis with 100 re-samplings (bootstrap values > 50% are indicated as black circles). Sequences were affiliated to the canonical nifH clusters I and III. Numbers of sequences in each cluster are indicated in a heatmap. OTUs containing less than 1% of the normalized dataset were not phylogenetically designated. The scale bar indicates 0.2 amino acid substitutions per site.
A profile clustering network analysis was applied to achieve better insight into the differences between the diazotrophic communities of the three medicinal plant rhizospheres (Fig. 4). OTUs equally distributed among all three rhizospheres were neglected in this network. The rhizosphere profiles revealed more similar diazotrophic communities between M. chamomilla and C. officinalis which were dominated by potential root nodule bacteria. NifH sequences of OTUs identified as genera Ensifer, Rhizobium and Bradyrhizobium were found in significantly higher abundances in the rhizospheres of Asteraceae. Conversely, the rhizosphere of S. distichum was colonized in greater numbers by nitrogen-fixing Beta-, Gamma- and Deltaproteobacteria. Significantly higher read counts were obtained for OTUs identified as Ideonella, Dechloromonas, Azoarcus, Azospira and Azonexus. For each medicinal plant, several specific OTUs were found within the nitrogen-fixing community.
Figure 4.

Profile clustering network analysis of NifH sequence libraries of rhizosphere samples from M. chamomilla, C. officinalis and S. distichum at a dissimilarity level of 8%. The abundance values for OTUs with a mean read change between plants of more than 1% of the normalized dataset were used. If the ratio of mean OTU read numbers exceeded 2, the OTUs were regarded as altered and assigned to the respective profile. Node sizes of OTUs correspond to the relative abundance of the total dataset; nodes matching abundances of 0.5% and 10% were added as reference points. Distributions between plants are displayed by widths of connection lines. Significances (p ≤ 0.05) are indicated by colored node borders: red node borders indicate significances between connected and all not linked profiles, green is used for significances between Matricaria and Calendula, orange for significances between Calendula and Solanum, blue for significances between Matricaria and Solanum, and nodes with black borders for no significant differences. Black node labels indicate a similarity to the taxonomic node label (closest database match) of ≥ 95%, whereas gray node labels have a similarity < 95%.
In situ visualization of bacterial rhizosphere colonization
Complementary FISH–CLSM analyses revealed generally strong rhizoplanic colonization. High abundances of Alphaproteobacteria and in particular the order Rhizobiales were observed in the root systems of medicinal plants grown under arid desert farming conditions (Fig. 5; S4, S5, Supporting Information). Alphaproteobacteria were, among the root-associated bacteria, clearly dominant over Betaproteobacteria and were found as both single cells and in large colonies in close interaction with other undefined bacteria (Fig. S4, Supporting Information). A high proportion of Alphaproteobacteria were subsequently identified as Rhizobiales (Fig. 5). As suggested in Erlacher et al. (2015), the combination of the FISH probes RHIZ1244 and RHIZ3r yielded into a robust coverage of most Rhizobiales including the predominant taxa Rhizobiaceae and Bradyrhizobiaceae. The tendency of Rhizobiales to preferentially form small sub-clusters consisting of three to six cells prevailed over the participation in bacterial plaques or biofilm like structures (Fig. 5). We observed both, packed and dense colonization on the rhizoplane and bacteria in the endorhiza. Further, Rhizobiales were found predominantly in niches of the rhizoplane and in compartments of the endorhiza.
Figure 5.
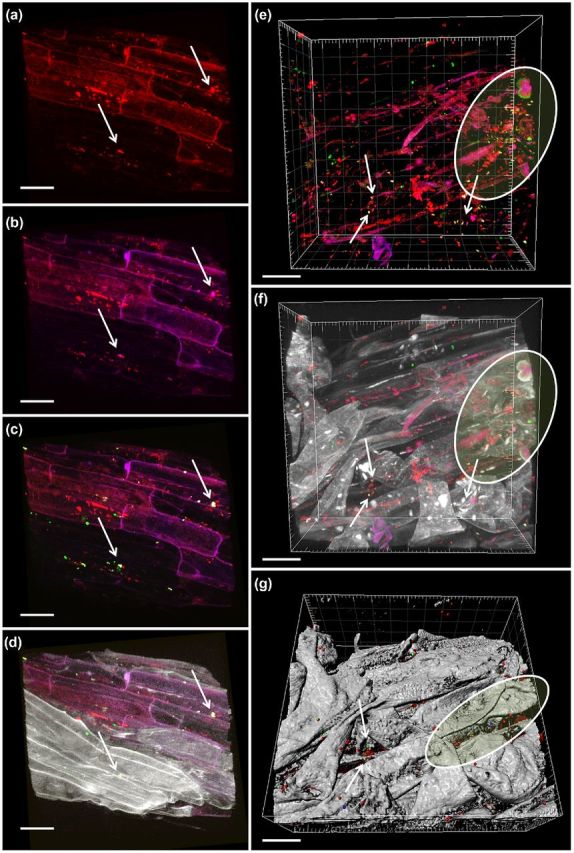
In situ visualization of bacterial colonization in the root system of M. chamomilla. Volume renderings (a–f) and three-dimensional reconstruction model made by Imaris (g) of confocal laser scanning microscopy stacks. Micrographs a–d show the efficacy of the FISH-probes and potential endo- and ectophytic root colonization patterns of Rhizobiales (white arrows). Panels e–g show mixed bacterial taxa including Rhizobiales and other Alphaproteobacteria colonizing niches (white marks) and the inner compartments of the root. white/green (g) = Rhizobiales; pink/blue (g) = Alphaproteobacteria; red = all bacteria; gray = root tissue. Scale bars = 20 μm. Probe set: RHIZ1244 (Cy5), RHIZ3r (Cy5), ALF968 (Alexa488), EUB338-MIX (Cy3). The colors red, green, and blue were assigned to the fluorochromes Cy3, Cy5 and Alexa488, respectively.
DISCUSSION
We observed a considerable shift in the diazotrophic soil community towards both higher abundance and diversity after long term organic desert agriculture, thus supporting our first hypothesis. The agricultural use of desert soil, especially through crop rotation with leguminous cover crops such as alfalfa or clover, enhances the presence and diversity of the nitrogen-fixing underground community. Nitrogen-fixing microorganisms in deserts play an indispensable role for both plant development and growth, yet desert soils are characterized by harsh environmental conditions, e.g. extreme temperatures, desiccation, high soil salinity, low nutrient levels, high UV radiation levels and physical instability caused by strong winds (Cary et al.2010) which can all be transformed into a more manageable environment by watering and cultivating plants. Native desert soil, however, still showed an impressive diversity and relatively high abundance in comparison to other ecosystems. Contrary to previous studies that describe desert microbial communities as solely structured through abiotic processes (Cary et al.2010), we additionally identified plants as important drivers for the functional gene pool diversity in arid soil ecosystems.
As stated in our second hypothesis, the high specificity for each plant species is in a functional gene. While specific structures of microbial communities were previously described (Berg and Smalla 2009), this specificity was confirmed for the structure of Arabidopsis cultivars using amplicon libraries (Bulgarelli et al.2012). In this study, we found evidence of the specificity of the diazotrophic communities associated with the three plant species. In general, diazotrophic communities in the medicinal plant-associated microenvironments (rhizosphere and endorhiza) were more similar between the two Asteraceae in comparison to the Solanum, and the nifH gene profiles revealed a higher abundance of Alphaproteobacteria associated with the roots of the Asteraceae. Conversely, the nightshade showed a higher proportion of Betaproteobacteria. Similarly, previous investigations of the total bacterial and fungal communities already revealed comparable colonization patterns between M. chamomilla and C. officinalis in comparison to S. distichum (Köberl et al.2013). Both Asteraceae are well-known medicinal plants that produce more similar bioactive metabolites, including several flavonoids, which could explain their similar microbiomes. The bioactive ingredients of the African S. distichum were all not yet identified (Bahgat et al.2008). Furthermore, M. chamomilla and C. officinalis are annual herbal medicinal plants, while Solanum distichum is a perennial plant, thus providing a longer timeframe to specifically select a stable associated microbiome.
Nitrogen-fixing alphaproteobacterial genera were also largely represented in both soils where plants primarily acquire their associated microbial community (Berg and Smalla 2009). The question remains, however, as to where the Beta- and Gammaproteobacteria originated as they were identified neither in the desert nor in agriculturally conditioned bulk soil. Transmission through seeds or pollen would be possible, as recent studies on pumpkin flowers showed that the pollen grain surfaces are densely colonized by Beta- and Gammaproteobacteria (Fürnkranz et al.2012). Even though plant seeds are well-known carriers of seed-borne pathogens, they can also transmit beneficial microorganisms (Hardoim et al.2012). A very rare abundance of Beta- and Gammaproteobacteria within diazotrophic communities of bulk arid soils combined with a recruitment by root exudates and the favorable conditions of plant rhizospheres could explain this phenomenon as well.
In general, all microenvironments were primarily inhabited by proteobacterial nitrogen fixers, and the desert soil was noticeably dominated by Rhizobiales. Similar results were reported by López-Lozano et al. (2012) for soil from the Chihuahuan Desert in Mexico investigated using nifH gene clone libraries. However through the use of deep sequencing, we detected a much higher diversity: NifH sequences from soils and rhizosphere reads could be classified at a genetic dissimilarity level of 8% into 361 OTUs and 400 OTUs, respectively. Moreover, pyrosequencing techniques for nifH gene sequencing allow for the analysis of more sequences than those analyzed for the global study published a few years before (Gaby and Buckley 2011). The high NifH diversity primarily encompasses bacteria, as the only suggestion for archaeal nitrogen fixation in these unique desert habitats was found with Methanocella in the endorhiza, especially from C. officinalis. The nifH genes are commonly found in many methanogens, but not all methanogens are capable of nitrogen fixation. A complete nif operon that enables the organism to undergo nitrogen fixation was recently detected in the genome of Methanocella conradii HZ254 (found as closest database match in this study) (Lü and Lu 2012), however the genome of the closely related species M. paludicola SANAE only contains genes similar to nifH that are neither part of an operon with other nif genes nor associated with the nitrogen fixation function (Sakai et al.2011). Through comparisons with the metagenomically reconstructed genome sequence of the Rice Cluster I archaeon RC-IMRE50 for which a full component of the genes for nitrogenase was found (Erkel et al.2006), Sakai et al. (2011) have already discussed an inter-species physiological difference between the nitrogen fixation capabilities among the members of the order Methanocellales.
With the same primer pair, we calculated much higher abundances of nifH genes in the rhizospheres of the investigated medicinal plants in Egypt than Hai et al. (2009) in the rhizosphere of Sorghum bicolor grown in Burkina Faso. Only a fraction of the medicinal plant-associated nitrogen fixers were able to invade the root, compete with other well-adapted root endophytes, and successfully colonize the inner tissue. Abundances calculated for endorhizas totaled 58%–71% of the appropriate rhizosphere, and the diversity of nitrogen-fixing microorganisms in the inner part of the root was noticeably lower. All habitats influenced by desert agriculture contained a higher amount of potential nitrogen fixers, whereby in particular the rotation with legume plants is a considerable source of rhizobia. The rhizobial life-cycle typically includes a nitrogen-fixing endosymbiosis within legume root-nodules and a free-living saprophytic persistence in soil; however, the detection of endosymbiotic life of rhizobia in non-legumes (Yanni et al.2001; Chi et al.2010) demonstrated that some rhizobia have evolved a three-component life-cycle including a beneficial plant growth-promoting endocolonization within non-legume roots when grown in rotation with leguminous hosts. As hypothesized, endophytic colonization by different rhizobial strains could also be shown for the investigated non-leguminous medicinal plant roots through nifH-specific fingerprints and Rhizobiales-specific FISH microscopy.
An immense diversity and high abundance of diazotrophic communities were detected in all investigated arid habitats, thus strongly supporting their important role in native and agricultural desert ecosystems. The uniform distribution of dominant Rhizobiales across plant roots and the frequent occurrence in the endorhiza also contributes to their important role as symbiotic partners for their plant hosts.
Supplementary Material
Acknowledgments
We would like to thank Ibrahim Abouleish, winner of the Alternative Nobel Prize, and his family for their generous hospitality in Sekem. We are grateful to Angela Hofmann (Cairo), Massimiliano Cardinale and Christin Zachow (Graz) for their valuable advice and support. Furthermore, we thank Kornelia Smalla (Braunschweig) for inspiring discussions and Meg Starcher (Washington/Graz) for critically reading the manuscript. The authors gratefully acknowledge support from NAWI Graz.
SUPPLEMENTARY DATA
FUNDING
This work was supported by the EU-Egypt Innovation Fund [RDI MED/2009/214-418 and ENPI/2014/342-707] and the Austrian Science Fund FWF [J 3638 to MK], co-funded by the European Commission.
Conflict of interest. None declared.
REFERENCES
- Amann RI, Binder BJ, Olson RJ, et al. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microb. 1990;56:1919–25. doi: 10.1128/aem.56.6.1919-1925.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahgat A, Abdel-Aziz H, Raafat M, et al. Solanum indicum ssp. distichum extract is effective against L-NAME-induced hypertension in rats. Fundam Clin Pharm. 2008;22:693–9. doi: 10.1111/j.1472-8206.2008.00627.x. [DOI] [PubMed] [Google Scholar]
- Bais HP, Weir TL, Perry LG, et al. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol. 2006;57:233–66. doi: 10.1146/annurev.arplant.57.032905.105159. [DOI] [PubMed] [Google Scholar]
- Berg G, Smalla K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol. 2009;68:1–13. doi: 10.1111/j.1574-6941.2009.00654.x. [DOI] [PubMed] [Google Scholar]
- Bragina A, Berg C, Müller H, et al. Insights into functional bacterial diversity and its effects on Alpine bog ecosystem functioning. Sci Rep. 2013;3:1955. doi: 10.1038/srep01955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragina A, Maier S, Berg C, et al. Similar diversity of Alphaproteobacteria and nitrogenase gene amplicons on two related Sphagnum mosses. Front Microbiol. 2012;2:275. doi: 10.3389/fmicb.2011.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgarelli D, Rott M, Schlaeppi K, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–5. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- Cardinale M, Vieira de Castro J Jr, Müller H, et al. In situ analysis of the bacterial community associated with the reindeer lichen Cladonia arbuscula reveals predominance of Alphaproteobacteria. FEMS Microbiol Ecol. 2008;66:63–71. doi: 10.1111/j.1574-6941.2008.00546.x. [DOI] [PubMed] [Google Scholar]
- Cary SC, McDonald IR, Barrett JE, et al. On the rocks: the microbiology of Antarctic Dry Valley soils. Nat Rev Microbiol. 2010;8:129–38. doi: 10.1038/nrmicro2281. [DOI] [PubMed] [Google Scholar]
- Chao A, Bunge J. Estimating the number of species in a stochastic abundance model. Biometrics. 2002;58:531–9. doi: 10.1111/j.0006-341x.2002.00531.x. [DOI] [PubMed] [Google Scholar]
- Chi F, Yang P, Han F, et al. Proteomic analysis of rice seedlings infected by Sinorhizobium meliloti 1021. Proteomics. 2010;10:1861–74. doi: 10.1002/pmic.200900694. [DOI] [PubMed] [Google Scholar]
- Daims H, Brühl A, Amann R, et al. The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol. 1999;22:434–44. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- Erkel C, Kube M, Reinhardt R, et al. Genome of rice cluster I archaea – the key methane producers in the rice rhizosphere. Science. 2006;313:370–2. doi: 10.1126/science.1127062. [DOI] [PubMed] [Google Scholar]
- Erlacher A, Cernava T, Cardinale M, et al. Rhizobiales as functional and endosymbiontic members in the lichen symbiosis of Lobaria pulmonaria L. Front Microbiol. 2015;6:53. doi: 10.3389/fmicb.2015.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnelid H, Andersson AF, Bertilsson S, et al. Nitrogenase gene amplicons from global marine surface waters are dominated by genes of non-cyanobacteria. PLoS One. 2011;6:e19223. doi: 10.1371/journal.pone.0019223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnelid H, Bentzon-Tilia M, Andersson AF, et al. Active nitrogen-fixing heterotrophic bacteria at and below the chemocline of the central Baltic Sea. ISME J. 2013;7:1413–23. doi: 10.1038/ismej.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP – Phylogeny inference package (version 3.2) Cladistics. 1989;5:164–6. [Google Scholar]
- Fish JA, Chai B, Wang Q, et al. FunGene: the functional gene pipeline and repository. Front Microbiol. 2013;4:291. doi: 10.3389/fmicb.2013.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürnkranz M, Lukesch B, Müller H, et al. Microbial diversity inside pumpkins: microhabitat-specific communities display a high antagonistic potential against phytopathogens. Microb Ecol. 2012;63:418–28. doi: 10.1007/s00248-011-9942-4. [DOI] [PubMed] [Google Scholar]
- Gaby JC, Buckley DH. A global census of nitrogenase diversity. Environ Microbiol. 2011;13:1790–9. doi: 10.1111/j.1462-2920.2011.02488.x. [DOI] [PubMed] [Google Scholar]
- Gaby JC, Buckley DH. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One. 2012;7:e42149. doi: 10.1371/journal.pone.0042149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai B, Diallo NH, Sall S, et al. Quantification of key genes steering the microbial nitrogen cycle in the rhizosphere of sorghum cultivars in tropical agroecosystems. Appl Environ Microb. 2009;75:4993–5000. doi: 10.1128/AEM.02917-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardoim PR, Hardoim CC, van Overbeek LS, et al. Dynamics of seed-borne rice endophytes on early plant growth stages. PLoS One. 2012;7:e30438. doi: 10.1371/journal.pone.0030438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köberl M, Müller H, Ramadan EM, et al. Desert farming benefits from microbial potential in arid soils and promotes diversity and plant health. PLoS One. 2011;6:e24452. doi: 10.1371/journal.pone.0024452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köberl M, Ramadan EM, Adam M, et al. Bacillus and Streptomyces were selected as broad-spectrum antagonists against soilborne pathogens from arid areas in Egypt. FEMS Microbiol Lett. 2013;342:168–78. doi: 10.1111/1574-6968.12089. [DOI] [PubMed] [Google Scholar]
- Kropf S, Heuer H, Grüning M, et al. Significance test for comparing complex microbial community fingerprints using pairwise similarity measures. J Microbiol Meth. 2004;57:187–95. doi: 10.1016/j.mimet.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, et al. ClustalW and ClustalX version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- López-Lozano NE, Eguiarte LE, Bonilla-Rosso G, et al. Bacterial communities and the nitrogen cycle in the gypsum soils of Cuatro Ciénegas Basin, Coahuila: a Mars analogue. Astrobiology. 2012;12:699–709. doi: 10.1089/ast.2012.0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy A, Maixner F, Wagner M, et al. probeBase–an online resource for rRNA-targeted oligonucleotide probes: new features 2007. Nucleic Acids Res. 2007;35:D800–4. doi: 10.1093/nar/gkl856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü Z, Lu Y. Complete genome sequence of a thermophilic methanogen, Methanocella conradii HZ254, isolated from Chinese rice field soil. J Bacteriol. 2012;194:2398–9. doi: 10.1128/JB.00207-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luske B, van der Kamp J. Louis Bolk Instituut & Soil and More International; 2009. Carbon sequestration potential of reclaimed desert soils in Egypt. http://orgprints.org/16438/1/2192.pdf (30 June 2015, date last accessed) [Google Scholar]
- McKay DL, Blumberg JB. A review of the bioactivity and potential health benefits of chamomile tea (Matricaria recutita L.) Phytother Res. 2006;20:519–30. doi: 10.1002/ptr.1900. [DOI] [PubMed] [Google Scholar]
- Manz W, Amann R, Ludwig W, et al. Phylogenetic oligodeoxynucleotide probes for the major subclasses of proteobacteria: problems and solutions. Syst Appl Microbiol. 1992;15:593–600. [Google Scholar]
- Oldroyd GE. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat Rev Microbiol. 2013;11:252–63. doi: 10.1038/nrmicro2990. [DOI] [PubMed] [Google Scholar]
- Orr CH, James A, Leifert C, et al. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl Environ Microb. 2011;77:911–9. doi: 10.1128/AEM.01250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing; 2011. http://www.r-project.org (30 June 2015, date last accessed) [Google Scholar]
- Raymond J, Siefert JL, Staples CR, et al. The natural history of nitrogen fixation. Mol Biol Evol. 2004;21:541–54. doi: 10.1093/molbev/msh047. [DOI] [PubMed] [Google Scholar]
- Rösch C, Mergel A, Bothe H. Biodiversity of denitrifying and dinitrogen-fixing bacteria in an acid forest soil. Appl Environ Microb. 2002;68:3818–29. doi: 10.1128/AEM.68.8.3818-3829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S, Takaki Y, Shimamura S, et al. Genome sequence of a mesophilic hydrogenotrophic methanogen Methanocella paludicola, the first cultivated representative of the order Methanocellales. PLoS One. 2011;6:e22898. doi: 10.1371/journal.pone.0022898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieger F, Tebbe CC. A new approach to utilize PCR-single-strand-conformation polymorphism for 16S rRNA gene-based microbial community analysis. Appl Environ Microb. 1998;64:4870–6. doi: 10.1128/aem.64.12.4870-4876.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE. The mathematical theory of communication. 1963. MD Comput. 1997;14:306–17. [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, et al. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–2. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, et al. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thayanukul P, Zang K, Janhom T, et al. Concentration-dependent response of estrone-degrading bacterial community in activated sludge analyzed by microautoradiography-fluorescence in situ hybridization. Water Res. 2010;44:4878–87. doi: 10.1016/j.watres.2010.07.031. [DOI] [PubMed] [Google Scholar]
- Ukiya M, Akihisa T, Yasukawa K, et al. Anti-inflammatory, anti-tumor-promoting, and cytotoxic activities of constituents of marigold (Calendula officinalis) flowers. J Nat Prod. 2006;69:1692–6. doi: 10.1021/np068016b. [DOI] [PubMed] [Google Scholar]
- Wallner G, Amann R, Beisker W. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry. 1993;14:136–43. doi: 10.1002/cyto.990140205. [DOI] [PubMed] [Google Scholar]
- Wang Q, 3rd Quensen JF, Fish JA, et al. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. MBio. 2013;4:e00592–13. doi: 10.1128/mBio.00592-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Kloepper JW, Ryu CM. Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci. 2009;14:1–4. doi: 10.1016/j.tplants.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Yanni YG, Rizk RY, Abd El-Fattah FK, et al. The beneficial plant growth-promoting association of Rhizobium leguminosarum bv. trifolii with rice roots. Aust J Plant Physiol. 2001;28:845–70. [Google Scholar]
- Yeager CM, Kornosky JL, Housman DC, et al. Diazotrophic community structure and function in two successional stages of biological soil crusts from the Colorado Plateau and Chihuahuan Desert. Appl Environ Microb. 2004;70:973–83. doi: 10.1128/AEM.70.2.973-983.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousuf B, Kumar R, Mishra A, et al. Differential distribution and abundance of diazotrophic bacterial communities across different soil niches using a gene-targeted clone library approach. FEMS Microbiol Lett. 2014;360:117–25. doi: 10.1111/1574-6968.12593. [DOI] [PubMed] [Google Scholar]
- Zani S, Mellon MT, Collier JL, et al. Expression of nifH genes in natural microbial assemblages in Lake George, New York, detected by reverse transcriptase PCR. Appl Environ Microb. 2000;66:3119–24. doi: 10.1128/aem.66.7.3119-3124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr JP, Jenkins BD, Short SM, et al. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ Microbiol. 2003;5:539–54. doi: 10.1046/j.1462-2920.2003.00451.x. [DOI] [PubMed] [Google Scholar]
- Zehr JP, McReynolds LA. Use of degenerate oligonucleotides for amplification of the nifH gene from the marine cyanobacterium Trichodesmium thiebautii. Appl Environ Microb. 1989;55:2522–6. doi: 10.1128/aem.55.10.2522-2526.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr JP, Turner PJ. Nitrogen fixation: nitrogenase genes and gene expression. In: Paul JH, editor. Methods in Microbiology. New York: Academic Press; 2001. pp. 271–85. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.