

Abstract
Histopathological changes of the gastric mucosa after Helicobacter pylori infection, such as atrophy, metaplasia, and dysplasia, are considered to be precursors of gastric cancer, yet the mechanisms of histological progression are unknown. The aim of this study was to analyze the histopathological features of the gastric mucosa in mice infected with H. pylori strain PMSS1 in relation to gastric stem cell marker expression. C57BL/6J mice infected with PMSS1 were examined for histopathological changes, levels of proinflammatory cytokines, and expression of stem cell markers. Histopathological gastritis scores, such as atrophy and metaplasia, and levels of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β), were increased after PMSS1 infection. Expression levels of the cell proliferation and stem cell markers CD44 and SOX9 were also significantly increased in PMSS1-infected mice. Importantly, almost all metaplastic cells induced by PMSS1 infection expressed SOX9. When IL-1 receptor (IL-1R) knockout mice were infected with PMSS1, metaplastic changes and expression levels of stem cell markers were significantly decreased compared with those in wild-type (WT) mice. In conclusion, H. pylori infection induced the expression of cytokines and stem cell markers and histopathological metaplasia in the mouse gastric mucosa. SOX9 expression, in particular, was strongly associated with metaplastic changes, and these changes were dependent on IL-1 signaling. The results suggested the importance of SOX9 in gastric carcinogenesis.
INTRODUCTION
Helicobacter pylori, a microaerophilic, spiral-shaped, Gram-negative bacterium, was first isolated in 1983 by Warren and Marshall (1). H. pylori colonizes the human gastric epithelium, causing atrophic gastritis and potentially triggering histological progression to carcinoma (2–4). Epidemiological studies have shown that cag pathogenicity island (PAI)-positive H. pylori strains are more likely to cause atrophic gastritis and gastric cancer than are cag PAI-negative strains (5–7).
The cag PAI, a cluster of ∼30 genes encoding a type IV secretion system (T4SS), is a major virulence factor of H. pylori. Studies of host cell signaling by H. pylori strains with the cag PAI revealed that important intracellular signaling cascades, including nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK), were especially activated by these types of strains (8, 9). Consequent upregulation and secretion of interleukin-8 (IL-8) from epithelial cells recruit activated neutrophils and monocytes into the lamina propria, where they secrete proinflammatory cytokines such as IL-1β and tumor necrosis factor alpha (TNF-α) (10, 11).
In animal models of H. pylori infection, the SS1 strain, which contains cagA genes, has been widely employed (12). However, it was recently revealed that the SS1 strain had a nonfunctional cag PAI and did not induce IL-8 or translocate CagA (13). In contrast, it was reported previously that the original human isolates, designated pre-mouse SS1 (PMSS1), has a functional cag PAI (14). In this report, an isogenic mutant of PMSS1 lacking an essential component of the T4SS (ΔcagE), did not induce the pathological changes typically observed in PMSS1-infected stomachs.
During chronic H. pylori infection and histological gastritis, two types of mucous cell metaplasia develop in the human stomach epithelium and represent putative preneoplastic lesions: goblet cell intestinal metaplasia (IM) and spasmolytic polypeptide-expressing metaplasia (SPEM) (15). Both IM and SPEM are present in the stomach after diagnosis of gastric cancer and are recognized as useful histological markers for gastric cancer risk (16, 17). SPEM has characteristic deep antral gland cells or Brunner's glands and expresses trefoil factor family 2 (TFF2) and MUC6, while IM demonstrates clear lineage characteristics of the intestines, with expression of TFF3 and MUC2 (18–20).
In normal corpus gastric units, progenitor cells located in the gland neck give rise to four types of epithelial cells, including pepsinogen-secreting zymogenic chief cells, acid-producing parietal cells, and two types of mucous cells; surface mucous cells with TFF1 and MUC5AC expression; and mucous neck cells with TFF2 and MUC6 expression (18, 19). Parietal cell atrophy causes increased proliferation of normal stem/progenitor cells in the isthmus (21). Clinically, the distributions of chronic parietal cell atrophy and SPEM have been associated with gastric cancer in 90% of resected stomachs (22, 23), suggesting that SPEM could represent a surrogate marker of gastric cancer risk. However, the molecular mechanisms leading to SPEM and the expansion of progenitors in H. pylori-induced gastritis are largely unknown.
Tissue stem cells, which are classically defined by vital self-renewal and multipotency properties, play pivotal roles in carcinogenesis (24). Several molecules, including Lgr5, SOX2, Troy, CD44, DCAMKL-1, and SOX9, have been identified as potential gastric stem cell and progenitor cell markers (24–29). Recently, Khurana et al. and Bertaux-Skeirik et al. reported that the expression of CD44 was increased in atrophic gastritis and metaplasia induced by H. pylori infection (21). However, the expression of the other stem cell markers in H. pylori-induced gastritis is largely unknown.
In this study, a mouse model of cag PAI-positive H. pylori infection was examined to understand the expression of stem cell markers in the development of SPEM.
MATERIALS AND METHODS
Animals.
Six- to ten-week-old C57BL/6J male mice were purchased from CLEA Japan Inc. (Tokyo, Japan), and IL-1 receptor knockout (IL-1R KO) mice on a C57BL/6J background were purchased from the Jackson Laboratory (Sacramento, CA, USA). Mice were maintained in a specific-pathogen-free environment in the Animal Care Facility of The University of Tokyo under conditions required by institutional guidelines. All animal experiments were carried out according to protocols approved by the Ethics Committee of The University of Tokyo.
Bacteria.
The H. pylori PMSS1 and SS1 strains were obtained from Anne Müller at The University of Zürich (14) and from A. H. Mitchell at The University of New South Wales (30), respectively. The H. pylori strains were grown on brucella broth (Becton Dickinson, Franklin Lakes, NJ, USA) containing 7.5% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) under microaerobic conditions at 37°C for 48 h. Colonies were harvested from plates of brucella broth containing 7.5% fetal bovine serum under microaerobic conditions for 24 h.
Infection of mice with H. pylori.
Mice were fasted for 24 h and inoculated intragastrically with a 0.1-ml bacterial suspension containing ∼1 × 108 CFU/ml in phosphate-buffered saline (PBS) by using a feeding needle. The mice were inoculated with the bacteria three times during a 7-day period. Control mice received sterile PBS. Animals were sacrificed at 12, 24, and 48 weeks postinfection (wpi). The stomachs were removed at each time point and divided longitudinally into two halves. One half of the stomach was fixed in 10% formalin for histopathological analysis, and the other half was used to assess colonization and to extract protein, DNA, and RNA.
Assessment of H. pylori colonization.
Bacterial culture and colony counting were performed as described previously (31). Stomach strips were homogenized in 0.3 ml PBS. Serial dilutions of each sample were spread onto brucella broth agar plates. After a 3- to 5-day incubation at 37°C under a microaerobic atmosphere, the numbers of H. pylori colonies were counted. H. pylori colonization was quantified by determining the number of CFU per stomach tissue sample.
RNA extraction and real-time PCR.
Total RNA was extracted from stomach strips by using the NucleoSpin RNA II kit (TaKaRa Bio Inc., Otsu, Japan) and converted to cDNA by using the ImProm-II reverse transcriptase system (Promega, Madison, WI, USA) according to the manufacturer's instructions. IL-1β, TNF-α, SOX9, CD44, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were amplified by using FastStart Universal SYBR green Master (Roche, Basel, Switzerland) in an ABI 7000 real-time PCR fluorescence reader (Life Technologies, Carlsbad, CA, USA), using specific primers. GAPDH levels were used for normalization.
Protein extraction and enzyme-linked immunosorbent assays.
Protein extraction from mouse gastric samples was performed by tissue disruption using a homogenizer with lysis buffer (50 mmol/liter Tris-HCl [pH 7.6], 1% Triton X-100, 5 mmol/liter EDTA, 1 mmol/liter Na3VO4) with cOmplete Mini protease inhibitor tablets (Roche). Samples were centrifuged at 15,000 rpm for 15 min, the supernatant was recovered, and total protein was determined by using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). IL-1β (mouse IL-1β ELISA Max standard set; BioLegend, San Diego, CA, USA) and TNF-α (mouse TNF-α Quantikine immunoassay; R&D Systems, Minneapolis, MN, USA) enzyme-linked immunosorbent assays (ELISAs) were performed by using gastric protein extracts according to the manufacturers' protocols.
Histological examination and grading of gastritis.
Sections stained with hematoxylin and eosin were used to determine the degree of gastritis, based on a previously described method (32). The severity of gastric pathology was graded on a scale of 0 to 6 for chronic inflammation, atrophy, metaplasia, and hyperplasia. All sections were graded blindly.
Immunohistochemistry.
Sections were deparaffinized and rehydrated in a graded series of ethanol solutions. The sections were subjected to antigen retrieval by boiling for 20 min in 1 mmol/liter sodium citrate (pH 6.0) in a microwave oven. After cooling, the slides were exposed to 3% hydrogen peroxide for 5 min before incubation with the appropriate blocking agent for 30 min. After washing with 0.1% Tween 20 in PBS (PBS-T), the slides were incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-TNF-α antibody (1:200; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-IL-1β antibody (1:200; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-Ki67 (1:200; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-PCNA (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rat monoclonal anti-CD44 (1:100; AbD Serotec, Oxford, United Kingdom), rabbit polyclonal anti-SOX9 (1:100; Millipore, Bedford, MA, USA), and mouse monoclonal anti-TFF2 (1:200; kindly provided by S. Nomura at The University of Tokyo). The primary antibody was probed with a biotinylated secondary antibody and detected by using an avidin/biotin detection system (Vectastain ABC kit; Vector Laboratories Ltd., Peterborough, United Kingdom) according to the manufacturer's instructions. Slides were developed with Histofine Simple Stain DAB solution (Nichirei Biosciences, Tokyo, Japan) and counterstained with Mayer's hematoxylin. Alcian blue at pH 2.5 (Sigma-Aldrich, St. Louis, MO, USA) was used according to the manufacturer's instructions.
Immunofluorescence staining.
Sections were deparaffinized, rehydrated, and blocked with 10% goat serum in PBS-T for 1 h. The slides were then incubated overnight at 4°C with the following antibodies: fluorescein isothiocyanate (FITC)-conjugated Griffonia simplicifolia lectin II (GSII) (1:3,000; Vector Laboratories, Burlingame, CA, USA), mouse monoclonal anti-H/K-ATPase (1:200; Medical & Biological Laboratories, Nagoya, Japan), mouse monoclonal anti-MIST1 and goat polyclonal anti-pepsin C (1:200; Santa Cruz Biotechnology), rabbit polyclonal anti-Helicobacter pylori (1:50; Dako, Santa Clara, CA, USA), rat monoclonal anti-mouse F4/80 (1:500; AbD Serotec), and purified rat anti-mouse Ly-6G and Ly-6C (1:200; Becton Dickinson). After washing with PBS-T, secondary antibodies (Alexa Fluor 555-conjugated goat anti-rabbit IgG antibody, Alexa Fluor 555-conjugated rabbit anti-goat IgG antibody, Alexa Fluor 488-conjugated goat anti-mouse IgG antibody, and Alexa Fluor 488-conjugated goat anti-mouse IgM antibody [1:1,000; Molecular Probes, Carlsbad, CA, USA]) were applied for 1 h. Nuclear counterstaining was performed by using Hoechst 33342 (1:1,000; Dojindo Molecular Technologies, Kumamoto, Japan). The slides were mounted by using fluorescence mounting medium (Dako) and examined under a fluorescence microscope.
Statistical analysis.
All results were expressed as mean values ± standard errors of the means (SEM) for each sample. Differences were analyzed by using the Mann-Whitney U test. Statistical significance was set at a P value of <0.05.
RESULTS
H. pylori with the cag PAI induces severe metaplastic changes in infected mice.
Male C57BL/6J mice were infected with the H. pylori PMSS1 or SS1 strain. Colonization levels and histopathological scores were assessed at 12 wpi. No significant difference in colonization between the two strains was observed (Fig. 1A). Both strains induced gastritis, but chronic infection with PMSS1 induced marked pathological changes, such as neutrophil infiltration to the lamina propria and submucosa, compared with infection of mice with SS1 (Fig. 1B). Chronic inflammation, atrophy, metaplasia, and hyperplasia scores were significantly higher in PMSS1-infected mice than in mice infected with SS1 (P < 0.001) (Fig. 1C). Next, proinflammatory cytokine expression in the stomach of H. pylori-infected mice was examined. The gastric tissues of mice infected with PMSS1 had significantly higher levels of TNF-α and IL-1β mRNAs than did those of SS1-infected mice (P < 0.05) (Fig. 1D).
FIG 1.
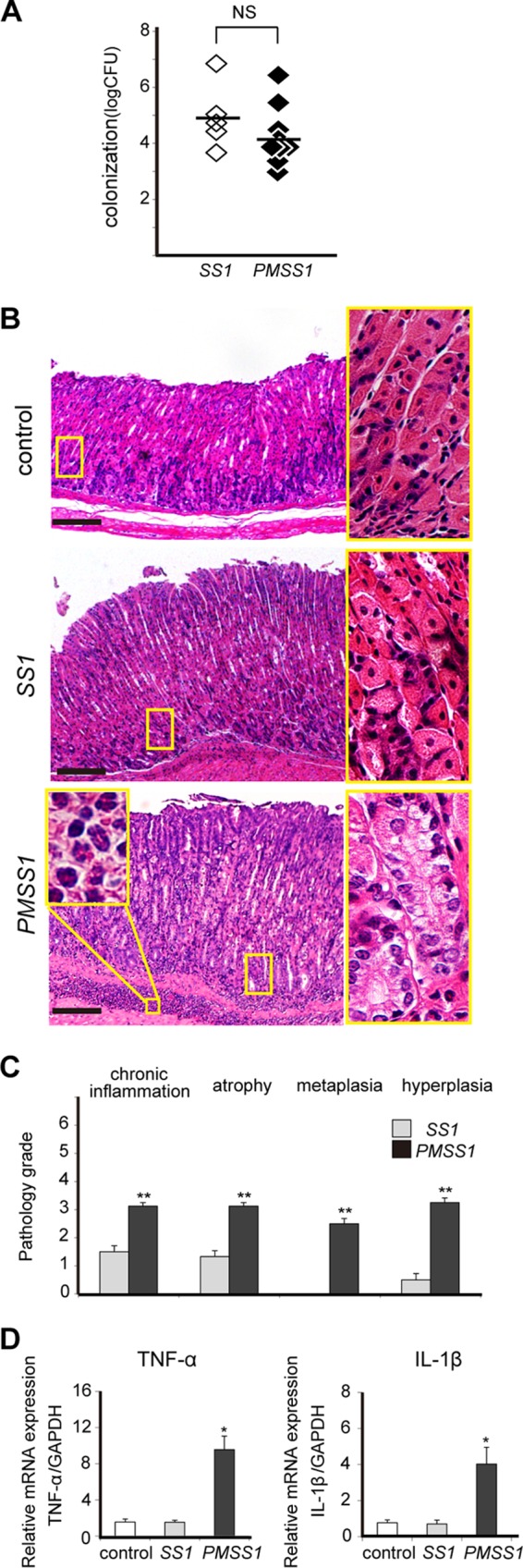
The PMSS1 strain induces severe gastritis in C57BL/6 mice. C57BL/6 mice were infected at 6 weeks of age with the SS1 or PMSS1 strain and sacrificed at 12 wpi. (A) CFU per stomach. NS, not significant. (B) Low-magnification (×40) (left) and high-magnification (×200) (right) micrographs of gastric tissues of one representative mouse. A magnified view (magnification, ×400) of neutrophil infiltration into the submucosa is shown in the inset. Bar = 500 μm. (C) Pathology scores for stomachs infected with SS1 or PMSS1 were assigned by the use of four parameters: chronic inflammation, atrophy, metaplasia, and hyperplasia. Data represent the means of the scores ± SEM (n = 6 to 8). **, P < 0.001 compared with SS1-infected stomachs. (D) Gastric TNF-α and IL-1β mRNA expression levels determined by real-time reverse transcription-PCR. Each mRNA level is standardized to the GAPDH level. *, P < 0.05 compared with uninfected controls.
As PMSS1 induced more severe metaplastic changes than did SS1 after 12 weeks, mice were evaluated over longer time periods of PMSS1 infection. Colonization levels of PMSS1 tended to decrease over time, although the difference was not significant (Fig. 2A). Histopathologically, mice with PMSS1 infection showed progression of metaplasia and hyperplasia (Fig. 2B and C), whereas chronic inflammation and atrophy were shown to regress after 24 wpi. The stomach tissues of PMSS1-infected mice were also analyzed for proinflammatory cytokines. TNF-α and IL-1β mRNA and protein expression levels increased with PMSS1 infection in a time-dependent manner (P < 0.05) (Fig. 2D and E). Immunohistochemical (IHC) staining for TNF-α and IL-1β also showed that these cytokines were highly expressed in cells in the lamina propria or submucosal layer of PMSS1-infected stomachs (Fig. 2F and G). We then examined the cellular sources of these cytokines. As shown by immunofluorescence, TNF-α was produced in most F4/80-positive macrophages and Gr-1-positive neutrophils. As for IL-1β, most macrophages but not neutrophils were costained with IL-1β (Fig. 2H and I). These results indicated that the cag PAI-positive PMSS1-infected mouse model exhibits many important aspects of human gastritis, such as metaplasia and cytokine expression.
FIG 2.

Time course of PMSS1-induced gastritis. C57BL/6 mice were infected at 6 weeks of age with PMSS1 and sacrificed at the indicated times. (A) CFU per stomach over the course of infection (n = 4 to 10). (B) Low-magnification (×40) (left) and high-magnification (×200) micrographs of gastric tissue of one representative mouse per group. Bar = 500 μm. (C) Pathology scores were assigned as described in the legend of Fig. 1C. Data are depicted as means ± SEM (n = 4 to 8). (D) Gastric TNF-α and IL-1β mRNA expression levels determined by real-time reverse transcription-PCR compared with those in uninfected controls. Each mRNA level is standardized to the GAPDH level. (E) Gastric TNF-α and IL-1β protein expression levels determined by an ELISA. *, P < 0.05 compared with noninfected controls. (F and G) IHC staining with TNF-α and IL-1β in PMSS1-infected murine gastric mucosa at 24 wpi compared with that in the controls. Shown are low-magnification (×40) (middle) and high-magnification (×200) (left and right) micrographs of the gastric tissues of one representative mouse per group. Bar = 500 μm. (H and I) Analysis of TNF-α and IL-1β in PMSS1-infected mice by costaining with markers of the cellular sources. (H) TNF-α and F4/80 as well as TNF-α and Gr-1. (I) IL-1β and F4/80 as well as IL-1β and Gr-1. Shown are high-magnification (×200) micrographs of the gastric tissues of one representative mouse.
PMSS1 upregulates the expression of cell proliferative and metaplastic markers in the gastric epithelium.
PCNA and Ki67, which are indicative of proliferating cells, are normally expressed in the progenitor zone in the isthmus of the gastric glands. Their expression largely extended bidirectionally from the isthmus at 24 weeks in response to PMSS1 infection (Fig. 3A and B). Numbers of alcian blue-positive mucous cells were increased from the neck to the base of the gastric gland in PMSS1-infected mice after 24 wpi but not in uninfected mice (Fig. 3C). We also examined TFF2 expression and GSII labeling as an indicator of metaplastic change. In control stomachs, TFF2 expression as well as GSII labeling were observed in neck lesions. In PMSS1-infected stomachs, TFF2 expression significantly increased to the base of the gastric gland (Fig. 3D). Double staining of H. pylori-infected stomachs showed that TFF2-expressing or GSII-labeled cells were mostly Ki67 or PCNA positive in their nuclei (Fig. 3E and F). We also examined the location of infected bacteria. H. pylori can be detected not only at the gastric surface but also in the middle of the gastric gland with GSII-labeled metaplastic cells (Fig. 3G). These data suggested that H. pylori infection induced cell proliferation and SPEM formation in murine gastric tissues, consistent with data from multiple previous studies (33–35).
FIG 3.

PMSS1 upregulates expression of cell proliferation and metaplasia markers in the murine stomach. (A and B) IHC staining with PCNA and Ki67 reveals cell proliferation in PMSS1-infected murine gastric mucosa at 24 wpi compared with the controls. (C and D) Histochemical (alcian blue) and IHC (TFF2) staining reveals metaplastic changes in PMSS1-infected murine gastric mucosa at 24 wpi compared with the controls. Shown are low-magnification (×40) (left) and high-magnification (×200) (right) micrographs of the gastric tissues of one representative mouse. (E and F) Analysis of cell proliferation markers in control and PMSS1-infected mice by costaining with metaplastic markers. (E) Ki67 and TFF2; (F) PCNA and GSII. Shown are low-magnification (×100) (left) and high-magnification (×200) (right) micrographs of the gastric tissues of one representative mouse. Bar = 500 μm. (G) IHC analysis of H. pylori and GSII in PMSS1-infected murine gastric mucosa at 24 wpi. Shown are high-magnification (×200) micrographs of the gastric tissues of one representative mouse.
PMSS1 upregulates the expression of stem cell markers in murine gastric tissues.
Next, the expression of putative gastric stem cell markers in the gastric epithelium of mice with or without H. pylori infection was examined. CD44, a type I transmembrane glycoprotein that serves as the receptor for an extracellular matrix component (hyaluronic acid), is a cell surface marker of gastric cancer stem cells (26). In our study, we found CD44-positive cells only at the squamocolumnar junction but not in the corpus epithelium of control mouse stomachs (Fig. 4A, top). After PMSS1 infection, CD44 was expressed at the base of the corpus glands (Fig. 4A, bottom), and there was a significant increase in CD44 expression compared with that in control mice, as reported in previous studies (P < 0.05) (36, 37) (Fig. 4B and C).
FIG 4.

PMSS1 upregulates the expression of stem cell markers in murine gastric tissues. (A and D) IHC staining of CD44 and SOX9 in PMSS1-infected mice compared with control mice. Shown are low-magnification (×40) (left) and high-magnification (×200) (right) micrographs of the gastric tissues of one representative mouse per group. Bar = 500 μm. (B and E) Numbers of CD44-positive or SOX9-positive cells in PMSS1-infected mice at the indicated times. HPF, high-power field. (C and F) Time course of CD44 and SOX9 mRNA expression in PMSS1-infected mice compared with control mice. CD44 and SOX9 mRNA levels are standardized to the GAPDH level. Data are depicted as the means ± SEM (n = 4 to 10). *, P < 0.05; **, P < 0.001 (compared with noninfected controls).
SOX9 has also been associated with stem cells and pluripotent, mitotically active progenitor cells in many organs. SOX9 expression in normal gastric mucosa was located predominantly at the isthmus of the corpus and at the base of the gland of the antrum (Fig. 4D, top). After PMSS1 infection, SOX9-positive cells extended beyond their normal location in the neck to the base of the gland of the gastric corpus (Fig. 4D, bottom). The murine stomach after PMSS1 infection displayed a significant increase in SOX9 expression compared with that in control mice (P < 0.05) (Fig. 4E and F). These results indicated that H. pylori infection induced the expansion of stem cell markers in mice.
SOX9 expression increases along with development of SPEM and fundic gland atrophy.
In IHC staining, the location of SOX9-positive cells closely resembled that of alcian blue- and TFF2-positive metaplasia (Fig. 5A). As seen after double staining (Fig. 5B), SOX9 expression was uniformly observed in GSII-positive cells. In addition, consistent with data shown in Fig. 3E and F, SOX9-positive cells mostly coexpressed the cell proliferation marker PCNA (Fig. 5C). Thus, SOX9-positive cells expanded after H. pylori infection seemed to be proliferative cells with a metaplastic marker.
FIG 5.

IHC analysis of gastric epithelial and stem cell markers. (A) Comparison of staining for SOX9 expression and metaplastic markers, such as alcian blue and TFF2. Shown are low-magnification (×100) micrographs of the gastric tissues of PMSS1-infected mice. (B) IHC analysis of SOX9 and GSII in PMSS1-infected stomachs. (C to I) Tissues from control or PMSS1-infected mice at 24 wpi were analyzed by IHC staining. Shown are low-magnification (×100) (left) and high-magnification (×200) (right) micrographs of the gastric tissue of one representative mouse per group. Bar = 500 μm. PCNA and SOX9 (C), SOX9 and TFF2 (D), H/K-ATPase and SOX9 (E), pepsin C and SOX9 (F), Mist1 and SOX9 (G), CD44 and GSII (H), and CD44 and SOX9 (I) were used for IHC analysis.
To further characterize proliferative SOX9-positive cells, the expression of several gastric gland-specific markers was determined. As shown in Fig. 5D, SOX9 expression almost corresponded to the cells positive for the SPEM marker TFF2 in H. pylori-infected mice. In noninfected mice, SOX9 and TFF2 expressions were limited to the isthmus of the gastric gland. After H. pylori infection, these cells appeared to expand to the whole gland. In contrast, SOX9 expression was almost exclusive from H/K-ATPase expression, which is characteristic of parietal cells (Fig. 5E). Pepsin C- or MIST1-positive cells were found at the base of the gastric gland in noninfected mice, which were negative for SOX9 expression (Fig. 5F and G, top). H. pylori infection significantly reduced the pepsin C- or MIST1-positive chief cell lineages in the stomach, and colocalization with SOX9 was not observed (Fig. 5F and G, bottom). These results suggested that SOX9 expression increases along with fundic gland atrophy and SPEM expansion.
When we examined CD44-expressing cells, CD44- and GSII-double-positive cells were found only in the bottom of gastric glands. However, many GSII-positive cells in the middle of the gland were CD44 negative in PMSS1-infected mouse stomachs (Fig. 5H). Similarly, CD44- and SOX9-double-positive cells were found only in the base of gastric glands, while SOX9-single-positive cells extended to the middle of glands (Fig. 5I). Thus, SOX9 but not CD44 expression corresponded well to SPEM cells.
Role of IL-1 signaling in PMSS1-infected mice.
As there were increased levels of proinflammatory cytokines such as IL-1β as well as histopathological gastritis in PMSS1-infected mouse stomachs, it was hypothesized that SOX9 expression may be driven by proinflammatory cytokines. IL-1R KO mice were inoculated with PMSS1, and the contribution of IL-1 signaling to SOX9 upregulation was evaluated at 12 wpi. No significant difference in colonization between wild-type (WT) and IL-1R KO mice was observed (Fig. 6A). We found mild gastritis in PMSS1-infected IL-1R KO mice compared to noninfected IL-1R KO mice (Fig. 6B, right). Compared to WT mice, IL-1R KO mice exhibited attenuated gastritis after PMSS1 infection (Fig. 6B, bottom). Histopathological scores of chronic gastritis, atrophy, metaplasia, and hyperplasia in IL-1R KO mice were statistically lower than those in WT mice (P < 0.05) (Fig. 6C). In addition, the gastric mucosa of IL-1R KO mice showed decreased levels of the proliferative marker PCNA and metaplastic markers such as alcian blue- or TFF2-positive cells (Fig. 6D to F). These data suggested that the loss of IL-1 signaling attenuated not only inflammation but also cell proliferation and metaplastic changes in PMSS1-infected gastric mucosa.
FIG 6.

Role of IL-1 signaling in PMSS1-induced gastritis and metaplasia. WT and IL-1R KO mice were infected with PMSS1 for 12 weeks. (A) Comparison of bacterial densities in each group. Shown are data for IL-1R KO mice (gray) and WT mice (black) (n = 8). (B) Histopathological changes in PMSS1-infected IL-1R KO mice or WT mice. Shown are low-magnification (×40) (left) and high-magnification (×200) (right) micrographs of the gastric tissues of one representative mouse. Bar = 500 μm. (C) Pathology scores were assigned as described in the legend of Fig. 1C. *, P < 0.05; **, P < 0.001 (compared with PMSS1-infected WT mice) (n = 6 to 8). (D to F) PCNA staining (D), alcian blue staining (E), and TFF2 staining (F) of PMSS1-infected WT and IL-1R KO mice. Shown are low-magnification (×40) (left) and high-magnification (×200) (right) micrographs of the gastric tissues of one representative mouse. Bar = 500 μm.
Finally, the expression of stem cell markers was examined with this model. CD44 and SOX9 expressions in the corpus were not upregulated in PMSS1-infected IL-1R KO mice compared with those in PMSS1-infected WT mice (Fig. 7A and B, top and middle). In particular, the SOX9 expression level was significantly decreased in PMSS1-infected IL-1R KO mice compared with PMSS1-infected WT mice (P < 0.05) (Fig. 7C and D). Indeed, in IL-1R KO mice, SOX9 expression did not expand from the isthmus, as was observed for uninfected IL-1R KO mice (Fig. 7B, bottom). The lack of SOX9 expression in PMSS1-infected IL-1R KO mice was similar to that in SS1-infected WT stomachs (Fig. 7E), in which upregulation of IL-1β was not observed (Fig. 1D). These results indicated that IL-1 signaling is important for SOX9 expression and metaplastic changes in H. pylori-infected gastric mucosa.
FIG 7.

Association of IL-1 signaling with stem cell markers in PMSS1-induced gastritis. (A and B) IHC staining of CD44 and SOX9 in PMSS1-infected WT and IL-1R KO mice and uninfected IL-1R KO mice (n = 6 to 8). Shown are low-magnification (×40) (left) and high-magnification (×200) (right) micrographs of the gastric tissues of one representative mouse per group. Bar = 500 μm. (C) Numbers of SOX9-positive cells in PMSS1-infected IL-1R KO mice compared with those in WT mice. *, P < 0.05 compared with infected WT mice. (D) SOX9 mRNA expression levels in gastric tissue were determined by real-time reverse transcription-PCR. SOX9 mRNA levels are standardized to the GAPDH level. *, P < 0.05. (E) SOX9 staining of tissues from WT mice infected with SS1 (n = 6). Shown are low-magnification (×40) (left) and high-magnification (×200) (right) micrographs of the gastric tissues of one representative mouse. Bar = 500 μm.
DISCUSSION
In this study, chronic H. pylori infection increased the expression of gastric stem cell markers in mice. In particular, SOX9 expression expanded from the isthmus in noninfected stomachs to the base of the gastric glands in H. pylori-infected stomachs in a time-dependent manner. The numbers of cells expressing SOX9 increased along with SPEM histological progression, which was characterized by TFF2 and alcian blue staining. Additionally, H. pylori-induced metaplastic changes were severely attenuated in IL-1 signaling-deficient mice, which also showed decreased SOX9 expression in the stomach. These results suggested that SOX9 plays a key role in the development of metaplastic changes in H. pylori-infected stomachs.
IM and SPEM were considered precursors of gastric cancer and useful surrogate markers of cancer risk in H. pylori-infected stomachs in many studies (17, 38, 39). Mice reportedly develop only SPEM in the fundic glands in H. pylori gastritis, while humans developed SPEM and IM in H. pylori infections and atrophic gastritis (40). The reason for these metaplastic differences between humans and mice has not been resolved. In our examinations, H. pylori infection did not induce CDX2 expression in mice according to IHC analysis and quantitative PCR (qPCR) (data not shown). As CDX2 is a critical transcription factor in the development of IM (18) and is negatively regulated by SOX9 (41), SOX9 upregulation resulting from H. pylori infection in the mouse fundic gland may account for the metaplastic differences observed.
Previous studies using models of mouse gastritis indicated that chronic Helicobacter felis infection induces SPEM in fundic glands, in addition to typical gastric atrophic pathologies such as parietal cell loss and decreased numbers of chief cells (34). Another study showed that hypergastrinemic transgenic mice with or without H. felis infection developed metaplasia and carcinoma in the corpus (42), although gastrin-deficient mice with H. felis infection exhibited only minimal changes in the corpus, indicating the importance of hypergastrinemia in the development of SPEM (43). Another study also reported a possible role for SPEM as a precursor of dysplasia in the mouse model (35). Importantly, eradication of Helicobacter early during infection in a mouse model inhibited the progression of metaplasia and dysplasia (44). Thus, a mechanistic analysis of the development of SPEM using mouse models would be useful for our understanding and prevention of gastric cancer.
In this study, H. pylori infection enhanced SOX9 expression in the mouse stomach. SOX9 is a transcription factor that controls the embryonic formation of various tissues and organs, including the digestive system (45–47). In the intestines, SOX9 expressed in the proliferative compartment under the control of Wnt signaling was suggested to represent a biomarker of crypt stem or progenitor cells (46). In the stomach, the exact location and definitive markers of gastric stem cells have not yet been established, but previous morphological and cell kinetic studies have suggested that multipotent stem cells reside in the isthmus region (21, 24, 29). SOX9 expression was also observed at the neck and isthmus of the fundic gland (48) (Fig. 4D), suggesting that SOX9 is a potential marker of gastric stem cells. SOX9 is highly expressed in human gastric metaplasia and carcinoma as well as in duodenal and colon cancers (48). Our findings indicating that H. pylori infection increased the number of SOX9-expressing stem cells and enhanced SPEM formation may represent important processes of gastric carcinogenesis. Interestingly, we found that gastric gland cells marked with SOX9 and TFF2 were mostly positive for cell proliferation markers (Fig. 3E and F and 5C). Thus, unlike normal differentiated cells, SPEM that develops after Helicobacter infection and is accompanied with inflammation possesses a proliferative ability, as was reported in previous studies (35, 49).
Another stem cell marker, CD44, was recently reported to play a functional role in H. pylori-induced epithelial cell proliferation and metaplasia (21, 36). Khurana et al. showed that CD44 expression was found in the isthmus in control mice, which expanded to the base after H. pylori infection. They found that atrophy induced CD44 expression, which regulates cell proliferation via STAT3 activation. Our study also showed that numbers of CD44-positive cells were significantly increased in PMSS1-infected mice compared with those in control mice, consistent with data from multiple previous studies (37, 50). We found that CD44-positive cells also expressed SOX9, especially at the base of the gastric gland (Fig. 5I) but not in the middle gland, where SOX9- and TFF2-double-positive cells reside (Fig. 5D). Thus, it may be possible that several different characteristics of H. pylori-infected stomach, such as metaplasia or proliferation, are independently associated with different stem cell markers, such as SOX9 and CD44. Our study demonstrated a close association between metaplasia and SOX9.
The role of IL-1 signaling in gastric carcinogenesis has been investigated intensively. Several reports suggested that inflammatory responses initiated by IL-1 play a pivotal role, and others showed that antiapoptotic pathways governed by IL-1 signaling were responsible for carcinogenic potential (51–53). Human single nucleotide polymorphism data also indicated the importance of IL-1β in the progression of gastric atrophy (5, 54). In this study, the cag PAI-positive H. pylori PMSS1 strain induced IL-1β expression, which was not observed after infection with the SS1 strain (Fig. 1E). In addition, the use of IL-1R KO C57BL/6J mice demonstrated that IL-1 signaling was necessary for inflammatory and metaplastic responses in the stomach but not for protection against infection. In our experiments, SOX9 expression was reduced in H. pylori-infected IL-1R KO mice compared with that in WT mice, in accordance with decreased SPEM in the pathology. These data suggest that IL-1 signaling enhances carcinogenesis via SOX9 expression in H. pylori-infected stomachs.
Regulation of the SOX9 transcript by IL-1 stimulation in the digestive system was not reported. We examined the effect of IL-1β on cultured gastric cancer cells or gastric organoids generated from normal mouse stomach, but neither SOX9 protein nor transcript levels were increased by stimulation (data not shown). For this reason, analysis of the mechanisms of SOX9 upregulation via IL-1 signaling in H. pylori-infected stomach remains an important area for future research. In this study, SOX9 expression correlated with the severity of inflammation and with the expansion of SPEM in the H. pylori-infected mouse model, in which severe gastric gland atrophy was evident, including the loss of differentiated parietal and chief cells. A similar association between SOX9 expression and metaplasia was reported for AGR2 KO mice, in which decreased numbers of differentiated gastric cells and increased numbers of proliferating cells were evident without inflammation or H. pylori infection (55). Thus, inflammation itself may have an indirect role in the enhancement of SOX9, which is essential for SPEM formation. It is also possible that IL-1 signaling enhanced SOX9 expression via atrophy, but not via inflammation, in H. pylori-infected mice.
In conclusion, this study indicated that cag PAI-positive H. pylori infection induced cytokine expression, stem cell marker expression, and histopathological metaplasia in the mouse gastric mucosa. SOX9 expression in particular was highly associated with metaplastic changes, and this change was dependent on IL-1 signaling. These results suggested the importance of SOX9 expression in H. pylori-induced gastric carcinogenesis.
ACKNOWLEDGMENTS
We thank Mitsuko Tsubouchi for technical assistance.
This work was partly supported by JSPS Kakenhi (grant numbers 22590679 and 26460934) and a research grant from the Fugaku Trust for Medical Research.
REFERENCES
- 1.Warren JR, Marshall BJ. 1983. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet i:1273–1275. [PubMed] [Google Scholar]
- 2.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. 2001. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 3.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. 1991. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 4.Forman D, Newell DG, Fullerton F, Yarnell JWG, Stacey AR, Wald N, Sitas F. 1991. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ 302:1302–1305. doi: 10.1136/bmj.302.6788.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, Capelinha AF, Quint W, Caldas C, van Doorn LJ, Carneiro F, Sobrinho-Simoes M. 2002. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst 94:1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 6.Parsonnet J, Friedman GD, Orentreich N, Vogelman H. 1997. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40:297–301. doi: 10.1136/gut.40.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaser MJ, Perezperez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. 1995. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55:2111–2115. [PubMed] [Google Scholar]
- 8.Mitsuno Y, Yoshida H, Maeda S, Ogura K, Hirata Y, Kawabe T, Shiratori Y, Omata M. 2001. Helicobacter pylori induced transactivation of SRE and AP-1 through the ERK signalling pathway in gastric cancer cells. Gut 49:18–22. doi: 10.1136/gut.49.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. 1999. Helicobacter pylori virulence and genetic geography. Science 284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 10.Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, Shiratori Y, Omata M. 2001. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem 276:44856–44864. doi: 10.1074/jbc.M105381200. [DOI] [PubMed] [Google Scholar]
- 11.Yamaoka Y, Kita M, Kodama T, Sawai N, Imanishi J. 1996. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology 110:1744–1752. doi: 10.1053/gast.1996.v110.pm8964399. [DOI] [PubMed] [Google Scholar]
- 12.Pritchard DM, Przemeck SM. 2004. Review article: how useful are the rodent animal models of gastric adenocarcinoma? Aliment Pharmacol Ther 19:841–859. doi: 10.1111/j.1365-2036.2004.01911.x. [DOI] [PubMed] [Google Scholar]
- 13.Barrozo RM, Cooke CL, Hansen LM, Lam AM, Gaddy JA, Johnson EM, Cariaga TA, Suarez G, Peek RM Jr, Cover TL, Solnick JV. 2013. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 9:e1003189. doi: 10.1371/journal.ppat.1003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Muller A. 2011. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140:199–209. doi: 10.1053/j.gastro.2010.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nozaki K, Ogawa M, Williams JA, Lafleur BJ, Ng V, Drapkin RI, Mills JC, Konieczny SF, Nomura S, Goldenring JR. 2008. A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology 134:511–522. doi: 10.1053/j.gastro.2007.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kusters JG, van Vliet AH, Kuipers EJ. 2006. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakitani K, Hirata Y, Watabe H, Yamada A, Sugimoto T, Yamaji Y, Yoshida H, Maeda S, Omata M, Koike K. 2011. Gastric cancer risk according to the distribution of intestinal metaplasia and neutrophil infiltration. J Gastroenterol Hepatol 26:1570–1575. doi: 10.1111/j.1440-1746.2011.06767.x. [DOI] [PubMed] [Google Scholar]
- 18.Barros R, Freund JN, David L, Almeida R. 2012. Gastric intestinal metaplasia revisited: function and regulation of CDX2. Trends Mol Med 18:555–563. doi: 10.1016/j.molmed.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Goldenring JR, Nam KT, Mills JC. 2011. The origin of pre-neoplastic metaplasia in the stomach: chief cells emerge from the Mist. Exp Cell Res 317:2759–2764. doi: 10.1016/j.yexcr.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reis CA, David L, Correa P, Carneiro F, de Bolos C, Garcia E, Mandel U, Clausen H, Sobrinho-Simoes M. 1999. Intestinal metaplasia of human stomach displays distinct patterns of mucin (MUC1, MUC2, MUC5AC, and MUC6) expression. Cancer Res 59:1003–1007. [PubMed] [Google Scholar]
- 21.Khurana SS, Riehl TE, Moore BD, Fassan M, Rugge M, Romero-Gallo J, Noto J, Peek RM Jr, Stenson WF, Mills JC. 2013. The hyaluronic acid receptor CD44 coordinates normal and metaplastic gastric epithelial progenitor cell proliferation. J Biol Chem 288:16085–16097. doi: 10.1074/jbc.M112.445551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halldorsdottir AM, Sigurdardottrir M, Jonasson JG, Oddsdottir M, Magnusson J, Lee JR, Goldenring JR. 2003. Spasmolytic polypeptide-expressing metaplasia (SPEM) associated with gastric cancer in Iceland. Dig Dis Sci 48:431–441. doi: 10.1023/A:1022564027468. [DOI] [PubMed] [Google Scholar]
- 23.Goldenring JR, Nomura S. 2006. Differentiation of the gastric mucosa. III. Animal models of oxyntic atrophy and metaplasia. Am J Physiol Gastrointest Liver Physiol 291:G999–G1004. doi: 10.1152/ajpgi.00187.2006. [DOI] [PubMed] [Google Scholar]
- 24.Mills JC, Shivdasani RA. 2011. Gastric epithelial stem cells. Gastroenterology 140:412–424. doi: 10.1053/j.gastro.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh SR. 2013. Gastric cancer stem cells: a novel therapeutic target. Cancer Lett 338:110–119. doi: 10.1016/j.canlet.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K, Nagano O, Matsuzaki J, Hibi T. 2012. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 12:764–777. doi: 10.1016/j.chom.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 27.Stange DE, Koo BK, Huch M, Sibbel G, Basak O, Lyubimova A, Kujala P, Bartfeld S, Koster J, Geahlen JH, Peters PJ, van Es JH, van de Wetering M, Mills JC, Clevers H. 2013. Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 155:357–368. doi: 10.1016/j.cell.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, Hochedlinger K. 2011. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9:317–329. doi: 10.1016/j.stem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, Abo A, Peters PJ, Wright N, Poulsom R, Clevers H. 2010. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 30.Thompson LJ, Danon SJ, Wilson JE, O'Rourke JL, Salama NR, Falkow S, Mitchell H, Lee A. 2004. Chronic Helicobacter pylori infection with Sydney strain 1 and a newly identified mouse-adapted strain (Sydney strain 2000) in C57BL/6 and BALB/c mice. Infect Immun 72:4668–4679. doi: 10.1128/IAI.72.8.4668-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. 1998. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology 115:642–648. doi: 10.1016/S0016-5085(98)70143-X. [DOI] [PubMed] [Google Scholar]
- 32.Sayi A, Kohler E, Hitzler I, Arnold I, Schwendener R, Rehrauer H, Muller A. 2009. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J Immunol 182:7085–7101. doi: 10.4049/jimmunol.0803293. [DOI] [PubMed] [Google Scholar]
- 33.Fox JG, Li X, Cahill RJ, Andrutis K, Rustgi AK, Odze R, Wang TC. 1996. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology 110:155–166. doi: 10.1053/gast.1996.v110.pm8536852. [DOI] [PubMed] [Google Scholar]
- 34.Nam KT, Lee HJ, Sousa JF, Weis VG, O'Neal RL, Finke PE, Romero-Gallo J, Shi G, Mills JC, Peek RM Jr, Konieczny SF, Goldenring JR. 2010. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 139:2028–2037. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nomura S, Baxter T, Yamaguchi H, Leys C, Vartapetian AB, Fox JG, Lee JR, Wang TC, Goldenring JR. 2004. Spasmolytic polypeptide expressing metaplasia to preneoplasia in H. felis-infected mice. Gastroenterology 127:582–594. doi: 10.1053/j.gastro.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 36.Bertaux-Skeirik N, Feng R, Schumacher MA, Li J, Mahe MM, Engevik AC, Javier JE, Peek RM Jr, Ottemann K, Orian-Rousseau V, Boivin GP, Helmrath MA, Zavros Y. 2015. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog 11:e1004663. doi: 10.1371/journal.ppat.1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bessede E, Staedel C, Acuna Amador LA, Nguyen PH, Chambonnier L, Hatakeyama M, Belleannee G, Megraud F, Varon C. 2014. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 33:4123–4131. doi: 10.1038/onc.2013.380. [DOI] [PubMed] [Google Scholar]
- 38.Weis VG, Goldenring JR. 2009. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer 12:189–197. doi: 10.1007/s10120-009-0527-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Correa P. 1988. A human model of gastric carcinogenesis. Cancer Res 48:3554–3560. [PubMed] [Google Scholar]
- 40.Ogawa M, Nomura S, Varro A, Wang TC, Goldenring JR. 2006. Altered metaplastic response of waved-2 EGF receptor mutant mice to acute oxyntic atrophy. Am J Physiol Gastrointest Liver Physiol 290:G793–G804. doi: 10.1152/ajpgi.00309.2005. [DOI] [PubMed] [Google Scholar]
- 41.Mutoh H, Sashikawa M, Sakamoto H, Tateno T. 2014. Cyclooxygenase 2 in gastric carcinoma is expressed in doublecortin- and CaM kinase-like-1-positive tuft cells. Gut Liver 8:508–518. doi: 10.5009/gnl13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, Raychowdhury R, Coffey RJ, Ito S, Varro A, Dockray GJ, Fox JG. 2000. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 118:36–47. doi: 10.1016/S0016-5085(00)70412-4. [DOI] [PubMed] [Google Scholar]
- 43.Takaishi S, Tu S, Dubeykovskaya ZA, Whary MT, Muthupalani S, Rickman BH, Rogers AB, Lertkowit N, Varro A, Fox JG, Wang TC. 2009. Gastrin is an essential cofactor for helicobacter-associated gastric corpus carcinogenesis in C57BL/6 mice. Am J Pathol 175:365–375. doi: 10.2353/ajpath.2009.081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai X, Carlson J, Stoicov C, Li H, Wang TC, Houghton J. 2005. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology 128:1937–1952. doi: 10.1053/j.gastro.2005.02.066. [DOI] [PubMed] [Google Scholar]
- 45.Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, Canamero M, Bujanda L, Schedl A, Cheah KS, Skotheim RI, Lothe RA, Lopez de Munain A, Briscoe J, Serrano M, Lovell-Badge R. 2012. Oncogenicity of the developmental transcription factor Sox9. Cancer Res 72:1301–1315. doi: 10.1158/0008-5472.CAN-11-3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, Hosokawa S, Elbahrawy A, Soeda T, Koizumi M, Masui T, Kawaguchi M, Takaori K, Doi R, Nishi E, Kakinoki R, Deng JM, Behringer RR, Nakamura T, Uemoto S. 2011. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 43:34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 47.Blache P, van de Wetering M, Duluc I, Domon C, Berta P, Freund JN, Clevers H, Jay P. 2004. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol 166:37–47. doi: 10.1083/jcb.200311021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sashikawa Kimura M, Mutoh H, Sugano K. 2011. SOX9 is expressed in normal stomach, intestinal metaplasia, and gastric carcinoma in humans. J Gastroenterol 46:1292–1299. doi: 10.1007/s00535-011-0443-5. [DOI] [PubMed] [Google Scholar]
- 49.Noto JM, Khizanishvili T, Chaturvedi R, Piazuelo MB, Romero-Gallo J, Delgado AG, Khurana SS, Sierra JC, Krishna US, Suarez G, Powell AE, Goldenring JR, Coffey RJ, Yang VW, Correa P, Mills JC, Wilson KT, Peek RM Jr. 2013. Helicobacter pylori promotes the expression of Kruppel-like factor 5, a mediator of carcinogenesis, in vitro and in vivo. PLoS One 8:e54344. doi: 10.1371/journal.pone.0054344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wada T, Ishimoto T, Seishima R, Tsuchihashi K, Yoshikawa M, Oshima H, Oshima M, Masuko T, Wright NA, Furuhashi S, Hirashima K, Baba H, Kitagawa Y, Saya H, Nagano O. 2013. Functional role of CD44v-xCT system in the development of spasmolytic polypeptide-expressing metaplasia. Cancer Sci 104:1323–1329. doi: 10.1111/cas.12236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shibata W, Takaishi S, Muthupalani S, Pritchard DM, Whary MT, Rogers AB, Fox JG, Betz KS, Kaestner KH, Karin M, Wang TC. 2010. Conditional deletion of IkappaB-kinase-beta accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology 138:1022–1034. doi: 10.1053/j.gastro.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakamoto K, Hikiba Y, Nakagawa H, Hayakawa Y, Yanai A, Akanuma M, Ogura K, Hirata Y, Kaestner KH, Omata M, Maeda S. 2010. Inhibitor of kappaB kinase beta regulates gastric carcinogenesis via interleukin-1alpha expression. Gastroenterology 139:226–238. doi: 10.1053/j.gastro.2010.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamanaka N, Morisaki T, Nakashima H, Tasaki A, Kubo M, Kuga H, Nakahara C, Nakamura K, Noshiro H, Yao T, Tsuneyoshi M, Tanaka M, Katano M. 2004. Interleukin 1beta enhances invasive ability of gastric carcinoma through nuclear factor-kappaB activation. Clin Cancer Res 10:1853–1859. doi: 10.1158/1078-0432.CCR-03-0300. [DOI] [PubMed] [Google Scholar]
- 54.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF Jr, Rabkin CS. 2000. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 55.Gupta A, Wodziak D, Tun M, Bouley DM, Lowe AW. 2013. Loss of anterior gradient 2 (Agr2) expression results in hyperplasia and defective lineage maturation in the murine stomach. J Biol Chem 288:4321–4333. doi: 10.1074/jbc.M112.433086. [DOI] [PMC free article] [PubMed] [Google Scholar]