

SUMMARY
Transporting and sensory epithelial cells shape apical specializations using protocadherin-based adhesion. In the enterocyte brush border, protocadherin function requires a complex of cytoplasmic binding partners, although the composition of this complex and logic governing its assembly remain poorly understood. We found that ankyrin repeat and sterile alpha motif domain containing 4B (ANKS4B) localizes to the tips of adherent brush border microvilli and is essential for intermicrovillar adhesion. ANKS4B interacts with USH1C and MYO7B, which link protocadherins to the actin cytoskeleton. ANKS4B and USH1C also bind to the MYO7B cargo-binding tail at distinct sites. However, a tripartite complex only forms if ANKS4B and MYO7B are first activated by USH1C. This study uncovers an essential role for ANKS4B in brush border assembly, reveals a hierarchy in the molecular interactions that drive intermicrovillar adhesion, and informs our understanding of diseases caused by mutations in USH1C and ankyrin repeat proteins, such as Usher syndrome.
Keywords: actin, myosin, intestine, brush border, epithelia, protrusion, Usher syndrome
Graphical Abstract
INTRODUCTION
Apical specializations composed of actin-supported membrane protrusions allow epithelial cells to interact with their external environment, enabling functions as diverse as solute absorption and mechanosensation (Crawley et al., 2014a; Pan and Zhang, 2012). In the case of the intestinal tract, enterocytes build apical microvilli, which play essential roles in the processing and uptake of nutrients, and defense against pathogenic microbes and toxic compounds that accumulate in the gut lumen. The surface of a single enterocyte is populated by hundreds of microvilli, forming an organelle known as the ‘brush border’ (BB). The destruction or malformation of BB microvilli is associated with numerous intestinal pathologies, including infection with attaching and effacing microbes and microvillus inclusion disease (Vallance et al., 2002; Wilson et al., 2001).
During differentiation, enterocytes organize microvilli into tightly packed, highly ordered arrays. This unique morphology increases the capacity for housing membrane-bound transporters, channels, and host defense enzymes, and thus, enhances the functional potential of the apical domain (Helander and Fandriks, 2014). We recently reported that microvillar packing is driven by BB-specific protocadherins (Crawley et al., 2014b), CDHR2 (also known as protocadherin-24) and CDHR5 (also known as μ-protocadherin or mucin-like protocadherin), which form heterophilic adhesion complexes between the tips of adjacent microvilli. Loss of either protocadherin disrupts BB assembly, leaving cells with disorganized microvilli that are reduced in number. Inside the cell, CDHR2 and CDHR5 interact with the multi-PDZ domain protein, USH1C (also known as harmonin), and the motor, myosin-7b (MYO7B), which are postulated to promote the localization of adhesion molecules to microvillar tips (Crawley et al., 2014b). This intermicrovillar adhesion complex (IMAC) is reminiscent of the ‘Usher complex’ that contains protocadherin-15 (PCDH15), cadherin-23 (CDH23), USH1C, and myosin-7a (MYO7A), which function together to form tip-links between adjacent stereocilia on the surface of mechanosensory hair cells in the inner ear (Pan and Zhang, 2012). Loss-of-function mutations in any of these components give rise to Type 1 Usher syndrome, a sensory disease at least partially caused by defects in stereocilia morphology and function (Ahmed et al., 2001; Bitner-Glindzicz et al., 2000; Bork et al., 2001; Verpy et al., 2000; Weil et al., 1995). Interestingly, Usher syndrome patients with mutations in USH1C, the only component common to both the IMAC and Usher complex, also present with intestinal dysfunction (Bitner-Glindzicz et al., 2000; Hussain et al., 2004), most likely explained by defects in intermicrovillar adhesion (Crawley et al., 2014b). Based on similarities in molecular composition and these functional links, the IMAC in the gut appears to represent a primordial form of the Usher complex found in the inner ear. However, mechanisms regulating the assembly of either complex remain poorly understood.
A fifth component of the Usher complex is USH1G (also known as SANS)(Weil et al., 2003), an ankyrin repeat scaffolding protein that interacts with MYO7A and USH1C (Wu et al., 2011; Yan et al., 2010). Mice lacking USH1G exhibit a loss of tip-links, defects in stereocilia morphology and organization, loss of mechanotransduction currents, and perturbations to hearing and balance (Caberlotto et al., 2011). However, Usher syndrome patients with mutations in USH1G do not present with intestinal symptoms, and initial studies on intermicrovillar adhesion did not detect USH1G expression in the gut (Crawley et al., 2014b). Another ankyrin repeat protein, ANKS4B, was previously identified in a screen for USH1C interacting partners (Johnston et al., 2004), although its role in intermicrovillar adhesion has not been explored.
Here we report ANKS4B is expressed in enterocytes where it exhibits striking ankyrin repeat-dependent localization to the tips of adherent microvilli, the site of IMAC function; cells lacking this factor are unable to organize microvilli during BB assembly. Analysis of ANKS4B interactions with other IMAC components revealed binding to both MYO7B and USH1C, which allowed us to reconstitute a stable tripartite complex. However, we uncovered an unexpected hierarchy of binding, where interactions between the MYO7B cargo-binding domain and ANKS4B were only possible following activation of these factors by USH1C. Thus, ANKS4B plays an essential role in BB assembly and USH1C regulates its integration into the IMAC. This work also suggests that actin-supported cell surface protrusions in functionally diverse epithelia are shaped by an evolutionarily conserved complex that includes, minimally, two heterophilic adhesion molecules, two scaffolding factors, and one actin-based motor.
RESULTS
ANKS4B is an USH1G-like molecule that targets to the tips of BB microvilli
In an effort to identify additional IMAC components, we searched the mouse enterocyte BB proteome for USH1G-like molecules and identified ANKS4B (McConnell et al., 2011). We first validated this candidate by examining expression and localization in intestinal and kidney tissues and epithelial cell lines. Confocal imaging of mouse duodenal tissue sections revealed high levels of endogenous ANKS4B, which exhibits striking enrichment at the distal tips of BB microvilli, the site of IMAC function (Fig. 1A and 1C). Similar ANKS4B localization was observed at the apical surface of kidney proximal tubule epithelial cells (Fig. S1A). We also examined ANKS4B localization in CACO-2BBE cells, which form a well-ordered apical BB similar to that observed in tissue (Peterson and Mooseker, 1992) and are amenable to super-resolution imaging using structured illumination microscopy (SIM). SIM of the apical domain of CACO-2BBE cells cultured for 12 days postconfluency (DPC) revealed marked enrichment of ANKS4B at the extreme distal tips of microvilli (Fig. 1B). Curve fitting of line scans from tissue and CACO-2BBE images revealed peak ANKS4B signal at 0.88 ± 0.12 (Fig. 1C) and 0.91 ± 0.09 (Fig. 1D), respectively, when plotted as a function of normalized microvillar length (where 0 = base and 1 = tip). In duodenal BB microvilli, which average ~2.5 μm in length, this corresponds to a ~300 nm zone of enrichment at the distal tips.
Figure 1. ANKS4B localization in native intestinal tissue and CACO-2BBE monolayers.

(A) Confocal microscopy of mouse duodenal tissue stained for ANKS4B (green) and villin (magenta, as a microvillar marker). Boxed region in merge denotes area in zoomed image panels. Arrows point to the distal tips of BB microvilli. Scale bar, 40 μm. (B) Super-resolution microscopy of 12 DPC CACO-2BBE cells stained for F-actin (magenta) and ANKS4B (green). Boxed regions denote areas in zoomed image panels. Arrows point to examples of distal tip enrichment of ANKS4B in clustering microvilli. Scale bar, 5 μm. (C and D) Line scan analysis of ANKS4B signal intensity parallel to the microvillar axis in tissue sections or CACO-2BBE cells, as visualized using confocal or super-resolution microscopy, respectively. Each plot shows a collection of 15 normalized scans. Curve fits revealed peak ANKS4B localization at 0.88 ± 0.12 (Fig. 1C) and 0.91 ± 0.09 (Fig 1D), respectively, as a function of normalized microvillar axis (where 0 = base and 1 = tip). N.U. = normalized units. (E) Quantification of microvillar clustering as a function of endogenous ANKS4B expression levels (n = 361 cells) in 12 DPC CACO-2BBE monolayers. *p < 0.0001, t test.
In differentiating CACO-2BBE cells, we also noted that ANKS4B tip targeting was most prominent in clustering microvilli (Fig. 1B, zooms). Because clustering indicates the presence of active intermicrovillar adhesion (Crawley et al., 2014b), these images suggest that ANKS4B is well positioned to play a role in this process, perhaps as a component of the IMAC. Indeed, analysis of ANKS4B expression levels relative to the appearance of microvillar clusters revealed a strong positive correlation (Fig. 1E).
ANKS4B targets to the BB using the ANKR domain
Like USH1G, ANKS4B is composed of an N-terminal ankyrin repeat (ANKR) domain, a central (CEN) unstructured region, and a C-terminal sterile-alpha motif (SAM) ending with a canonical class I PDZ-binding motif (PBM; Fig. 2A). To investigate the mechanism of ANKS4B targeting to the BB, we created stable CACO-2BBE cell lines overexpressing a range of EGFP-tagged truncation constructs representing different ANKS4B domains (Fig. 2A). Using confocal imaging, we calculated the targeting efficiency of each construct as a ratio of BB:cytosol EGFP signal (Fig. 2B-I and S1B). Similar to endogenous ANKS4B localization, full-length ANKS4B targeted to the apical BB (Fig. 2B,I). While neither the isolated CEN nor SAM domain displayed any apical targeting (Fig. 2D,E,I), concatenation of these domains increased targeting, though levels were still far below full-length (Fig. 2G,I). However, the isolated ANKR domain displayed robust targeting to the BB (Fig. 2C,I). Addition of the CEN domain to the ANKR domain slightly muted this localization, indicating that the CEN domain may partially mask the targeting surface of the ANKR domain (Fig. 2F,I). Nevertheless, the ANKS4B ANKR domain appears to be sufficient for targeting to the BB.
Figure 2. Mechanism of ANKS4B targeting to the BB.

(A) Constructs of ANKS4B used for CACO-2BBE stable cell line creation. (B-H) Confocal images of 12 DPC CACO- 2BBE monolayers stably expressing the various EGFP-tagged ANKS4B constructs outlined in A. Boxed regions denote areas in zoomed panels for each figure. Dashed lines represent the location where x-z sections were taken; x-z sections are shown below each en face image. Scale bars, 10 μm. (I) Scatterplot quantification of the BB:cytosol ratios of EGFP signal for all EGFP-tagged ANKS4B constructs. EGFP alone is included as a negative control. Bars indicate mean and SD. *p < 0.0001, t test.
Because ANKR domains are known to function as protein-protein interaction modules, often associating with membrane targets (Bennett and Chen, 2001), we sought to determine if the ANKS4B ANKR domain interacts with the cytoplasmic domain (CD) of either CDHR2 or CDHR5. Pull-down analyses using the recombinant CDs of CDHR2 and CDHR5 fused to GST incubated with COS7 cell lysates expressing EGFP-ANKR failed to reveal interactions between these molecules (Fig. S1C).
The ANKR domains of ANKS4B and USH1G are highly conserved (72% identical), which prompted us to examine the impact of USH1G-associated Usher syndrome mutations on the function of ANKS4B. The L48P mutation, which is located in the USH1G ANKR domain, was discovered in members of a German family exhibiting profound congenital deafness (Weil et al., 2003). How this mutation leads to defective cochlear function is not known, but L48P is expected to disrupt ANKR domain folding (Pan and Zhang, 2012). When we reconstituted L48P in the isolated ANKS4B ANKR domain and examined its subcellular localization in CACO-2BBE cells, we observed a marked reduction in BB targeting (Fig. 2H,I). This suggests that the mutant form of USH1G in Usher syndrome patients harboring L48P is probably unable to target to hair cell stereocilia.
ANKS4B is required for microvillar clustering and normal BB assembly
With robust localization at microvillar tips, ANKS4B is well positioned to play a role in intermicrovillar adhesion. To test this proposal, we assessed whether ANKS4B is required for microvillar clustering, intermicrovillar adhesion link formation, and normal BB assembly. To this end, we used lentivirus-mediated stable transduction to create knockdown (KD) CACO-2BBE cell lines lacking ANKS4B. Two independent cell lines created with different shRNA constructs exhibited a KD efficiency of greater than 90% (Fig. 3A,B). In both cases, scoring of 12 DPC CACO-2BBE monolayers showed that depletion of endogenous ANKS4B levels resulted in a dramatic loss of microvillar clustering relative to scramble, a phenotype similar to that observed following KD of other IMAC adhesion molecules, such as CDHR2 or CDHR5 (Fig. 3A,C). Importantly, expression of an EGFP-ANKS4B construct refractory to KD in either ANKS4B KD cell line rescued microvillar clustering, demonstrating that the observed KD phenotype was due to loss of ANKS4B (Fig. 3A,C).
Figure 3. Loss of ANKS4B in CACO-2BBE cells abolishes microvillar clustering and disrupts BB assembly.

(A) Confocal images of 12 DPC CACO-2BBE cells stably expressing either a scramble shRNA construct or two different shRNAs targeting ANKS4B (KD 45 and KD 80), stained for F-actin (magenta) and ANKS4B (green). The ANKS4B KD line expressing an EGFP-tagged “Rescue” variant of ANKS4B refractory to KD is stained for F-actin (magenta) and GFP (green). Boxed regions in merge images denote areas in zoomed panels in each case. Asterisks in zoom panels highlight cells with robust microvillar clustering. Scale bars, 15 μm (B) Western blot analysis of endogenous ANKS4B levels in lysates from 20 DPC non-transduced control, scramble shRNA control, and two independent shRNA ANKS4B KD stable CACO-2BBE lines (KD 45 and KD 80); GAPDH served as a loading control. (C) Quantification of microvillar clustering in non-transduced, scramble, ANKS4B KD, and ANKS4B rescue CACO-2BBE cell lines. For quantification of rescue cell lines, only EGFP-positive cells were scored. Non-transduced, n = 563 cells; scramble control, n = 525 cells; ANKS4B KD, n = 534 cells; ANKS4B KD rescue EGFP-ANKS4B, n = 156 cells. *p < 0.0001, t test. (D) Representative SEM images of 20 DPC CACO-2BBE cells stably expressing either a scramble shRNA construct (top) or an shRNA targeting ANKS4B (bottom). Scale bars, 1 μm. (E) Quantification of microvillar packing density in non-transduced, scramble and ANKS4B KD stably transduced 20 DPC CACO-2BBE monolayers. Area quantified and number of microvilli counted: non-transduced cells, 84.77 μm2, n = 3554; scramble control, 90.34 μm2, n = 3958; ANKS4B KD 45, 103.98 μm2, n = 2005; ANKS4B KD 80, 290.88 μm2, n = 3298; *p < 0.0001, t test. (F) Quantification of observable adhesion links at the distal tips of microvilli in non-transduced, scramble control, and ANKS4B KD CACO-2BBE lines. 100 microvilli were counted for each condition. *p < 0.0001, t test.
We next used scanning electron microscopy (SEM) to assess the impact of ANKS4B KD on the apical morphology of CACO-2BBE monolayers that were allowed to polarize for 20 days in cell culture; this is sufficient time for most cells in the monolayer to build well-formed BBs (Peterson and Mooseker, 1993). Relative to scramble shRNA-expressing control cells, ANKS4B KD monolayers exhibited severely disheveled BBs, with significantly fewer microvilli that were highly variable in length (Fig. 3D,E and S2A). SEM also revealed that intermicrovillar adhesion links were rare in ANKS4B KD monolayers (Fig. 3D,F). Together, these results suggest that ANKS4B is necessary for microvillar clustering, intermicrovillar adhesion link formation, and the assembly of a well-ordered BB during CACO-2BBE differentiation.
We also examined CACO-2BBE cell lines overexpressing EGFP-ANKS4B, but did not observe any significant impact on BB morphology (Fig. S2B). Thus, ANKS4B is not sufficient to drive BB assembly, and some other limiting factor probably mediates ANKS4B function in this context.
ANKS4B interacts with USH1C
ANKS4B was initially isolated in a yeast-two-hybrid screen for USH1C binding partners (Johnston et al., 2004). We first confirmed that USH1C and ANKS4B interact using in vitro pull-downs from COS7 cell lysates and then mapped the domains involved in binding. These experiments detected binding between the ANKS4B C-terminal SAM domain and the NPDZ1 supramodule of USH1C (Fig. 4A,B). This interaction was significantly potentiated, however, when using a larger ANKS4B fragment including both the CEN and SAM domains (Fig. 4B). Thus, ANKS4B uses its C-terminal domains to interact directly with USH1C, a bona fide IMAC component.
Figure 4. Analysis of binding interactions between ANKS4B, USH1C, and MYO7B.

(A) Mapping binding interactions between full length ANKS4B and sub-domains of USH1C; FLAG-tagged ANKS4B served as bait whereas V5-tagged fragments of USH1C served as prey. PD = pulldown. (B) Mapping binding interactions between full length USH1C and ANKS4B sub-domains; FLAG-tagged USH1C served as bait where as EGFP-tagged fragments of ANKS4B served as prey. (C) Confocal microscopy of ANKS4B (green) and villin (magenta) staining in duodenal tissue from wild type and USH1C KO mice. Scale bars, 10 μm. (D) Line scan analysis of ANKS4B signal intensity parallel to the microvillar axis as visualized in confocal images of wild type and USH1C KO tissue. N.U. = normalized units. (E) Analysis of binding interactions between full length USH1C and mutant forms of ANKS4B lacking a functional PBM; FLAG-tagged USH1C served as bait whereas myc-tagged ANKS4B variants served as prey. (F) Confocal images of 12 DPC CACO-2BBE monolayers stably expressing EGFP-ANKS4B (left) and EGFP-ANKS4B-ΔPBM (right). Scale bars, 10 μm. (G) Quantification of BB:cytosol EGFP signal intensity ratios for EGFP-ANKS4B, EGFP-ANKS4B-ΔPBM, and EGFP-alone. Bars indicate mean ± SD. *p < 0.0001, t test. (H) Cartoon depicting the mechanism of ANKS4B/USH1C interaction. (I) Mapping binding interactions between the MYO7B-MF1SH3 tail fragment and sub-domains of ANKS4B; FLAG-tagged MYO7B-MF1SH3 tail fragment served as bait whereas EGFP-tagged fragments of ANKS4B served as prey. (J) Pull-down analysis of interactions between the MYO7B-SH3MF2 tail fragment and sub-domains of USH1C; FLAG-tagged MYO7B-SH3MF2 tail served as bait whereas myc-tagged fragments of USH1C served as prey. (K and L) Cartoons depicting mechanisms of binding between MYO7B and ANKS4B or USH1C.
ANKS4B targeting to the BB requires USH1C
To examine the biological significance of ANKS4B binding to USH1C, we assessed the localization of endogenous ANKS4B in intestinal and kidney tissue sections from USH1C knockout (KO) mice (Tian et al., 2010). ANKS4B was almost completely absent from the BB in USH1C KO enterocytes and proximal tubule kidney epithelial cells (Fig. 4C, S3A,B). Line scan analysis of the distribution of ANKS4B along the enterocyte microvillar axis clearly showed that ANKS4B fails to accumulate at microvillar tips in the absence of USH1C (Fig. 4D). To determine if USH1C-dependent targeting is mediated by the C-terminal PBM of ANKS4B, we created mutants lacking this motif. Mutation of the C-terminal residue of the ANKS4B PBM (ANKS4B-L417R) decreased USH1C binding, while deletion of the entire PBM by removal of the last seven C-terminal residues (ANKS4B-ΔPBM) completely abolished binding (Fig. 4E). Despite containing an intact ANKR domain, the ANKS4B-ΔPBM construct failed to localize to the BB, exhibiting a targeting ratio that was comparable to EGFP alone (Fig. 4F,G and S3C). These data demonstrate that USH1C binds directly to ANKS4B (Fig. 4H) and this interaction is necessary for ANKS4B targeting to the BB. These results also suggest that in the context of full-length ANKS4B, ANKR domain targeting activity is suppressed unless USH1C is present and able to bind.
USH1C and ANKS4B bind to distinct regions of the MYO7B tail domain
Stable targeting of the IMAC to the tips of BB microvilli likely requires direct interactions with the underlying actin cytoskeleton. We previously identified MYO7B as the only actin-binding component of the IMAC and further showed that USH1C plays a key role in coupling microvillar protocadherins to this motor (Crawley et al., 2014b). MYO7B possesses a large C-terminal cargo-binding tail, consisting of two MyTH4-FERM (MF) domains with an intervening SH3 domain (Chen et al., 2001). To determine if ANKS4B is a MYO7B cargo, we assessed binding using a pull-down approach similar to that outlined above. Isolated MF domains of MYO7B were unstable and expressed poorly in COS7 cells. However, addition of the intervening SH3 domain had a marked effect on stabilizing both domains. Therefore, we co-expressed Flag-tagged constructs of the first (MF1SH3) and second (SH3MF2) MyTH4-FERM SH3 domain fragments with EGFP-tagged ANKS4B truncation constructs and performed pull-down analyses. All ANKS4B constructs containing the CEN domain bound specifically to the MF1SH3 fragment of MYO7B (Fig. 4I), with the isolated CEN domain exhibiting the most robust interaction. However, ANKS4B failed to interact with the SH3MF2 fragment (Fig. S3D). This suggests the SH3 domain is not involved in binding between ANKS4B and MYO7B, and also indicates that the two MF domains of MYO7B are not simply redundant cargo binding modules. This specificity prompted us to further investigate binding between USH1C and MYO7B. Interestingly, we found that USH1C interacts with the SH3MF2 fragment, but not MF1SH3 (Fig. S3E). We previously demonstrated that USH1C interacts with MYO7B via its second PDZ domain, although this binding interaction is substantially weaker than that observed with full-length USH1C (Crawley et al., 2014b). We performed more detailed domain mapping of this binding interaction and found that the region encoding PDZ2, the proposed coiled-coil domain, and PDZ3 (PDZ2CCPDZ3) is necessary for robust binding to MYO7B (Fig. 4J). Thus, ANKS4B and USH1C bind to distinct regions of the MYO7B cargo-binding tail domain (Fig. 4K,L). Moreover, USH1C uses separate domains to interact with ANKS4B and MYO7B, namely NPDZ1 and PDZ2CCPDZ3, respectively.
MYO7B cargo binding is a sequential process
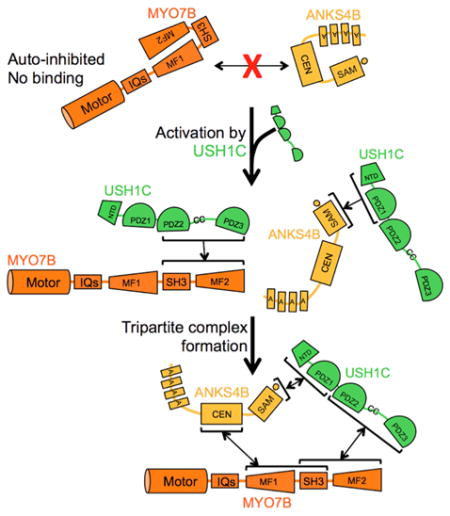
We next investigated whether ANKS4B, USH1C and MYO7B could form a stable tripartite complex in vitro. We first performed pull-downs using the full-length tail of MYO7B. Unexpectedly, we found that neither ANKS4B nor any of its truncated fragments were able to interact with the full-length tail of MYO7B (Fig. S3F). This suggested that, in the context of the full-length tail, the ANKS4B binding site in MF1 might be masked and unavailable for interaction. In contrast, we observed robust binding between USH1C and the full-length tail of MYO7B (Fig. S3E). We then asked if USH1C binding to MYO7B was necessary for ANKS4B incorporation into the complex. Pull-downs revealed that ANKS4B was only recovered with full-length MYO7B tail when USH1C was co-expressed (Fig. 5A). To test whether binding of USH1C to MYO7B MF2 unmasks the ANKS4B binding site found in MF1, we repeated our pull-downs using versions of ANKS4B and USH1C that cannot interact directly. We co-expressed the isolated PDZ2CCPDZ3 fragment of USH1C (unable to bind ANKS4B) and the isolated CEN domain of ANKS4B (unable to bind USH1C) along with full-length tail of MYO7B. Pull-down analyses demonstrated robust tripartite complex formation between these fragments (Fig. 5B). Based on these data, we conclude that in the absence of USH1C, the cargo binding tail of MYO7B rests in an auto-inhibited state where the ANKS4B binding site in MF1 is masked. However, binding of USH1C to MF2 activates the tail such that MF1 becomes available for binding to ANKS4B (Fig. 5C). Together, these interaction studies implicate ANKS4B as a multivalent IMAC component and position USH1C as a critical activator of IMAC assembly.
Figure 5. Tripartite complex formation between ANKS4B, USH1C, and MYO7B.

(A) Pull-downs of FLAG-tagged MYO7B full tail fragment co-expressed with myc-tagged USH1C and EGFP-tagged ANKS4B variants reveals formation of a stable tripartite complex. (B) Pull-downs of FLAG-tagged MYO7B full tail fragment, co-expressed with EGFP-tagged ANKS4B-CEN domain and myc-tagged fragments of USH1C indicate that a stable tripartite complex still forms even when ANKS4B and USH1C are unable to interact directly. (C) Cartoon summary of the regulated assembly of the tripartite complex composed of MYO7B, ANKS4B, and USH1C.
ANKS4B is required for IMAC assembly
Given that ANKS4B interacts with multiple IMAC components, loss of this factor might disrupt IMAC assembly, which could explain the KD phenotype described above (Fig. 3A-F). To examine the impact of ANKS4B KD on IMAC formation in cells, we stained KD cell lines with probes specific for IMAC components and imaged samples using confocal microscopy. Strikingly, we observed a near-complete loss of all IMAC components from the BB of ANKS4B KD cells (Fig. 6A and S4). Western blot analysis of KD lysates revealed that levels of CDHR2, CDHR5, USH1C, and MYO7B were all reduced to <10% of controls (Fig. 6B,C). Interestingly, actin levels were also significantly reduced in both ANKS4B KD cell lines, an observation consistent with the reduced microvillar density observed in our SEM analysis (Fig. 6C and 3E). These data indicate that ANKS4B plays an essential role in IMAC assembly, most likely by functioning as a multivalent scaffold, which promotes targeting of IMAC components to the BB and stabilizes the resulting adhesion complexes.
Figure 6. ANKS4B is required for IMAC assembly and localization at microvillar tips.

(A) Confocal images of 12 DPC CACO-2BBE cells stably expressing either a scramble shRNA construct or an shRNA targeting ANKS4B, stained for F-actin (magents) and either CDHR2, CDHR5, USH1C, or MYO7B (green). Boxed regions denote the area in zoomed image panels. Scale bars, 15 μm. (B) Western blot analysis of endogenous CDHR2, CDHR5, USH1C, MYO7B, and actin levels in lysates from 20 DPC scramble control, or two independent shRNA ANKS4B KD stable CACO-2BBE lines. GAPDH was used as a loading control. (C) Quantification of endogenous CDHR2, CDHR5, USH1C, MYO7B and actin levels in lysates from 20 DPC scramble control, or two independent shRNA ANKS4B KD stable CACO-2BBE lines. Values are expressed as expression level relative to scramble control cell line.
DISCUSSION
USH1C regulates ANKS4B function
Our data suggest a model for IMAC assembly where a key event is regulated formation of a stable tripartite complex between ANKS4B, USH1C, and MYO7B (Fig. 7). In the absence of USH1C, ANKS4B appears to exist in a dormant state, unable to target to the BB. Indeed, despite containing an intact ANKR targeting domain, endogenous ANKS4B is lost from the BB in USH1C KO mice, suggesting that USH1C is needed to activate apical targeting. Moreover, a construct of ANKS4B lacking the C-terminal PBM motif (required for USH1C binding) no longer targets to the BB in CACO-2BBE cells. Further deletion of the C-terminal region to eliminate the entire SAM-PBM domain, however, rescues the ability of the fragment to target to correctly. This suggests that USH1C binding to ANKS4B might relieve SAM-PBM mediated auto-inhibition of ANKR targeting activity. Studies aimed at how such intramolecular interactions are controlled by USH1C will be key for understanding the regulation of ANKS4B function.
Figure 7. Model summarizing interactions between IMAC components revealed in this and previous studies.

MYO7B tail domain templates sequential IMAC assembly
Our biochemical analyses uncovered an unexpected layer of regulation controlling IMAC assembly. Specifically, we found that ANSK4B was unable to interact with the full-length cargo binding tail of MYO7B, although it exhibited robust binding to isolated MF1SH3. This suggests that the ANKS4B binding site found in MF1 is masked in the context of the full-length tail, i.e. the MYO7B tail domain may exist in an auto-inhibited state. Including USH1C in binding reactions enabled ANKS4B to bind to the MYO7B tail. When these experiments were repeated with fragments of ANKS4B and USH1C that were unable to interact with each other, we observed a similar result suggesting that USH1C binding to the MYO7B tail domain is the key activating event in tripartite complex formation. Previous studies of Drosophila and human MYO7A (the functional homolog of MYO7B in the Usher complex) demonstrated that the tail folds back to inhibit the activity of the motor domain (Sakai et al., 2015; Umeki et al., 2009; Yang et al., 2009). This inhibition is mediated by interactions between the motor-IQ domain and both the N- and C-terminal regions of the tail domain of MYO7A (Umeki et al., 2009). Although the molecular details are still unclear, relief of motor inhibition is thought to occur through calcium binding to calmodulin light chains bound to the neck domain, actin binding to MF2, or binding of cargo to MF2 (Sakai et al., 2011; Umeki et al., 2009; Yang et al., 2009). Although it is unknown whether MYO7B motor activity is regulated in a manner similar to MYO7A, our results suggest a mechanism for sequential, regulated cargo binding, which is entirely independent of the motor domain. We propose that the MYO7B tail can adopt an auto-inhibited conformation that does not require physical contacts with the motor domain. Moreover, such auto-inhibition appears to impart a sequence for cargo docking, where USH1C binding to MF2 must occur before ANKS4B is allowed to bind to MF1. Going forward, it will be important to determine how such regulation of cargo binding impacts MYO7B motor activity and targeting to microvillar tips. It will also be important to elucidate mechanisms that control the availability, activity, and targeting of USH1C to the BB.
ANKR domain mutations in human disease
As we were unable to detect binding between the ANKS4B ANKR domain and other known IMAC components, this domain likely binds to an as-of-yet unidentified factor, perhaps a BB membrane protein. Interestingly, a number of mutations linked to Usher syndrome have been mapped to the USH1G ANKR domain, including the missense mutation, L48P (Weil et al., 2003). When L48P is reconstituted in the ANKR domain of ANKS4B, we observed diminished BB targeting. Given the high level of homology that exists between the ANKR domains of ANKS4B and USH1G, we propose that the USH1G ANKR domain functions to target this molecule to stereocilia in hair cells, and that the L48P mutation probably disrupts targeting in Usher syndrome patients. Identification of ANKR domain binding partners will be a critical goal of future studies.
ANKS4B in IMAC formation and BB assembly
Our data clearly shows that ANKS4B plays a critical role in BB assembly during enterocyte differentiation. Disruption of ANKS4B in CACO-2BBE cells had a severe impact on microvillar growth and organization, effects that are likely related to defects in IMAC formation. Indeed, in the absence of ANKS4B all other known IMAC components fail to accumulate within the BB. Together these results indicate that ANKS4B serves as a critical scaffold, which is necessary for stabilizing the localization of other IMAC components. In this light, ANKS4B may serve as another important point of regulation for IMAC formation and function. Elucidation of upstream signals that control ANKS4B function will be critical for understanding how this factor and its interacting partners contribute to apical morphogenesis.
In summary
The identification of ANKS4B as an USH1G-like molecule in the IMAC further reinforces the emerging paradigm that epithelial cells from diverse tissues with divergent functions rely on a conserved adhesion-based mechanism for shaping their apical surface during differentiation. The fact that the adhesion complexes from the gut and the inner ear display remarkable homology in their molecular composition should allow for meaningful cross-pollination of information between systems, and provide a wider range of experimental avenues for interrogating how genetic perturbations in complex components result in human diseases such as Usher syndrome. Indeed, the results reported here may shed light on molecular mechanisms underlying sensory dysfunction and enteropathy exhibited by patients with Type 1C Usher syndrome (Bitner-Glindzicz et al., 2000; Hussain et al., 2004; Verpy et al., 2000), and provide insight on the molecular pathology of disease associated with mutations in USH1G (Weil et al., 2003).
EXPERIMENTAL PROCEDURES
Cell culture and stable line generation
CACO-2BBE, COS7, and HEK293FT cells were cultured at 37°C and 5% CO2 in DMEM with high glucose and 2 mM L-glutamine. Media was supplemented with 20% FBS for CACO-2BBE cells and 10% FBS for COS7 and HEK293FT cells. For lentivirus transduction, CACO-2BBE cells were grown to 90% confluency in T25 flasks. Before lentiviral infection, the media was supplemented with 8 μg/ml polybrene. After 12 hours of incubation with lentivirus, the cells were reseeded into T75 flasks and grown for 3 days. Cells were then reseeded into T182 flasks with media containing 50 μg/ml of puromycin or 1 mg/ml of G418 and grown to select for stable integration. For stable cell lines transduced with constructs cloned into the pINDUCER20-EGFP-C1 vector, 1 μg/ml of doxycycline was included in the growth media at all times.
Molecular biology
The human cDNA constructs used in this study were as follows: ANKS4B, GI: 148664245; USH1C, GI: 663071173; MYO7B, GI: 122937511. DNA encoding the various domain constructs of ANKS4B, USH1C and MYO7B were generated by PCR and TOPO cloned into the pCR8 entry vector (Invitrogen). The resulting entry vectors were then shuttled into destination expression vectors using standard Gateway protocols. The domain boundaries for the ANKS4B constructs used are as follows: ANKR (aa 1-252), CEN (aa 253-346), SAM (aa 348-417), ANKRCEN (aa 1-346), CENSAM (aa 253-417) and ANKS4B-ΔPBM (aa 1-410). The domain boundaries for the USH1C constructs used are as follows: NPDZ1 (aa 1-193), PDZ2 (aa 194-354), PDZ2CC (aa 194-432), PDZ3 (aa 360-533), CCPDZ3 (aa 275-533), NPDZ12 (aa 1-354) and PDZ2CCPDZ3 (aa 194-533). The domain boundaries for the MYO7B constructs used are as follows: MYO7B full-length tail (aa 916-2116), MF1SH3 (aa 916-1542), SH3MF2 (aa 1501-2116). The pINDUCER20 lentiviral vector (Meerbrey et al., 2011) was modified to include an EGFP-tag upstream of the Gateway recombination cassette, to create pINDUCER20-EGFP-C1. This vector was used for the generation of stable cell lines expressing EGFP-fusion protein. The pKMyc was a kind gift from Ian Macara (Addgene plasmid # 19400) and was adapted to Gateway technology using the Gateway Conversion kit to generate pKMyc-GW. Vectors used for expression of FLAG and V5-tagged proteins in this study have been previously described (Crawley et al., 2014b). Knockdown shRNA clones targeting ANKS4B were expressed in the pLKO.1 vector, and correspond to TRC clones TRCN0000122345 and TRCN0000139080 (Sigma). A non-targeting scramble shRNA cloned into the pLKO.1 vector was used as a control (Addgene; plasmid 1864). For rescue experiments, the Quikchange site-directed mutagenesis kit (Agilent; Cat#200523) was used to introduce refractory silent mutations into the regions of ANKS4B targeted by the shRNA constructs. The primers sets used for mutagenesis were the following: ANKS4B RF45 sense TCCTCTACTGCAGATTATCTCCAAAGCTTCCAAGTTCCCATGGTAG, ANKS4B RF45 antisense CTACCATGGGAACTTGGAAGCTTTGGAGATAATCTGCAGTAGAGGA, and ANKS4B RF80 sense GCCTCTGATGCTCCTTGTCTTTGTAAGAGTTCACTGGGCA GTTTAAAACCAA, ANKS4B RF80 antisense TTGGTTTTAAACTGCCCAGTGAACTCT TACAAAGACAAGGAGCATCAGAGGC. The constructs encoding the cytoplasmic domains of CDHR2 and CDHR5 fused to GST have been previously described (Crawley et al., 2014b).
Light microscopy
Cells and tissue sections were imaged using a Leica TCS SP5 or a Nikon A1R laser-scanning confocal microscope. Super-resolution microscopy was performed using an Applied Precision DeltaVision OMX (GE Healthcare). Paraffin-embedded duodenal tissue sections from wild type and USH1C KO mice were deparaffinized using Histo-clear solution (Fisher), rehydrated in a descending graded ethanol series and subjected to an antigen retrieval step which involved boiling the sample slides for 1 hour in a 10mM Tris pH 9, 0.5mM EGTA solution. Samples were then washed three times and stained using anti-ANKS4B (1:50; Santa Cruz Cat#sc241821) and villin (1:50; Santa Cruz Cat#sc58897) followed by Alexa Fluor-488 donkey anti-rabbit (1:200; Invitrogen) and Alexa Fluor-568 donkey anti-mouse (1:200; Invitrogen). Sections were washed three times and then dehydrated in an ascending graded ethanol series and mounted with Prolong anti-fade reagent (Invitrogen). Cells were washed once in warm PBS and incubated briefly with 0.02% Saponin (Sigma) in warm PBS, then fixed for 15 mins with 4% paraformaldehyde in PBS containing 0.1% Triton (Sigma). After fixation, cells were washed three times with PBS and blocked overnight in 5% BSA in PBS. Immunostaining was performed using anti-ANKS4B (1:25; Sigma Cat#SAB1302033), anti-CDHR2 (1:75; Sigma Cat#HPA012569), anti-CDHR5 (1:250; Sigma Cat#HPA009081), anti-USH1C (1:70; Sigma Cat#HPA027398), anti-MYO7B (1:25; Sigma Cat#HPA039131) or anti-GFP (1:200; Aves labs Cat#GFP1020) at 37°C for 2 hours, after which coverslips were washed three times with PBS and Alexa Fluor-488 donkey anti-rabbit (1:200) secondary antibody or Alexa Fluor-488 goat anti-chicken (1:200) along with Alexa Fluor-568 Phalloidin (1:200) were applied for 1 hour at room temperature. Coverslips were then washed five times in PBS and mounted with Prolong anti-fade reagent (Invitrogen). All images shown are en face maximum projections through the full height of the BB, with the exception of x-z sections, which are single plane confocal images.
Electron microscopy
All EM reagents were purchased from Electron Microscopy Sciences. CACO-2BBE cells used for SEM were seeded at a density of 100,000 cells/well into 0.4 μm 12 mm Transwell-COL inserts (Corning Inc.) and allowed to grow for 20 days. Samples were then processed for SEM as described previously (Heintzelman and Mooseker, 1990).
Image analysis
All image analysis was performed using ImageJ (NIH). For line scan analysis, a line was drawn parallel to the microvillar axis using F-actin signal (visualized with phalloidin staining) as a reference and the intensity of the ANKS4B signal along that line was recorded. Intensity values were normalized by the maximum gray scale value for an 8-bit image (i.e. 255), whereas all microvillar lengths were normalized so that the base of the microvillus was equal to 0 and the tip was equal to 1. Normalized line-scans were then plotted together and fit to a single Lorentzian using non-linear regression (Prism v.6, GraphPad). The resulting fit revealed the position of peak ANKS4B localization (LP) and distribution width (S.D.) relative to the microvillar axis. For analysis of microvillar clustering, cells from 12 DPC monolayers were stained for F-actin using Alexa Fluor-568 Phalloidin (1:200) and either endogenous ANKS4B (1:25; Sigma Cat#SAB1302033) for non-transduced and pLKO.1 stable cell lines or GFP (1:200; Aves labs Cat#GFP1020) for GFP-fusion stable cell lines. Individual cells were scored for microvillar clustering as previously described (Crawley et al., 2014b). For EGFP-fusion stable cell lines, only EGFP-positive cells were scored. For analysis of construct enrichment in microvilli, we calculated the ratio of BB to cytosolic signal intensity as we previously described (Mazerik and Tyska, 2012).
Pull-down assays
COS7 cells were grown in T75 flasks to 90% confluency and transfected with pull-down constructs using Lipofectamine 2000 according to the manufacturer’s protocol. After 48 hours, cell were recovered by a cell scraper, washed once in PBS, and lysed using ice-cold Cellytic M buffer (Sigma) containing 2mM ATP, 1× cOmplete ULTRA protease inhibitor cocktail (Roche) and 1mM Pefabloc SC (Roche). Cell lysates were centrifuged at 16,000×g and the soluble material recovered and incubated with a 50 μl bed volume of either pre-equilibrated anti-Flag M2 resin (Sigma) or glutathione resin (Sigma) with GST fused to the CDs of the protocadherins. Resins were incubated with cell lysates for two hours, rocking at 4°C, pelleted by a low sp eed spin, washed four times using RIPA buffer supplemented with 2mM ATP, 1× cOmplete ULTRA protease inhibitor cocktail (Roche) and 1mM Pefabloc SC (Roche) and eluted by boiling in 2×SDS buffer to recover bound material. Resin bound material was detected by either staining with Coomassie blue or western analysis with the following antibody dilutions: mouse anti-Flag M2 (1:1000; Sigma Cat#F3165), mouse anti-V5 (1:5000; Invitrogen Cat#R960-25), or mouse anti-myc 9E10 (1:1000; Vanderbilt Antibody and Protein Resource Core). All pull-down assays were repeated at least three times in independent experiments and results shown are representative.
Western blot analysis of CACO-2BBE knockdown cell lines
CACO-2BBE ANKS4B knockdown cell lines were seeded into T75 flasks and allowed to grow for 20 DPC. Cells were then washed once with PBS and recovered by a cell scraper, pelleted at low speed, washed again in PBS, and lysed using ice-cold Cellytic M buffer (Sigma) containing 2mM ATP, 1× cOmplete ULTRA protease inhibitor cocktail (Roche) and 1mM Pefabloc SC (Roche). The resulting cell lysates were centrifuged at 16,000×g and the soluble material recovered and boiled in the presence of SDS sample buffer at a 2× final concentration for 5 minutes. Samples were then separated on 4–12% Bis-Tris gels (Novex) and western analysis performed using the following antibody dilutions: ANKS4B (1:200; Sigma Cat#HPA043523), anti-CDHR2 (1:100; Sigma Cat#HPA012569), anti-CDHR5 (1:500; Sigma Cat#HPA009081), anti-USH1C (1:250; Sigma Cat#HPA027398), anti-MYO7B (1:100; Sigma Cat#HPA039131), anti-GAPDH (1:1000, Cell Signaling Cat#2118L) and anti-actin (1:1000, Sigma Cat#A2066). Quantification of expression levels was performed using ImageJ software (NIH) with the signal from GAPDH used to normalize the loading values for samples.
Statistical analysis
All graphs were generated and statistical analyses performed using either SigmaPlot v. 12 (Systat) or Prism v.6 (GraphPad). For all figures, error bars represent standard deviation. Unpaired t tests were employed determine statistical significance between reported values.
Supplementary Material
Highlights.
Ankyrin repeat protein ANKS4B is found at the tips of brush border microvilli
ANKS4B is a component of the protocadherin-based intermicrovillar adhesion complex
ANKS4B is essential for adhesion complex formation and normal brush border assembly
PDZ domain protein USH1C regulates ANKS4B function in intermicrovillar adhesion
Acknowledgments
We thank all members of the Tyska laboratory and the Vanderbilt Epithelial Biology Center for advice and support. Electron microscopy was performed through use of the VUMC Cell Imaging Shared Resource. This work was supported by American Heart Association (AHA) predoctoral fellowships (DAS and NGL), AHA postdoctoral fellowship (SWC), Vanderbilt University Medical Center's Digestive Disease Research Center (P30DK058404), and NIH DK075555 and DK095811 (MJT).
Footnotes
AUTHOR CONTRIBUTIONS
SWC designed experiments, performed experiments, analyzed data, and wrote the manuscript. MJT supervised the study, analyzed data, and wrote the manuscript. MLW, NGL, and DAS helped with data collection. All authors contributed to editing the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, Chen L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol. 2001;13:61–67. doi: 10.1016/s0955-0674(00)00175-7. [DOI] [PubMed] [Google Scholar]
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, et al. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet. 2000;26:56–60. doi: 10.1038/79178. [DOI] [PubMed] [Google Scholar]
- Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberlotto E, Michel V, Foucher I, Bahloul A, Goodyear RJ, Pepermans E, Michalski N, Perfettini I, Alegria-Prevot O, Chardenoux S, et al. Usher type 1G protein sans is a critical component of the tip-link complex, a structure controlling actin polymerization in stereocilia. Proc Natl Acad Sci U S A. 2011;108:5825–5830. doi: 10.1073/pnas.1017114108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Hasson T, Zhang DS, Schwender BJ, Derfler BH, Mooseker MS, Corey DP. Myosin-VIIb, a novel unconventional myosin, is a constituent of microvilli in transporting epithelia. Genomics. 2001;72:285–296. doi: 10.1006/geno.2000.6456. [DOI] [PubMed] [Google Scholar]
- Crawley SW, Mooseker MS, Tyska MJ. Shaping the intestinal brush border. J Cell Biol. 2014a;207:441–451. doi: 10.1083/jcb.201407015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley SW, Shifrin DA, Jr, Grega-Larson NE, McConnell RE, Benesh AE, Mao S, Zheng Y, Zheng QY, Nam KT, Millis BA, et al. Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell. 2014b;157:433–446. doi: 10.1016/j.cell.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzelman MB, Mooseker MS. Assembly of the brush border cytoskeleton: changes in the distribution of microvillar core proteins during enterocyte differentiation in adult chicken intestine. 1990;15:12–22. doi: 10.1002/cm.970150104. [DOI] [PubMed] [Google Scholar]
- Helander HF, Fandriks L. Surface area of the digestive tract - revisited. Scand J Gastroenterol. 2014;49:681–689. doi: 10.3109/00365521.2014.898326. [DOI] [PubMed] [Google Scholar]
- Hussain K, Bitner-Glindzicz M, Blaydon D, Lindley KJ, Thompson DA, Kriss T, Rajput K, Ramadan DG, Al-Mazidi Z, Cosgrove KE, et al. Infantile hyperinsulinism associated with enteropathy, deafness and renal tubulopathy: clinical manifestations of a syndrome caused by a contiguous gene deletion located on chromosome 11p. J Pediatr Endocrinol Metab. 2004;17:1613–1621. doi: 10.1515/jpem.2004.17.12.1613. [DOI] [PubMed] [Google Scholar]
- Johnston AM, Naselli G, Niwa H, Brodnicki T, Harrison LC, Gonez LJ. Harp (harmonin-interacting, ankyrin repeat-containing protein), a novel protein that interacts with harmonin in epithelial tissues. Genes Cells. 2004;9:967–982. doi: 10.1111/j.1365-2443.2004.00776.x. [DOI] [PubMed] [Google Scholar]
- Mazerik JN, Tyska MJ. Myosin-1A targets to microvilli using multiple membrane binding motifs in the tail homology 1 (TH1) domain. J Biol Chem. 2012;287:13104–13115. doi: 10.1074/jbc.M111.336313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell RE, Benesh AE, Mao S, Tabb DL, Tyska MJ. Proteomic analysis of the enterocyte brush border. Am J Physiol Gastrointest Liver Physiol. 2011;300:G914–926. doi: 10.1152/ajpgi.00005.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108:3665–3670. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Zhang M. Structures of usher syndrome 1 proteins and their complexes. Physiology (Bethesda) 2012;27:25–42. doi: 10.1152/physiol.00037.2011. [DOI] [PubMed] [Google Scholar]
- Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- Peterson MD, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. I. Ultrastructural analysis of cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:445–460. doi: 10.1242/jcs.105.2.445. [DOI] [PubMed] [Google Scholar]
- Sakai T, Jung HS, Sato O, Yamada MD, You DJ, Ikebe R, Ikebe M. Structure and Regulation of the Movement of Human Myosin VIIA. J Biol Chem. 2015;290:17587–17598. doi: 10.1074/jbc.M114.599365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Umeki N, Ikebe R, Ikebe M. Cargo binding activates myosin VIIA motor function in cells. Proc Natl Acad Sci U S A. 2011;108:7028–7033. doi: 10.1073/pnas.1009188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Liu XZ, Han F, Yu H, Longo-Guess C, Yang B, Lu C, Yan D, Zheng QY. Ush1c gene expression levels in the ear and eye suggest different roles for Ush1c in neurosensory organs in a new Ush1c knockout mouse. Brain Res. 2010;1328:57–70. doi: 10.1016/j.brainres.2010.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeki N, Jung HS, Watanabe S, Sakai T, Li XD, Ikebe R, Craig R, Ikebe M. The tail binds to the head-neck domain, inhibiting ATPase activity of myosin VIIA. Proc Natl Acad Sci U S A. 2009;106:8483–8488. doi: 10.1073/pnas.0812930106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance BA, Chan C, Robertson ML, Finlay BB. Enteropathogenic and enterohemorrhagic Escherichia coli infections: emerging themes in pathogenesis and prevention. Can J Gastroenterol. 2002;16:771–778. doi: 10.1155/2002/410980. [DOI] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, et al. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000;26:51–55. doi: 10.1038/79171. [DOI] [PubMed] [Google Scholar]
- Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003;12:463–471. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- Wilson W, Scott RB, Pinto A, Robertson MA. Intractable diarrhea in a newborn infant: microvillous inclusion disease. Can J Gastroenterol. 2001;15:61–64. doi: 10.1155/2001/743925. [DOI] [PubMed] [Google Scholar]
- Wu L, Pan L, Wei Z, Zhang M. Structure of MyTH4-FERM domains in myosin VIIa tail bound to cargo. Science. 2011;331:757–760. doi: 10.1126/science.1198848. [DOI] [PubMed] [Google Scholar]
- Yan J, Pan L, Chen X, Wu L, Zhang M. The structure of the harmonin/sans complex reveals an unexpected interaction mode of the two Usher syndrome proteins. Proc Natl Acad Sci U S A. 2010;107:4040–4045. doi: 10.1073/pnas.0911385107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Baboolal TG, Siththanandan V, Chen M, Walker ML, Knight PJ, Peckham M, Sellers JR. A FERM domain autoregulates Drosophila myosin 7a activity. Proc Natl Acad Sci U S A. 2009;106:4189–4194. doi: 10.1073/pnas.0808682106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.