

Abstract
Listeria monocytogenes is a bacterial pathogen which establishes intracellular parasitism in various cells, including macrophages and nonhematopoietic cells, such as hepatocytes. It has been reported that several proinflammatory cytokines have pivotal roles in innate protection against L. monocytogenes infection. We found that a proinflammatory cytokine, interleukin 22 (IL-22), was expressed by CD3+ CD4+ T cells at an early stage of L. monocytogenes infection in mice. To assess the influence of IL-22 on L. monocytogenes infection in hepatocytes, cells of a human hepatocellular carcinoma line, HepG2, were treated with IL-22 before L. monocytogenes infection in vitro. Gene expression analysis of the IL-22-treated HepG2 cells identified phospholipase A2 group IIA (PLA2G2A) as an upregulated antimicrobial molecule. Addition of recombinant PLA2G2A to the HepG2 culture significantly suppressed L. monocytogenes infection. Culture supernatant of the IL-22-treated HepG2 cells contained bactericidal activity against L. monocytogenes, and the activity was abrogated by a specific PLA2G2A inhibitor, demonstrating that HepG2 cells secreted PLA2G2A, which killed extracellular L. monocytogenes. Furthermore, colocalization of PLA2G2A and L. monocytogenes was detected in the IL-22-treated infected HepG2 cells, which suggests involvement of PLA2G2A in the mechanism of intracellular killing of L. monocytogenes by HepG2 cells. These results suggest that IL-22 induced at an early stage of L. monocytogenes infection enhances innate immunity against L. monocytogenes in the liver by stimulating hepatocytes to produce an antimicrobial molecule, PLA2G2A.
INTRODUCTION
Listeria monocytogenes is an intracellular bacterial pathogen which establishes infection in cytoplasm of various cells, including macrophages and nonhematopoietic cells, such as hepatocytes and epithelial cells (1, 2). Gamma interferon (IFN-γ)-dependent acquired immune responses have pivotal role in protection against L. monocytogenes infection (3, 4). At the level of innate immunity, macrophages are important effector cells, and involvement of IFN-γ produced by NK cells and gamma/delta T-cell receptor (TCRγδ) T cells has also been reported (5–7). Recently, a proinflammatory cytokine produced by TCRγδ T cells, interleukin 17A (IL-17A), was identified as a new effector cytokine in innate immunity to L. monocytogenes infection in the liver (8–10).
“Inflammatory” lymphocytes producing proinflammatory cytokines (IL-17A, IL-17F, and/or IL-22) have been identified. CD4+ T cells termed Th17 or inflammatory Th (Thi) cells are characterized by the production of IL-17A (11–13), which induces neutrophilic inflammation and production of antimicrobial peptides, such as β-defensins and S100A molecules (14). Lymphocytes of the innate immune system, such as TCRγδ T cells and type 3 innate lymphoid cells (ILC3), which lack expression of antigen-specific receptor, were also reported to produce IL-17A (15–18). IL-17A participates in protective immunity against extracellular pathogens, including Klebsiella pneumonia, Staphylococcus aureus, and Candida albicans, via induction of neutrophils (19, 20). The importance of IL-17A produced by TCRγδ T cells in protective immunity against intracellular pathogens such as Listeria monocytogenes and Mycobacterium tuberculosis has also been reported (8, 9, 15, 21), although the precise mechanism of IL-17A-dependent protection remains to be clarified. Another inflammatory cytokine, IL-22, was also reported to be produced by CD4+ T cells, γδ T cells, and ILC3 cells (22).
IL-22 is a proinflammatory cytokine of the IL-10 family. The IL-22 receptor, consisting of IL-22R1 and IL-10R2, chains is expressed by nonhematopoietic cells in the liver, intestine, lung, and skin (22). Ligand binding to the IL-22 receptor induces expression of antimicrobial peptides such as β-defensins, RegIII proteins, and S100 proteins, and cytokines and chemokines, such as IL-6, granulocyte colony-stimulating factor (G-CSF), CXCL1, and CXCL5, just as IL-17A does (22–24). IL-22 has been reported to induce expression of acute-phase proteins, including amyloid A, lipopolysaccharide (LPS)-binding protein, and haptoglobin (25, 26). It has been reported that IL-22 participates in protective immunity against extracellular bacterial infection in the intestine and lung through induction of antimicrobial and proinflammatory molecules (27, 28).
In the present study, we found that IL-22 expression was induced in the livers of L. monocytogenes-infected mice at an early stage of infection, and the innate IL-22-producing cells were identified as CD4+ T cells. Based on this observation, the influence of IL-22 on L. monocytogenes infection of hepatocytes was analyzed using the human hepatocellular carcinoma line HepG2. The data demonstrated that treatment of HepG2 cells with IL-22 induced expression of an antimicrobial phospholipase, phospholipase A2 group IIA (PLA2G2A), suggesting a new pathway of IL-22-mediated innate immunity against bacterial pathogens.
MATERIALS AND METHODS
Mice.
C57BL/6 mice and C3H/HeJ mice were purchased from Japan SLC (Hamamatsu, Japan), maintained under conventional conditions, and used at 8 to 12 weeks of age. Experiments were conducted according to the Institutional Ethical Guidelines for Animal Experiments of the University of the Ryukyus under approval of the Animal Experiments Safety and Ethics Committee of the University of the Ryukyus.
Microorganisms and in vivo infection.
L. monocytogenes strain EGD was isolated from spleen homogenates of L. monocytogenes-infected mice, grown in tryptic soy broth (Difco, Detroit, MI), resuspended in phosphate-buffered saline (PBS), and stored at −70°C in small aliquots until use. C57BL/6 and C3H/HeJ mice were infected intraperitoneally (i.p.) with 5 × 104 and 5 × 103 CFU of L. monocytogenes, respectively, doses which correspond to 1/10 of the 50% lethal dose for each mouse strain.
Isolation of LMNC and flow cytometry (FCM).
Liver mononuclear cells (LMNC) were isolated from the livers of L. monocytogenes-infected C57BL/6 mice as described previously (8, 9). The LMNC were stimulated with 25 ng/ml phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, St. Louis, MO) and 1 μg/ml of the calcium ionophore A23187 (Sigma-Aldrich) for 4 h in the presence of brefeldin A (GolgiPlug; BD, Oxnard, CA). The cultured cells were collected and treated with anti-CD16/32 monoclonal antibody (MAb) (2.4G2) to block Fc receptor-mediated nonspecific Ab binding, followed by staining of surface molecules. The MAb used for surface staining was as follows: allophycocyanin (APC)-Cy7-conjugated anti-CD3 (BioLegend. San Diego, CA), Alexa Fluor 700-conjugated anti-CD4 (BioLegend), peridinin chlorophyll protein (PerCP)-Cy5-conjugated anti-TCRCδ (BioLegend), phycoerythrin (PE)-Cy7-conjugated NKp46 (BioLegend), and PE-conjugated anti-CD117/c-Kit (BD) MAbs. The cells were then intracellularly stained with APC-conjugated anti-IL-22 MAb (clone IL22JOP; eBioscience, San Diego, CA) using a Cytofix/Cytoperm kit (BD, Oxnard, CA) according to the manufacturer's instructions. The stained cells were analyzed using a FACSCanto flow cytometer (BD).
In vitro infection.
HepG2 (human hepatocellular carcinoma) cells were incubated in antibiotic-free Dulbecco's minimal essential medium (MEM) (Wako, Osaka, Japan) with 10% fetal bovine serum (FBS) (Equitech Bio, Kerrville, TX) at 37°C, 5% CO2 in 24-well or 96-well culture plates at 1 × 106 cells/ml in the presence or absence of human recombinant IL-22 (rIL-22) (10 ng/ml) and/or rIL-17A (50 ng/ml) (PeproTech, Rocky Hill, NJ) for 48 h. The cells were then infected with L. monocytogenes at a multiplicity of infection (MOI) of 10 for 1 h, followed by incubation with gentamicin at 5 μg/ml to kill extracellular bacteria. In some experiments, HepG2 cells were plated in 96-well culture plates and incubated overnight, and then human recombinant lipocalin-2 (rLCN-2) (50 μg/ml) (Sigma-Aldrich) or rPLA2G2A (R&D Systems, Minneapolis, MN) was added into the culture just before in vitro infection with L. monocytogenes at an MOI of 10. After infection for 1 h, the cells were cultured for 3 h with gentamicin, washed three times with warm PBS, and lysed with distilled water, and serial dilutions of the lysates were plated on nutrient agar plates to measure bacterial numbers in the infected cells.
Bactericidal activity of HepG2 culture supernatants.
To analyze bactericidal activity in the culture supernatants of HepG2 cells, the cells were cultured with 10 ng/ml of IL-22 for 48 h, and culture supernatants were collected. Forty microliters of the supernatants was mixed with 10 μl of PBS containing 2,000 CFU of L. monocytogenes, incubated for 1 h at 37°C, and plated on nutrient agar plates after serial dilution to determine bacterial count. To analyze the influence of PLA2G2A, the specific PLA2G2A inhibitor LY315920 (AdooQ BioScience, Irvine, CA) (29) was added at 20 μg/ml to the culture supernatants.
Microarray and real-time reverse transcription-PCR (RT-PCR) analysis.
RNA was extracted from HepG2 cells by using TRIzol reagent (Life Technologies, Carlsbad, CA). For microarray analysis, L. monocytogenes-infected HepG2 cells cultured with or without rIL-22 plus rIL-17A for 24 h before the infection were used. RNA was amplified by using an amino allyl aRNA kit (Life Technologies), labeled with Cy5 or Cy3 monoreactive dye (GE Healthcare, Pittsburgh, PA), hybridized with 3D-Gene DNA Tip (Human Oligochip 25k; Toray, Kamakura, Japan), and analyzed. The data were expressed as the ratio of the signal of the cytokine-treated cells to that of the non-cytokine-treated HepG2 cells.
For RT-PCR analysis, RNA extracted from HepG2 cells or the livers of L. monocytogenes-infected mice was reverse transcribed with reverse transcriptase (SuperScript Vilo cDNA synthesis kit; Invitrogen, Carlsbad, CA) and amplified with Taq polymerase premixed with SYBR green using the Step One real-time PCR system (Applied Biosystems, Foster City, CA). PCR primers used were as follows: human glyceraldehyde 3-phosphate dehydrogenase (hGAPDH) sense, GTC AGC CGC ATC TTC TTT TG; hGAPDH antisense, GCA ACA ATA TCC ACT TTA CCA GAG; hLCN2 sense, AGA CAA AGA CCC GCA AAA G; hLCN2 antisense, TGG CAA CCT GGA ACA AAA G; hPLA2G2A sense, GGA AAG GAA GCC GCA CTC AGT TAT; hPLA2G2A antisense, CAC ATC CAC GTT TCT CCA GAC GTT; murine β-actin sense, CAT CCG TAA AGA CCT CTA TGC CAA C; murine β-actin antisense, ATG GAG CCA CCG ATC CAC A; murine PLA2G2A (mPLA2G2A) sense, GGC TGT GTC AGT GCG ATA AA; mPLA2G2A antisense, TTT ATC ACC GGG AAA CTT GG; mIL-22 sense, TTC CAG CAG CCA TAC ATC GTC; mIL-22 antisense, CTT CCA GGG TGA AGT TGA GCA.
Immunofluorescent staining.
HepG2 cells were incubated with or without 10 ng/ml of IL-22 in tissue culture dishes (Nunc, Roskilde, Denmark) at 1× 105 cells/ml for 2 days. Culture medium was then replaced with fresh medium containing 5 × 106 CFU/ml of L. monocytogenes. The cells were incubated for 1 h, then washed with PBS to remove uninfected bacteria, and fixed on the dishes with 3.7% formaldehyde in PBS for 15 min. The fixed cells were then treated with permeabilization solution consisting of 0.1% Triton X and 0.05% bovine serum albumin (BSA) in PBS for 15 min. The cells were stained for 1 h at room temperature with goat anti-human/mouse PLA2G2A antibody (Santa Cruz, Dallas, TX) at a 50× dilution and biotin-conjugated rabbit anti-L. monocytogenes antibody (Serotec, Oxford, United Kingdom) at a 100× dilution, followed by secondary staining with Alexa Fluor 568-conjugated donkey anti-goat IgG and Alexa Fluor 488-conjugated streptavidin (Molecular Probes, Eugene, OR). The bottoms of the dishes with stained cells were cut, mounted with ProLong Gold with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes), and analyzed with a C2 confocal laser-scanning microscope (Nikon, Tokyo, Japan).
The livers of C3H/HeJ mice were frozen in OCT (optimal cutting temperature) compound (Sakura Finetek, Tokyo, Japan), sliced using a microtome at 20 μm, stained with anti-human/mouse PLA2G2A antibody, mounted as described above, and analyzed with a C2 confocal-scanning microscope.
Statistics.
To compare the data for two groups, Student's t test was applied. To compare more than three groups, one-way analysis of variance with a Tukey-Kramer multiple-comparison test was used. All the statistical analyses were carried out using GraphPad InStat software (GraphPad Software, La Jolla, CA). A P value of <0.05 was considered to indicate a significant difference.
Microarray data accession number.
The entire data set from the microarray experiments can be found under the NCBI Gene Expression Omnibus (GEO) accession number GSE71248.
RESULTS
L. monocytogenes infection induced IL-22 expression in the livers of C57BL/6 mice at an early stage of the infection.
We have reported that a proinflammatory cytokine, IL-17A, plays an important role in protective innate immunity against L. monocytogenes infection in the livers of C57BL/6 mice (8, 9). Since another proinflammatory cytokine, IL-22, is frequently produced in parallel to IL-17A (14, 22, 24), we analyzed IL-22 expression in the liver of the mice at an early stage after L. monocytogenes infection. On day 5 of infection, IL-22 expression increased in the livers of the infected mice (Fig. 1A). To determine numbers of IL-22-producing cells in the liver, LMNC were analyzed by FCM on day 5 of the infection. Very few IL-22-expressing cells were detected in CD3− fractions (Fig. 1B), including NKp46+ and CD117/c-Kit+ cells (data not shown), which suggests that ILC3 cells were not the major IL-22-producing cells. In contrast, cells expressing IL-22 at high levels were detected mainly in the CD3+ fraction, and the majority of the CD3+ IL-22high cells expressed CD4 (Fig. 1B) but not TCRCδ (data not shown). The results suggest that the CD3+ CD4+ TCRαβ+ Th22-type cells were important in innate IL-22 production in L. monocytogenes-infected livers. From these observations, we hypothesized that IL-22 is a cytokine which enhances innate protective immunity along with IL-17A against L. monocytogenes infection in the liver.
FIG 1.

Expression of IL-22 in the livers of L. monocytogenes-infected C57BL/6 mice. (A) On days 0, 1, and 5 of infection, the IL-22 mRNA level in the livers was analyzed by RT-PCR. The data are representative of two analyses with three mice in a group. *, P < 0.005 compared to the value on day 0. (B) Lymphocytes in the liver of the L. monocytogenes-infected mice were collected, stimulated with PMA and ionomycin in the presence of brefeldin A, and stained for CD3 and CD4 on their surfaces followed by intracellular staining of IL-22. CD3+ IL-22+ cells are indicated by red dots. The data demonstrate a representative profile of a mouse from two independent experiments with 6 individual mice.
Induction of antimicrobial molecules by IL-22 and IL-17A on cells of the human hepatocellular carcinoma line HepG2.
IL-22 was reported to induce production of antimicrobial molecules by nonhematopoietic cells and synergized with IL-17A (14). It is therefore possible that the expression of IL-22 in the presence of IL-17A in the liver enhances innate protective immunity of nonhematopoietic cells against L. monocytogenes infection via induction of antimicrobial molecules. To address this possibility, gene expression profiles were compared by DNA microarray analysis between L. monocytogenes-infected HepG2 cells pretreated with IL-22 and IL-17A and those without cytokine pretreatment. Expression of 44 genes was reproducibly increased in the cytokine-treated HepG2 cells compared to the non-cytokine-treated HepG2 cells (Fig. 2). Among the products of these genes, serum amyloid A (SAA), haptoglobin (HP), and LPS-binding protein (LBP) are classified as acute-phase proteins, which confirms induction of the inflammatory response in HepG2 cells by IL-22 and IL-17A (25, 26). Genes encoding the antimicrobial molecules LCN2 and PLA2G2A were identified (Fig. 2). LCN2 is a siderophore-binding antimicrobial protein which suppresses bacterial growth by inhibiting iron uptake of siderophore-producing bacteria (30). PLA2G2A is a secretory phospholipase which can penetrate the negatively charged cell wall of Gram-positive bacteria and degrade cell membrane phospholipids to kill bacteria (31, 32).
FIG 2.

Genes induced by IL-22 and IL-17A in L. monocytogenes-infected HepG2 cells. HepG2 cells were cultured in the presence or absence of IL-22 and IL-17A before L. monocytogenes infection. Gene expression profiles of the two groups were compared by microarray analysis. Genes whose expression increased more than 2 times in the cytokine-treated group in two independent experiments are listed, and the mean of the increased level is shown. Genes encoding the antimicrobial molecules LCN2 and PLA2G2A are highlighted in bold.
We next analyzed expression of PLA2G2A and LCN2 on IL-22- and/or IL-17A-treated HepG2 cells by a real-time RT-PCR method. As shown in Fig. 3A, L. monocytogenes infection alone induced expression of neither PLA2G2A nor LCN2 on HepG2 cells, while pretreatment of HepG2 cells with IL-22 plus IL-17A before L. monocytogenes infection induced PLA2G2A and LCN2 expression. Even in the absence of L. monocytogenes infection, IL-22 treatment of HepG2 cells induced both PLA2G2A and LCN2 expression, while IL-17A treatment induced LCN2 but not PLA2G2A (Fig. 3B). Combination of IL-22 and IL-17A synergistically enhanced expression of LCN2, while no synergistic effect was observed in PLA2G2A expression. The results demonstrate that PLA2G2A is induced by IL-22 alone, while LCN2 expression was induced by both IL-22 and IL-17A.
FIG 3.

Expression of PLA2G2A by IL-22-stimulated HepG2 cells. Expression of PLA2G2A and LCN2 were analyzed by RT-PCR on untreated HepG2 cells (control), untreated L. monocytogenes-infected HepG2 cells, or IL-22+IL-17A-pretreated L. monocytogenes-infected HepG2 cells (A) and on HepG2 cells treated with IL-17A alone, IL-22 alone, or both (B). Bars and error bars represent means and standard deviations. *, P < 0.001 compared to untreated control. §, P < 0.001 compared to IL-17A alone or IL-22 alone. The data are representative of at least two independent experiments.
IL-22 induces a PLA2G2A-dependent innate protective response of HepG2 cells against L. monocytogenes infection.
It was reported that purified PLA2G2A killed L. monocytogenes in vitro (31). In contrast, LCN2 may not be effective in control of L. monocytogenes infection, because L. monocytogenes does not use a siderophore-dependent iron uptake mechanism (33). Consistent with this estimation, addition of rPLA2G2A to the culture of HepG2 cells suppressed L. monocytogenes infection, while rLCN2 showed no influence on bacterial burden of HepG2 cells (Fig. 4A). To verify involvement of PLA2G2A in the IL-22-induced protection of HepG2 cells, bactericidal activity of culture supernatants from IL-22-stimulated HepG2 cells was analyzed. The IL-22-stimulated culture supernatants killed L. monocytogenes, and the activity was abrogated by a specific PLA2G2A inhibitor, LY315920 (Fig. 4B). The data suggest that IL-22 induces PLA2G2A production of hepatocytes, which kills L. monocytogenes extracellularly.
FIG 4.

Anti-Listeria activity of PLA2G2A in IL-22-treated HepG2 cells. (A) HepG2 cells were incubated in the presence or absence of rPLA2G2A (left) or rLCN2 (right) before L. monocytogenes infection. The infected cells were then incubated for 1 h, extracellular bacteria were killed by addition of gentamicin, and the samples were incubated for 3 h more. Intracellular bacterial number was analyzed after the culture. (B) HepG2 cells were incubated with IL-22 for 2 days, and culture supernatants were collected. L. monocytogenes was added to control medium or IL-22-conditioned supernatants with or without the PLA2G2A-specific inhibitor LY315920 (LY), mixtures were incubated for 1 h, and bacterial numbers were determined. Bars and error bars represent means and standard deviations. *, P < 0.001 compared to the untreated control. The data are representative of at least two independent experiments with four to six samples.
Intracellular distribution of PLA2G2A in L. monocytogenes-infected HepG2 cells.
Active uptake of PLA2G2A via binding to heparan sulfate proteoglycans was reported previously (32, 34). If the uptake of PLA2G2A resulted in its colocalization with L. monocytogenes, it would damage intracellular L. monocytogenes. To address this possibility, confocal laser scanning microscopy was carried out on colocalization of PLA2G2A and L. monocytogenes in IL-22-treated HepG2 cells. To avoid detection of L. monocytogenes which bound PLA2G2A in the culture medium, cells were washed with fresh medium before infection. The analysis showed granular distribution of intracellular PLA2G2A (Fig. 5A) and colocalization of the intracellular PLA2G2A with L. monocytogenes (Fig. 5A and B). This colocalization may not be due to interaction of L. monocytogenes and PLA2G2A in the medium, because L. monocytogenes organisms which did not colocalize with PLA2G2A were also detected (data not shown). This suggests that not only extracellular but also intracellular PLA2G2A may be involved in the IL-22-induced protective response of nonhematopoietic cells.
FIG 5.
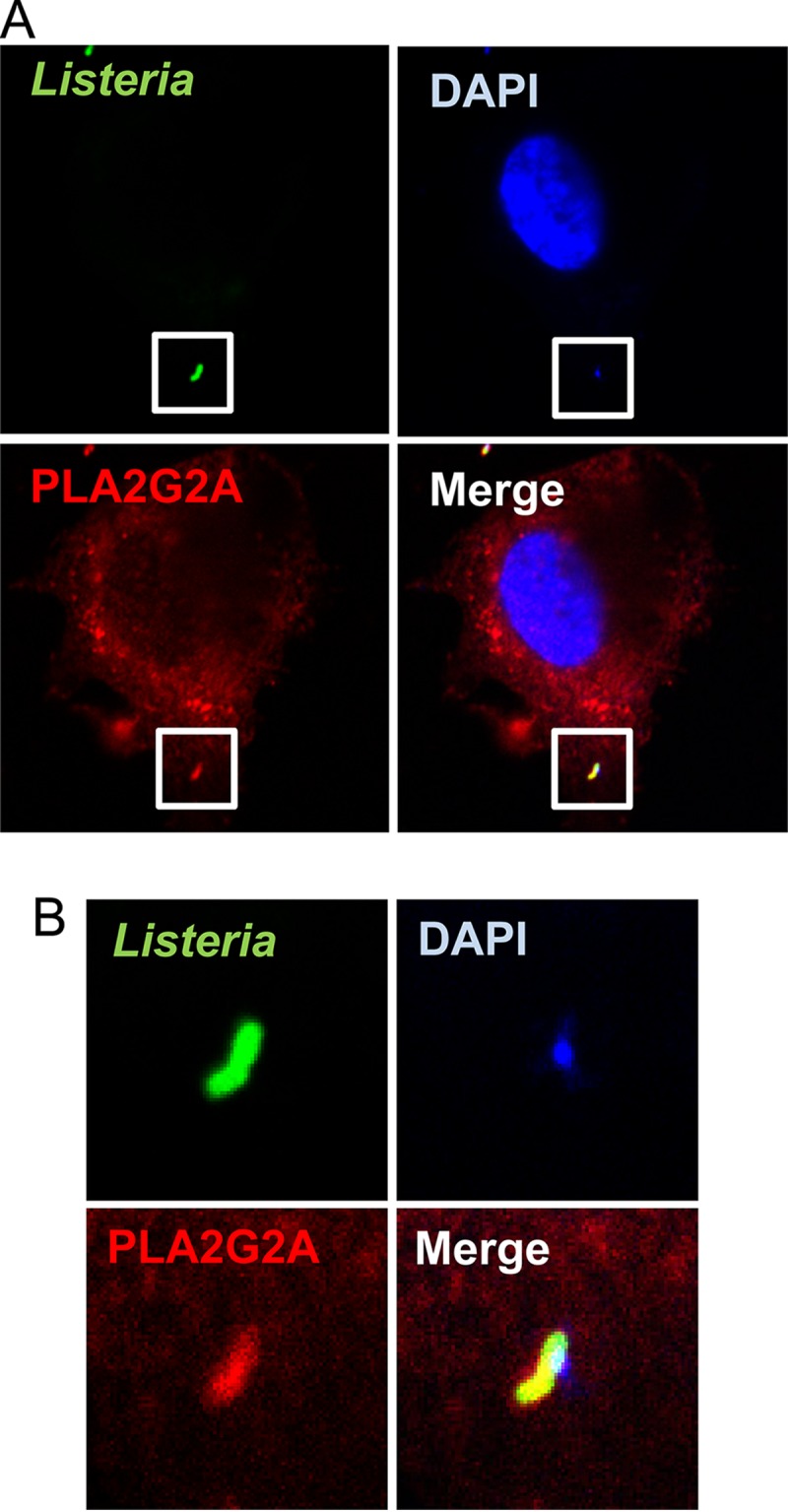
Intracellular colocalization of PLA2G2A and L. monocytogenes in HepG2 cells. IL-22-treated HepG2 cells were washed to remove PLA2G2A in supernatants, infected with L. monocytogenes, and incubated for 1 h. Extracellular bacteria were washed out, fixed, permeabilized, and stained to detect intracellular PLA2G2A and L. monocytogenes. The cells were mounted with medium containing DAPI to visualize cellular and bacterial DNA. The cells were observed by confocal microscopy with a 60× objective and 4× zoom (A). The insets (A) are magnified in panel B to show colocalization of PLA2G2A, L. monocytogenes, and DNA. The images show a cell with intracellular bacteria that is representative of three independent experiments.
Induction of PLA2G2A in the liver of L. monocytogenes-infected mice.
PLA2G2A gene of C57BL/6 mice was reported to be inactivated by frameshift mutation (35). To analyze the influence of IL-22 on PLA2G2A in mice, we therefore analyzed induction of PLA2G2A in the livers of C3H/HeJ mice after L. monocytogenes infection because a functional PLA2G2A gene was demonstrated in the mouse strain (36). As shown in Fig. 6A, L. monocytogenes infection induced expression of IL-22 on day 1 after the infection while its expression decreased on day 5, suggesting different kinetics of IL-22 induction between mouse strains. The kinetics of PLA2G2A expression was similar to that of IL-22 in that PLA2G2A expression increased on day 1 and decreased on day 5 (Fig. 6A); however, there was no statistical significance because of variability of expression level between mice. Interestingly, we found a positive correlation between IL-22 and PLA2G2A expression level (r = 0.8015, R2 = 0.6424, P = 0.0017) when the data for the all liver samples were plotted (Fig. 6B). Furthermore, when the livers of C3H/HeJ mice were stained with anti-PLA2G2A Ab before and 1 day after infection, the livers from infected mice showed more granular staining of PLA2G2A than those from uninfected mice (Fig. 6C). These results indicate that IL-22 expression is enhanced by L. monocytogenes infection in the C3H/HeJ mice and that the expression is correlated to PLA2G2A production.
FIG 6.

Expression of PLA2G2A in L. monocytogenes-infected C3H/HeJ mice. PLA2G2A-functional C3H/HeJ mice were infected with L. monocytogenes, and expression of IL-22 and PLA2G2A was analyzed. (A) Expression of IL-22 and PLA2G2A was analyzed by RT-PCR before and on days 1, 2, and 5 after L. monocytogenes infection. Each group consisted of 3 mice. *, P < 0.05 between the indicated groups. (B) Correlation of IL-22 and PLA2G2A expression of the liver. Individual RT-PCR data for livers from uninfected mice (day 0) and L. monocytogenes-infected mice on days 1, 2, and 5 are plotted. (C) The livers of uninfected mice (day 0) or L. monocytogenes-infected mice on day 1 were stained with anti-PLA2G2A Ab (red) and DAPI (blue) and analyzed by confocal laser scanning microscopy with a 60× objective and 3× zoom.
DISCUSSION
In the present report, we demonstrate that IL-22 stimulates nonhematopoietic human HepG2 cells to express a gene encoding PLA2G2A, an antimicrobial enzyme which shows bactericidal activity against Gram-positive bacteria. IL-22-activated HepG2 cells secreted anti-Listeria activity into culture medium, and the activity was abrogated by a PLA2G2A-specific inhibitor, which confirms the requirement for enzymatic activity of the PLA2G2A in protective innate immunity. Furthermore, colocalization of intracellular L. monocytogenes and PLA2G2A was demonstrated, which suggests the possibility of intracellular bactericidal activity of PLA2G2A. Since PLA2G2A was reported to bind to mammalian membrane heparin sulfate proteoglycan and actively internalized into cells (32, 34), it is possible that the PLA2G2A uptake after secretion is important in intracellular killing of L. monocytogenes. All the results suggest that IL-22 induces PLA2G2A production by nonhematopoietic cells, including hepatocytes, and participates in innate protective immunity against L. monocytogenes infection at both extracellular and intracellular locations.
IL-22 has been reported to enhance protective immunity by inducing antimicrobial molecules in bacterial infections. In a murine intestinal infection caused by Citrobacter rodentium, IL-22 was pivotal in protective immunity, and colon organ culture with IL-22 induced production of the antimicrobial peptides RegIIIγ, RegIIIβ, and S100A8/9 (27). IL-22 was also an important protective factor in the Klebsiella pneumoniae lung infection model and stimulated mouse tracheal epithelial cells to produce LCN2, which had an important role in innate protection against K. pneumoniae (28). Our data demonstrated that IL-22 stimulation of hepatocellular carcinoma HepG2 cells induced another antimicrobial molecule, PLA2G2A, and participated in innate immunity against Gram-positive bacteria.
IL-22-induced antimicrobial molecules have distinct target specificity to damage microbes. PLA2G2A selectively kills Gram-positive bacteria. It has been reported that PLA2G2A penetrates the cell walls of Gram-positive bacteria, including L. monocytogenes and Staphylococcus aureus, and digests bacterial cell membrane phospholipids to induce autolysis of the bacteria (31, 32). The cationic nature of PLA2G2A is important in the penetration of anionic bacterial cell walls. RegIIIγ also shows bactericidal activity against Gram-positive bacteria (37). In contrast, LCN2, which is induced by various inflammatory cytokines, including IL-17A and IL-22 (reference 28 and this report) suppress bacterial growth by competitively blocking siderophore-mediated iron uptake of bacteria (30). Therefore, IL-22 participates in induction of multiple antimicrobial molecules to protect the host against variable pathogens.
It has been reported that IL-22 is dispensable in protective immunity against L. monocytogenes in mice because IL-22-deficient mice or anti-IL-22 Ab-treated mice showed normal protective immunity, although IL-22 expression increased as a result of infection (38–40). In these studies, IL-22-deficient mice with the C57BL/6 background or Ab-treated C57BL/6 mice were used. Since PLA2G2A is not produced by C57BL/6 mice because of a frameshift mutation in the PLA2G2A gene (35), it is possible that IL-22-deficient C57BL/6 mice failed to show impaired innate immunity against L. monocytogenes because of the lack of PLA2G2A. In the PLA2G2A-functional C3H/HeJ mouse strain (36), we demonstrated that PLA2G2A expression was closely correlated with IL-22 expression. The results indicate a role for IL-22 in the induction of PLA2G2A, and they indicate that PLA2G2A would participate in protective immunity in mice with a functional PLA2G2A allele. Further experiments utilizing in vivo blocking of PLA2G2A and/or IL-22 would be necessary to clarify this issue.
ACKNOWLEDGMENTS
We thank Tomoko Shigeno and Naoko Teruya for excellent assistance with in vitro infection and gene expression analyses.
This work was supported by Cooperative Research Grant NEKKEN2014-2015 and by JSPS Grants-in-Aid for Scientific Research (KAKENHI), grant number 25293105 (to G.M.).
REFERENCES
- 1.Dramsi S, Biswas I, Maguin E, Braun L, Mastroeni P, Cossart P. 1995. Entry of Listeria monocytogenes into hepatocytes requires expression of InlB, a surface protein of the internalin multigene family. Mol Microbiol 16:251–261. doi: 10.1111/j.1365-2958.1995.tb02297.x. [DOI] [PubMed] [Google Scholar]
- 2.Ireton K. 2007. Entry of the bacterial pathogen Listeria monocytogenes into mammalian cells. Cell Microbiol 9:1365–1375. doi: 10.1111/j.1462-5822.2007.00933.x. [DOI] [PubMed] [Google Scholar]
- 3.Magee DM, Wing EJ. 1988. Cloned L3T4+ T lymphocytes protect mice against Listeria monocytogenes by secreting IFN-γ. J Immunol 141:3203–3207. [PubMed] [Google Scholar]
- 4.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Viček J, Zinkernagel RM, Aguet M. 1993. Immune response in mice that lack the interferon-γ receptor. Science 259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 5.Unanue ER. 1997. Inter-relationship among macrophages, natural killer cells and neutrohpils in early stage of Listeria resistance. Curr Opin Immunol 9:35–43. doi: 10.1016/S0952-7915(97)80156-2. [DOI] [PubMed] [Google Scholar]
- 6.Williams MA, Schmidt RL, Lenz LL. 2012. Early events regulating immunity and pathogenesis during Listeria monocytogenes infection. Trends Immunol 33:488–495. doi: 10.1016/j.it.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuzaki G, Yamada H, Kishihara K, Yoshikai Y, Nomoto K. 2002. Mechanism of murine Vγ1+ γδ T cell-mediated innate immune response against Listeria monocytogenes infection. Eur J Immunol 32:928–935. [DOI] [PubMed] [Google Scholar]
- 8.Hamada S, Umemura M, Shiono T, Hara H, Tanaka K, Mayuzumi H, Ohta T, Matsuzaki G. 2008. Importance of murine Vδ1 γδ T cells expressing interferon-γ and interleukin-17A in innate protection against Listeria monocytogenes. Immunology 125:170–177. doi: 10.1111/j.1365-2567.2008.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, Oshiro K, Okamoto Y, Watanabe H, Kawakami K, Roark C, Born WK, O'Brien R, Ikuta K, Ishikawa H, Nakae S, Iwakuta Y, Ohta T, Matsuzaki G. 2008. IL-17A produced by γδ T cells plays a critical role in innate immunity against Listeria monocytogenes infection in the liver. J Immunol 181:3456–3463. doi: 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheridan B, Romagnoli PA, Pham QM, Fu HH, Alonzo F III, Schubert WD, Freitag AN, Lefraoçois L. 2013. γδ T cells exhibit multifunctional and protective memory in intestinal tissues. Immunity 39:184–195. doi: 10.1016/j.immuni.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langrish CL, Chen Y, Blumenschein WB, Mattson J, Basham B, Sedgwick JD, MaClanahan T, Kastelein RA, Cua DJ. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bettelli E, Carrier Y, Gao W, Korn T, Storm TB, Oukka M, Weiner HL, Kuchroo VK. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 13.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. 2007. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 14.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y, Matsuzaki G. 2007. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guérin infection. J Immunol 178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 16.Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. 2007. Resident Vδ1+γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol 178:4466–4472. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- 17.Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, Eberl G. 2010. Lineage relationship analysis of RORγt+ innate lymphoid cells. Science 330:665–669. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- 18.Sutton CE, Mielke LA, Mills KH. 2012. IL-17-producing γδ T cells and innate lymphoid cells. Eur J Immunol 42:2221–2231. doi: 10.1002/eji.201242569. [DOI] [PubMed] [Google Scholar]
- 19.Ye P, Rodriguez FH, Kanaly S, Stockinger KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuzaki G, Umemura M. 2007. Interleukin-17 as an effector molecule of innate and acquired immunity against infections. Microbiol Immunol 51:1139–1147. doi: 10.1111/j.1348-0421.2007.tb04008.x. [DOI] [PubMed] [Google Scholar]
- 21.Lockhart E, Green AM, Flynn JL. 2006. IL-17 production is dominated by γδ T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol 177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 22.Dudakov JA, Hanash AM, van den Brink MRM. 2015. Interleukin-22: immunobiology and pathology. Annu Rev Immunol 33:747–785. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolk K, Kunz S, Witte E, Friedrich M, Asadulla K, Sabat R. 2004. IL-22 increases the innate immunity of tissues. Immunity 21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Rutz S, Eidenschenk C, Ouyang W. 2013. IL-22, not simply a Th17 cytokine. Immunol Rev 252:116–132. doi: 10.1111/imr.12027. [DOI] [PubMed] [Google Scholar]
- 25.Wolk K, Witte E, Hoffmann U, Doecke WD, Endesfelder S, Asadullah K, Sterry W, Volk HD, Wittig BM, Sabat R. 2007. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn's disease. J Immunol 178:5973–5981. doi: 10.4049/jimmunol.178.9.5973. [DOI] [PubMed] [Google Scholar]
- 26.Liang SC, Nickerson-Nutter C, Pittman DD, Carrier Y, Goodwin DG, Shields KM, Lambert AJ, Schelling SH, Medley QG, Ma HL, Collins M, Dunussi-Joannopoulos K, Fouser LA. 2010. IL-22 induces an acute-phase response. J Immunol 185:5531–5538. doi: 10.4049/jimmunol.0904091. [DOI] [PubMed] [Google Scholar]
- 27.Zheng Y, Valdez PA, Danilenko DM, Hu Y, SA SM, Gong Q, Abbas AR, Modrusan Z, Chilardi N, de Sauvage FJ, Ouyang W. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 28.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. 2008. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snyder DW, Bach NJ, Dillard RD, Draheim SE, Carlson DG, Fox N, Roehm NW, Armstrong CT, Chang CH, Hartley LW, Johnson LM, Roman CR, Smith RA, Song M, Fleisch JH. 1999. Pharmacology of LY315920/S-5920, [[3-(aminooxoacetyl)-2-sthyl-1-(phenylmethyl)-1H-indol-4-yl]oxy]acetate, a potent and selective secretory phospholipase A2 inhibitor: a new class of anti-inflammatory drugs, SPI. J Pharmacol Exp Ther 288:1117–1124. [PubMed] [Google Scholar]
- 30.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. 2004. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 31.Qu XD, Lehrer RI. 1998. Secretory phospholipase A2 is the principal bactericide for staphylococci and other gram-positive bacteria in human tear. Infect Immun 66:2791–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birts CN, Barton CW, Wilton DC. 2010. Catalytic and non-catalytic functions of human IIA phospholipase A2. Trends Biochem Sci 35:28–35. doi: 10.1016/j.tibs.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Jin B, Newton MC, Shao Y, Jiang X, Charbit A, Klebba PE. 2005. Iron acquisition systems for ferric hydroxamates, haemin and haemoglobin in Listeria monocytogenes. Mol Microbiol 58:1185–1198. [DOI] [PubMed] [Google Scholar]
- 34.Birts CN, Barton CH, Wilton DC. 2008. A catalytically independent physiological function for human acute phase protein group IIA phospholipase A2. J Biol Chem 283:5034–5045. doi: 10.1074/jbc.M708844200. [DOI] [PubMed] [Google Scholar]
- 35.Kennedy BP, Payette P, Mudgett J, Vadas P, Pruzanski W, Kwan M, Tang C, Rancourt DE, Cromlish WA. 1995. A natural distribution of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem 270:22378–22385. doi: 10.1074/jbc.270.38.22378. [DOI] [PubMed] [Google Scholar]
- 36.MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM. 1995. The secretory phospholipase A2 gene is a candidate for Mom1 locus, a major modifier of APCMin-induced intestinal neoplasia. Cell 81:957–966. doi: 10.1016/0092-8674(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 37.Mukherjee S, Zheng H, Derebe MG, Callenberg KM, Partch CL, Rollins D, Propheter DC, Rizo J, Grabe M, Jing QX, Hooper LV. 2014. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature 505:103–107. doi: 10.1038/nature12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. 2007. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27:647–659. doi: 10.1016/j.immuni.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graham AC, Carr KD, Sieve AN, Indramohan M, Break TJ, Berg RE. 2011. IL-22 production is regulated by IL-23 during Listeria monocytogenes infection but is not required for bacterial clearance or tissue protection. PLoS One 6:1–9. doi: 10.1371/journal.pone.0005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reynders A, Yessaad N, Manh TPV, Dalod M, Fenis A, Aubry C, Nikitas G, Escalière B, Renauld JC, Dussurget O, Cossart P, Lecuit M, Vivier E, Tomasello E. 2011. Identity, regulation and in vivo function of gut NKp46+RORγt+ and NKp46+RORγt− lymphoid cells. EMBO J 30:2934–2947. doi: 10.1038/emboj.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]