

Abstract
The liver is frequently affected in patients with active brucellosis. In the present study, we identified a virulence factor involved in the modulation of hepatic stellate cell function and consequent fibrosis during Brucella abortus infection. This study assessed the role of BPE005 protein from B. abortus in the fibrotic phenotype induced on hepatic stellate cells during B. abortus infection in vitro and in vivo. We demonstrated that the fibrotic phenotype induced by B. abortus on hepatic stellate (LX-2) cells was dependent on BPE005, a protein associated with the type IV secretion system (T4SS) VirB from B. abortus. Our results indicated that B. abortus inhibits matrix metalloproteinase 9 (MMP-9) secretion through the activity of the BPE005-secreted protein and induces concomitant collagen deposition by LX-2 cells. BPE005 is a small protein containing a cyclic nucleotide monophosphate binding domain (cNMP) that modulates the LX-2 cell phenotype through a mechanism that is dependent on the cyclic AMP (cAMP)/protein kinase A (PKA) signaling pathway. Altogether, these results indicate that B. abortus tilts LX-2 cells to a profibrogenic phenotype employing a functional T4SS and the secreted BPE005 protein through a mechanism that involves the cAMP and PKA signaling pathway.
INTRODUCTION
Brucellosis is a worldwide zoonosis characterized by hepatomegaly, splenomegaly, and peripheral lymphadenopathy. It is a chronic and debilitating infection caused by Gram-negative facultative intracellular bacteria that infect domestic and wild animals and that can be transmitted to humans (1, 2). The frequency of liver involvement in active brucellosis ranges from 5% to 52% or more (1). However, although numerous studies have focused on brucellar liver histopathology (1), the pathogenic mechanisms of liver disease caused by Brucella have not been completely investigated at the molecular and cellular levels.
Liver fibrosis is a wound-healing response to chronic hepatic injury, which may be caused by alcohol abuse, hepatitis virus infection, or nonalcoholic steatohepatitis, and it is characterized by an excessive accumulation of extracellular matrix proteins in the liver (3, 4). An early event in the development of liver fibrosis is the activation of hepatic stellate cells (HSCs), the major cell type responsible for increased synthesis of extracellular matrix proteins (5). An elevated level of transforming growth factor β1 (TGF-β1) is also observed in the damaged liver, and it has a close correlation with fibrogenic changes in HSCs and liver tissue (6–8). In addition, decreased matrix metalloproteinase 9 (MMP-9) expression was observed in alcoholic liver fibrosis (9). This fibrogenic phenotype involves alterations in the balance of MMPs and their natural inhibitors—tissue inhibitors of metalloproteinases (TIMPs). In particular, MMP-2 and MMP-9 (gelatinase A and B, respectively) are important in regulating fibrogenesis and scar degradation. They can degrade a variety of collagens, including basement membrane (type IV collagen), denatured fibrillar type I collagen (gelatin), and type V collagen (10). Collagen type I is the prototype constituent of the fibril-forming matrix in fibrotic liver (11–13), and TGF-β1, derived from paracrine and autocrine sources, remains the classic fibrogenic cytokine (14, 15).
Previously, we demonstrated that infection of HSCs with Brucella abortus induces a series of events characterized by inhibition of MMP-9 secretion, induction of collagen deposition, and increased secretion of TIMP-1. These phenomena were dependent on TGF-β1 induction (16). However, the molecular mechanisms exerted by Brucella to activate this fibrogenic phenotype of HSCs have not yet been identified.
Type IV secretion systems (T4SS) are multiprotein complexes that translocate nucleoproteins and/or protein substrates across the bacterial cell envelope to the host cell, generally by a contact-dependent mechanism (17). T4SS protein substrates have been shown to modulate various cellular processes in the host cell, including apoptosis, vesicular traffic, and ubiquitination (18, 19). As the Brucella T4SS encoded by the virB gene has been shown to be involved in the modulation of the immune response during infection (20–22), we decided to investigate whether the effect of Brucella infection on the activation of HSCs is dependent on the presence of a functional T4SS and/or its secreted proteins. To this end, LX-2 cells were infected with B. abortus or its isogenic mutants to determine the levels of production of MMPs, collagen deposition, and TGF-β1 secretion. The results of the study are presented here.
MATERIALS AND METHODS
Bacterial culture.
Brucella abortus S2308 or the isogenic B. abortus virB10 polar, B. abortus bpe005, B. abortus bpe275, and B. abortus bpe123 strains and the corresponding complemented mutants (23, 24) were grown overnight in 10 ml of tryptic soy broth (Merck, Buenos Aires, Argentina) with constant agitation at 37°C. Bacteria were harvested and the inocula were prepared as described previously (25). All live Brucella manipulations were performed in biosafety level 3 (BSL-3) facilities located at the Instituto de Investigaciones Biomédicas en Retrovirus y SIDA (INBIRS).
Construction of the Brucella abortus BPE005, BPE123, and BPE275 mutants.
Unmarked deletion mutants with mutations of the selected candidate genes (bpe005 and bpe275) were generated as follows. DNA fragments (∼500 bp) containing the flanking regions of BAB1_2005 and BAB1_1275 were amplified from B. abortus 2308 genomic DNA with modified primers carrying BamHI and EcoRI restriction sites at the 5′ and 3′ ends, respectively. The PCR amplicons were ligated to the BamHI and EcoRI sites of mobilizable suicide vector pK18mobsacB, and the resulting plasmid was transformed in Escherichia coli S17 λ-pir and subsequently conjugated to B. abortus 2308. Single recombinants were selected with kanamycin and replica plated in Trypticase soy agar (TSA) supplemented with 10% sucrose to counterselect the double recombinants. Deletion of the selected gene was confirmed by colony PCR and sequence analysis. Using these procedures, the B. abortus Δbpe005 and B. abortus Δbpe275 mutant strains were generated. To obtain the bpe123 deletion mutant, a DNA fragment of 500 bp coding for BPE123 was amplified by PCR using primers 20123 BamHI and 20123 SpeI. The PCR product was ligated to pGem-T-Easy (Promega), and the resulting plasmid was linearized with HindIII and blunt ended with T4 DNA polymerase (New England BioLabs). Linearized pGem-T-bpe123 was ligated to a HincII DNA fragment coding for a nonpolar kanamycin resistance cassette to generate pGem-Tbpe123::Kan. This plasmid was electroporated into B. abortus 2308, where it is incapable of autonomous replication. Homologous recombination events were selected using kanamycin resistance and ampicillin sensitivity. Deletion of the selected gene was confirmed by colony PCR and sequence analysis. To generate the complemented strain of the B. abortus bpe005 mutant, the construct pLF-bpe005 (containing BAB1_2005 fused to a 3× FLAG tag) was transferred by biparental conjugation to the B. abortus deletion mutant, and the resulting complemented strain was selected with ampicillin.
Cell culture.
The LX-2 cell line, a spontaneously immortalized human hepatic stellate cell line, was kindly provided by Scott L. Friedman (Mount Sinai School of Medicine, New York, NY). LX-2 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies/Invitrogen, Carlsbad, CA, USA) and supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2% (vol/vol) fetal bovine serum (FBS; Gibco/Invitrogen, Carlsbad, CA, USA). All cultures were grown at 37°C and 5% CO2.
Cellular infection.
LX-2 cells were infected with B. abortus S2308 or its isogenic mutants at different multiplicities of infection (MOI; 100 to 1,000). After the bacterial suspension was dispensed, the plates were centrifuged for 10 min at 2,000 rpm and then incubated for 2 h at 37°C under a 5% CO2 atmosphere. Cells were extensively washed with DMEM to remove extracellular bacteria and incubated in medium supplemented with 100 μg/ml gentamicin and 50 μg/ml streptomycin to kill extracellular bacteria. LX-2 cells were harvested at different times to determine cytokine production, MMP secretion, and collagen deposition. To determine the role of cyclic AMP (cAMP) in the inhibition of MMP secretion and collagen deposition, some infection experiments were performed in the presence of 0.001, 0.01, 0.1, and 1 μM dibutyryl cAMP (B2cAMP) (Sigma-Aldrich de Argentina SA, Buenos Aires, Argentina).
Zymography.
Gelatinase activity was assayed by the method of Hibbs et al. with modifications as described previously (25–27).
Measurement of cytokine concentrations.
Secretion of TGF-β1, interleukin-6 (IL-6), IL-8, and monocyte chemotactic protein 1 (MCP-1) in the supernatants was quantified by enzyme-linked immunosorbent assay (ELISA) (BD Biosciences). TIMP-1 was quantified in culture supernatants by ELISA (R&D Systems).
Assessment of collagen deposition using Sirius red staining.
Collagen deposition was quantified using Sirius red (Sigma-Aldrich, Argentina), a strong anionic dye that binds strongly to collagen molecules (28). Sirius red staining was performed as previously described (16).
Transfection experiments.
LX-2 cells were transfected with BPE005 plasmid DNA or pRSV-PKI plasmid (kind gift of J. Silvio Gutkind), and pCDNA3-c-myc (2 mg/ml of plasmid DNA in each well transfected for 5 h) was used as a control for analysis of transfection efficiency. Transfection was carried out with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) as described by the manufacturer. MMP secretion and collagen deposition were determined at the end of cultures by zymography and Sirius red staining, respectively. Transfection efficiency was determined with mouse anti-c-myc antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted in phosphate-buffered saline (PBS)–0.1% Tween for 30 min at room temperature and then with fluorescein isothiocyanate (FITC)-conjugated anti-mouse antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). DAPI (4′,6-diamidino-2-phenylindole) was used for nuclear staining for 30 min at room temperature. After they were washed in PBS, cells were mounted and then were analyzed by fluorescence microscopy. These assays determined the presence of more than 70% transfected cells.
Evaluation of hepatic fibrosis in a mouse model of infection.
Six- to 8-week-old female BALB/c mice were infected through the intraperitoneal route with 5 × 105 CFU of B. abortus S2308, B. abortus bpe005 mutant, or vehicle (PBS). Mice were sacrificed at 4 and 12 weeks postinfection. To determine the levels of CFU, TGF-β1, and collagen production in mice livers, a liver lobe from each mouse was excised and placed immediately into 1 ml of cold PBS. Liver extractions were performed by using a tissue homogenizer. Aliquots of homogenates were serially diluted in sterile PBS and plated for CFU determinations, the remained homogenates were centrifuged at 2,000 × g for 20 min at 4°C, and the supernatants were stored at −70°C until TGF-β1 and collagen measurements were performed. In another group of mice, histological examination of liver was carried out at week 4 postinfection after routine fixation and paraffin embedding. Five-micrometer-thick sections were cut and stained with hematoxylin and eosin and with Masson's trichrome stain. Masson's trichrome staining was conducted according to the manufacturer's instructions (Sigma-Aldrich). Collagen-positive areas were visualized by light microscopy and quantified using Image Pro-Plus 6.0 software (Media Cubernetics, Inc.). The animal housing and all animal manipulations were conducted in the animal BSL-3 facility at Instituto de Investigaciones Biotecnológicas, Universidad Nacional de San Martín (IIB-UNSAM), under the guidance of and using protocols approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL-UNSAM).
Statistical analysis.
Statistical analysis was performed with one-way analysis of variance (ANOVA), followed by a post hoc Tukey test using GraphPad Prism 4.0 software. Data were represented as means ± standard errors of the means (SEM).
RESULTS
B. abortus induces a profibrogenic phenotype in LX-2 cells in a VirB-dependent manner.
The type IV secretion system (T4SS) VirB has been shown to be involved in the capacity of different Brucella species to establish an intracellular replication niche (24). In addition, this system has been involved in the induction of inflammatory response during B. abortus infection (21, 22). Taking into account that the major mediators of liver fibrosis are liver injury and inflammation (29), we decided to test whether VirB is involved in the ability of B. abortus to replicate within LX-2 cells. To this end, LX-2 cells were infected with B. abortus and its isogenic B. abortus virB10 mutant, which has been shown to be incapable of replicating within several cell types (24, 30, 31). B. abortus intracellular replication in LX-2 cells was dependent on the presence of a functional T4SS. In addition, and as occurs in other cell types (32), the B. abortus RB51 rough mutant was also unable to replicate in LX-2 cells (Fig. 1A).
FIG 1.

The B. abortus virB10 mutant (ΔvirB10) does not induce a profibrogenic phenotype in LX-2 cells. (A) LX-2 cells were infected with B. abortus (Ba), B. abortus RB51 (RB51), or the B. abortus bpe005 mutant (Δbpe005) or with the ΔvirB10 mutant at an MOI of 1,000, and CFU levels were determined 2, 4, 6, 24, 48, and 72 h postinfection. (B) MMP-9 production assessed by zymography at 24 h postinfection, (C) Densitometric analysis of results from three independent experiments performed as described for panel B. (D to H) ELISA determinations of levels of IL-6 (D), MCP-1 (E), IL-8 (F), TIMP-1 (G), and TGF-β1 (H) were performed in culture supernatants from LX-2 cells infected for 24 h. (I) Collagen deposition was assessed by quantification of Sirius red at 7 days postinfection. MMP-9 activity, cytokine secretion, and collagen deposition analyses were performed using LX-2 cells infected at an MOI of 1,000. PMA, phorbol myristate acetate. Data are given as the means ± SEM of results from experiments performed in duplicate. Data shown are from a representative experiment of three performed. **, P < 0.01; ***, P < 0.001 (versus noninfected [N.I.]).
The persistence of an infectious stimulus might drive liver fibrosis because its presence could induce marked alterations in a variety of immune and structural cells. In this context, hepatic stellate cells have a fundamental role in tissue homeostasis and injury repair through the production of extracellular matrix proteins (33). We have demonstrated that infection of LX-2 cells with Brucella inhibits the spontaneous secretion of MMP-9 (as determined by ELISA and by measurement of its activity by zymography) and induces collagen deposition (16). To determine whether intracellular replication is critical for inhibition of MMP-9 secretion and for induction of collagen deposition, experiments were performed using the B. abortus virB10 mutant and the RB51 rough mutant. Inhibition of MMP-9 secretion induced by B. abortus was dependent on the expression of a functional T4SS, since the levels of MMP-9 activity did not differ significantly between LX-2 cells infected with a B. abortus virB10 mutant and uninfected controls. However, this phenomenon was not dependent on intracellular replication, since RB51 was able to inhibit MMP-9 secretion by LX-2 cells (Fig. 1B and C), indicating that truncated bacterial intracellular replication is not a mandatory step in the inhibition of MMP-9 secretion in LX-2 cells. In addition, collagen deposition and TGF-β1 secretion were also dependent on the expression of a functional T4SS and are not related to bacterial replication, since the levels of collagen deposition and TGF-β1 secretion did not differ significantly between LX-2 cells infected with a B. abortus virB10 mutant and uninfected controls whereas B. abortus RB51-infected cells showed increased TGF-β1 secretion and collagen deposition (Fig. 1H and I). In contrast, IL-6, IL-8, MCP-1, and TIMP-1 secretion was induced by wild-type B. abortus, B. abortus RB51, and B. abortus virB10 mutant infection (Fig. 1D to G). However, levels of IL-6, IL-8, MCP-1, and TIMP-1 (Fig. 1C to F) were lower for the B. abortus virB10 mutant than for the other Brucella strains tested. The reduction of the levels of all these mediators was not due to bacterium-induced cell death. Measured at an MOI of 1,000 by Hoechst staining, by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay, or by annexin V-phosphatidylinositol staining, B. abortus infection did not induce apoptosis of LX-2 cells (data not shown).
These results indicated that expression of TGF-β1, the main profibrogenic cytokine, is modulated by the T4SS system of B. abortus. However, this system is not involved in the modulation of expression of IL-6 and of chemokines MCP-1 and IL-8 (attractants for neutrophils and monocytes, respectively), indicating that T4SS could not modulate the inflammatory infiltrate.
The participation of MMPs and their specific inhibitors, TIMPs, together with collagen deposition, is implicated in the formation and recovery processes of liver fibrosis (33). However, although the B. abortus wild type and the virB10 mutant modulate the expression of MMP-9 in different ways, both strains induce TIMP-1 secretion.
Together, these results indicate that upon infection of LX-2 cells, B. abortus inhibits MMP-9 secretion and induces concomitant collagen and TGF-β1 secretion in a VirB-dependent manner.
VirB-dependent effector protein BPE005 is responsible for the profibrogenic phenotype induced by B. abortus.
Recently, secreted proteins of B. abortus have been identified that require a functional VirB system to be translocated into the host cells (23). B. abortus mutants of three of them (BPE005, BPE275, and BPE123) were obtained, and their potential profibrogenic effect on LX-2 cells was studied. To this end, the levels of secretion of MMP-9 and TGF-β1 and collagen deposition were determined using LX-2 cells that were infected with B. abortus bpe005, bpe275, and bpe123 mutants. B. abortus bpe005 mutant bacteria infect and replicate in LX-2 cells (Fig. 1A), and the results mimic the LX-2 responses mediated by the B. abortus virB10 mutant. In contrast, the B. abortus bpe275 and bpe123 mutants inhibited MMP-9 activity and increased TGF-β1 secretion and collagen deposition in a manner similar to that seen with B. abortus wild-type infection (Fig. 2).
FIG 2.

BPE005 inhibits MMP-9 secretion and induces collagen deposition and TGF-β1 secretion in LX-2 cells. (A and B) LX-2 hepatic stellate cells were infected with B. abortus (Ba) or its isogenic bpe123 (Δbpe123), bpe275 (Δbpe275), and bpe005 (Δbpe005) mutants, and at 24 h postinfection, supernatants were harvested to analyze TGF-β1 secretion by ELISA (A) and MMP-9 production by zymography (B) in culture supernatants from LX-2 cells infected at an MOI of 1,000. (C) Densitometric analysis of results from three independent experiments performed as described for panel B. (D) Collagen deposition was assessed by quantification of Sirius red at 7 days postinfection. PMA, phorbol myristate acetate. Data are given as the means ± SEM of results from experiments performed in duplicate. Data shown are from a representative experiment of three performed. **, P < 0.01; ***, P < 0.001 (versus noninfected [N.I.]).
Together, these results indicate that upon infection of LX-2 cells, B. abortus inhibits MMP-9 secretion and induces concomitant TGF-β1 secretion and collagen deposition and that these phenomena are dependent on the presence of T4SS effector protein BPE005.
Expression of BPE005 in LX-2 cells mimics the profibrogenic effect caused by B. abortus infection.
In order to determine whether the observed profibrogenic effect caused by B. abortus infection of LX-2 cells is dependent on BPE005 protein expression, cells were transfected with a eukaryotic expression vector harboring the bpe005 gene and the levels of MMP-9 activity, collagen deposition, and TGF-β1 secretion were determined. Expression of BPE005 protein in LX-2 cells was able to inhibit MMP-9 activity and to induce collagen deposition and TGF-β1 secretion in a manner similar to that seen with B. abortus infection (Fig. 3). This result corroborated that this effector protein is the protein responsible for triggering the profibrogenic phenotype observed in LX-2 cells.
FIG 3.
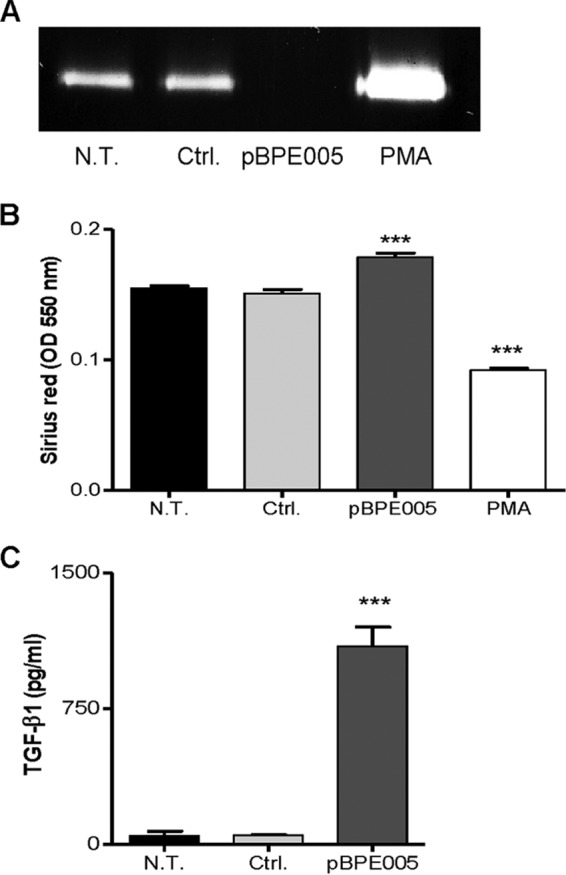
BPE005 protein mimics the B. abortus effect on MMP-9, collagen deposition, and TGF-β1 secretion by LX-2 hepatic stellate cells. LX-2 cells were transfected with BPE005 plasmid DNA (pBPE005) or with pcDNA3-c-myc as a control (Ctrl.). (A) MMP-9 production was assessed by zymography in culture supernatants harvested 48 h after transfection. (B) At 5 days posttransfection, collagen deposition was analyzed by Sirius red quantification. (C) ELISA determination of TGF-β1 levels in supernatants of LX-2 after 24 h of transfection. PMA, phorbol myristate acetate. Data are given as the means ± SEM of results from experiments performed in duplicate. Data shown are from a representative experiment of three performed. ***, P < 0.001 (versus nontransfected [N.T.]).
Butiril-cAMP restores the ability of BPE005-transfected LX2 cells to secrete MMP-9 and inhibits TGF-β1 secretion.
BPE005 is a protein containing a cyclic nucleotide monophosphate binding domain. Although its function is unknown, the predicted structure suggests that it might have an effect on cAMP-dependent signaling pathways in the host cell (23). Previous studies (34, 35) revealed that MMP-9, TGF-β, and collagen secretion could be regulated via activation of the cAMP/protein kinase A (cAMP/PKA) pathway. Our hypothesis is that B. abortus, through the activity of BPE005 protein, could alter this pathway, modulating cellular responses in LX-2 cells. Experiments were then conducted to evaluate whether cAMP could restore the effect triggered by BPE005 in LX-2 cells. To this end, LX-2 cells were transfected with the plasmid carrying bpe005 in the presence or absence of dibutyryl cyclic AMP (B2cAMP). This treatment completely reversed the inhibition of MMP-9 activity and TGF-β1 secretion induced by BPE005 expression in LX-2 cells (Fig. 4A and B). To corroborate this finding during B. abortus infection, LX-2 cells were infected with B. abortus in the presence of different concentrations of B2cAMP. Treatment with B2cAMP reversed the inhibitory effect induced by B. abortus infection on MMP-9 production by LX-2 cells in a dose-dependent manner (Fig. 4C). Our results indicated that cAMP could be involved in the mechanisms by which B. abortus could drive LX-2 responses to a profibrotic phenotype.
FIG 4.
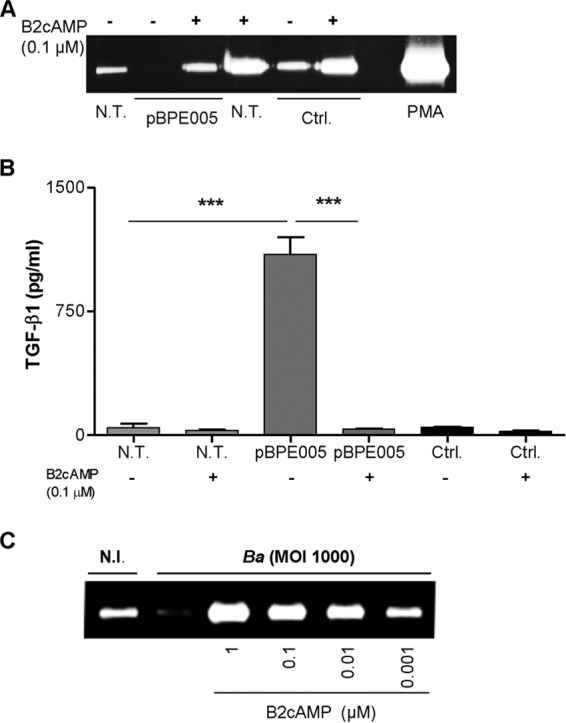
Butiril-cAMP restores the ability of BPE005-transfected LX2 cells to secrete MMP-9 and inhibits TGF-β1 secretion. (A and B) LX-2 cells were transfected with BPE005 plasmid DNA (pBPE005) or with pcDNA3-c-myc (Ctrl.) in the presence or absence of B2cAMP. At 24 h posttransfection, MMP-9 production was assessed by zymography (A) and TGF-β1 secretion by ELISA (B). (C) Cells were infected with B. abortus (Ba) at an MOI of 1,000 in the presence or absence of B2cAMP. At 24 h after infection, supernatants were harvested and MMP-9 levels were determined by zymography. Data are given as the means ± SEM of results from experiments performed in duplicate. Data shown are from a representative experiment of three performed. ***, P < 0.001 (versus nontransfected [N.T.]) or transfected and treated [pBPE005, B2cAMP]).
The PKA signaling pathway is involved in the profibrogenic response of LX-2 cells upon B. abortus infection.
The action of cAMP in eukaryotic cells was thought to occur mainly via activation of protein kinase A (PKA) and PKA-mediated changes in protein expression and function (36, 37). Therefore, experiments were conducted to determine whether PKA was involved in the profibrogenic response of the cells during B. abortus infection. To this end, LX-2 cells were infected with B. abortus or its isogenic bpe005 mutant in the presence or absence of KT5720 (PKA inhibitor) and MMP-9 and TGF-β1 production were evaluated. KT5720 treatment induced the inhibition of MMP-9 secretion in LX-2 cells infected with B. abortus bpe005 mutant. KT5720 treatment also increased the inhibition of MMP-9 production in B. abortus wild-type-infected LX-2 cells (Fig. 5A).
FIG 5.
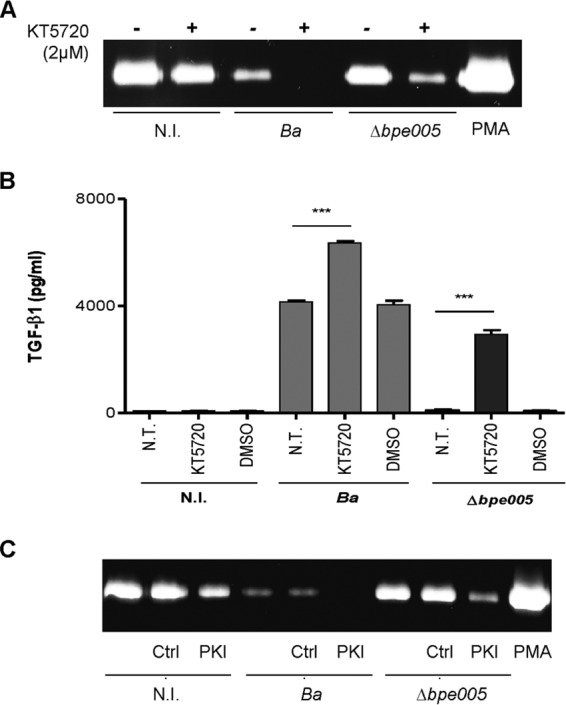
The PKA signaling pathway is involved in the modulation of LX-2 cells in response to B. abortus infection. (A and B) LX-2 cells were infected with B. abortus S2308 (Ba) or its isogenic bpe005 mutant (Δbpe005) in the presence or absence of KT5720 (PKA inhibitor) (2 μM), and 24 h after infection, MMP-9 levels were determined by zymography (A) and TGF-β1 production by ELISA (B). (C) Noninfected LX-2 cells and B. abortus- or Δbpe005 mutant-infected LX-2 cells were transfected with pRSV-PKI plasmid (PKI) or with pcDNA3-c-myc (Ctrl.), and MMP-9 production was assessed by zymography. PMA, phorbol myristate acetate. Data are given as the means ± SEM of results from experiments performed in duplicate. Data shown are from a representative experiment of three performed. ***, P < 0.001 (versus nontreated [N.T.]).
In addition, KT5720 treatment induced TGF-β1 secretion in LX-2 cells infected with the wild-type B. abortus strain or the mutant strain (Fig. 5A and B). To corroborate these results, LX-2 cells were transfected with pRSV-PKI plasmid, an expression vector containing heat-stable inhibitor PKI of cAMP-dependent PKA (38). Transfection with PKI of LX2 cells infected with the B. abortus bpe005 mutant induced inhibition of MMP-9 secretion; in addition, transfection with PKI of LX-2 cells infected with the B. abortus wild type increased the inhibition of MMP-9 secretion even more (Fig. 5C). Taken together, these results indicated that the cAMP/PKA signaling pathway is involved in the modulation of responses induced during B. abortus infection.
Infection of mice with the B. abortus bpe005 mutant induced a diminished fibrotic lesion in livers.
Finally, to determine the in vivo relevance of our hypothesis, BALB/c mice were infected with B. abortus and its isogenic bpe005 mutant.
As shown in Fig. 6A, CFU levels recovered from livers after 4 weeks of infection in mice infected with the B. abortus wild type and in those infected with the B. abortus bpe005 mutant were similar, indicating that the differences in the fibrotic phenotype were not dependent on bacterial replication.
FIG 6.

The B. abortus bpe005 mutant induced a mild fibrotic lesion in livers. (A) Liver colonization by B. abortus (Ba) or the B. abortus bpe005 mutant (Δbpe005) following intraperitoneal infection of mice. (B) Representative photomicrographs of liver sections from control mice, B. abortus (Ba)-infected mice, and B. abortus bpe005 isogenic mutant (Δbpe005)-infected mice (n = 5) stained with Masson's trichrome staining. (C to E) Collagen-positive areas were quantified using Image Pro-Plus 6.0 software (C), collagen production was determined by quantification of Sirius red (D), and TGF-β1 secretion was measured by ELISA in liver extracts (E) 4 weeks after infection. *, P < 0.1; **, P < 0.01 (versus control).
Masson's trichrome staining revealed that the level of fibrotic patches is lower in mice infected with the B. abortus bpe005 mutant than in those infected with the B. abortus wild type (Fig. 6B and C). Moreover, the levels of collagen, measured by Sirius red staining, were significantly increased in the livers of mice infected with wild-type B. abortus compared with the livers of mice infected with the B. abortus bpe005 mutant (Fig. 6D). In addition, these parameters correlated with the lower increase in TGF-β1 secretion in livers from B. abortus bpe005 mutant-infected mice than in those infected with the B. abortus wild type (Fig. 6E). These results indicated that BPE005 plays a key role in modulation of fibrosis during B. abortus infection.
DISCUSSION
The liver is frequently affected in patients with active brucellosis. Different histology patterns can be observed in liver involvement in brucellosis, the most common being (i) granuloma formation with inflammatory infiltrations and (ii) parenchymal necrosis (39). In any event, he persistence of an infectious stimulus might drive liver fibrosis because its presence could induce marked alterations in a variety of immune and structural cells, leading to a healing phenotype which is characterized by the deposition of extracellular matrix (40).
Hepatic stellate cells are recognized as the main source of liver fibrosis (5). Liver fibrosis is characterized by the deposition of extracellular matrix. An infectious stimulus, such as B. abortus infection, and the sustained injury might drive fibrosis (40).
We have previously demonstrated that B. abortus bacteria infect and replicate in hepatic stellate cells in vitro; in vivo, however, the cells would not represent a replicative niche but could be infected transiently or stimulated by Brucella antigens or by cytokines present in the inflammatory milieu generated by the infection. B. abortus infection inhibits basal levels of MMP-9 secretions and induces collagen deposition and TIMP-1 secretion by hepatic stellate cells. These phenomena were dependent on TGF-β1 induction (16).
As occurs in other bacterial infections (41, 42), a fibrotic phenotype may contribute to the persistence of infection, leading to poor penetration of antibiotics and immune mediators into the lesion. In addition, a fibrotic response could contribute to granuloma formation. The compact structure of granuloma successfully prevents the dissemination of the microorganisms; however, a negative facet of the granuloma is that the lesion may harbor within its burnt-out or calcified structure residual viable bacteria.
On the other hand, taking into account the role played by MMPs in infectious disease, orchestrating the recruitment of innate inflammatory cells and regulating their effector functions subsequent to cellular activation (43), the inhibition of MMP-9 secretion induced by B. abortus infection in hepatic stellate cells could contribute to partially inhibit the immune response, contributing to the chronicity of infection.
In the present study, we demonstrated that the ability of B. abortus to activate hepatic stellate cells is dependent on the presence of a functional T4SS and the secreted protein BPE005. Bacteria use T4SS for genetic exchange and to deliver effector molecules to eukaryotic target cells. The T4SS system plays a crucial role in the intracellular replication of B. abortus (44). Recently, it has been demonstrated that Brucella bacteria are able to modulate the host cell secretory pathway via multiple T4SS effector proteins (45). Yet BPE005 inhibits MMP-9 secretion but does not affect the secretion of other cytokines and collagen deposition. Therefore, our results indicate that BPE005 could have a specific role in the inhibition of MMP-9 secretion; however, we could not rule out other different molecular mechanisms that could be implicated such as the alteration of MMP-9 expression or the inhibition of the host secretory pathway. Also, it has been demonstrated that Brucella bacteria could use this secretion system to translocate effector proteins into host cytosol or even nucleic acids to further activate the inflammasome in a way that involved NLRP3 (22). In addition, it has been demonstrated that NLRP3 inflammasome activation in hepatocytes and nonparenchymal liver cells results in the induction of proinflammatory signaling and hepatocyte pyroptotic cell death (46). Then, the T4SS from B. abortus could be involved in the modulation of immune response in liver through activation of inflammasomes with concomitant fibrosis induction to repair inflammatory damage, depending on the secreted effector and the target cell. This response may therefore enable Brucella survival by subverting the immune response in vivo through an altered acute inflammatory response, resulting in impaired bacterial clearance and the establishment of chronic disease.
The modulation of collagen deposition and MMP secretion in HSCs involved the cAMP/PKA signaling pathway, as previously demonstrated in other cell types (47–49). It has been demonstrated that cAMP is a key messenger of many hormones and neuropeptides, some of which modulate the composition of extracellular matrix, and, recently, cAMP/PKA signaling was hypothesized to be involved in the proliferation and activation of rat HSCs (50–53). The action of cAMP in eukaryotic cells was thought to occur mainly via activation of PKA, and this molecule mediates changes in protein expression and function (36, 37). cAMP inhibits TGF-β-induced collagen synthesis in fibroblasts; in fact, several studies have shown that elevation of intracellular cAMP levels with phosphodiesterase inhibitors such as pentoxifylline inhibits fibroblast growth and collagen synthesis induced by serum and/or fibroblast-activating cytokines (54–56). It has been reported that, in this way, cAMP-elevating agents have the potential to act as antifibrotic therapeutics in a mechanism(s) that involves TGF-β (57). The ability of TGF-β to stimulate the proliferation of extracellular matrix production by cultured fibroblasts is well documented (58, 59).
We demonstrated that the capacity of B. abortus to induce fibrosis upon infection is dependent on the modulation of the cAMP/PKA signaling pathway via the BPE005 effector protein, since the observed effect on the fibrotic phenotype of HSCs was increased by adding an inhibitor of PKA, KT5720, or by transfecting cells with pRSV-PKI plasmid. In addition, the effect was also dependent on the presence of cAMP since the phenomenon was restored when we performed the infection with B. abortus in the presence of B2-cAMP. We demonstrated that the T4SS BPE005 effector plays a main role in the modulation of fibrosis as revealed by experiments performed with the B. abortus bpe005 mutant or with cells transfected with a plasmid encoding BPE005 protein.
BPE005 is a protein containing a cyclic nucleotide monophosphate binding domain. Although its function is unknown, the predicted structure suggests that it might have an effect on cAMP-dependent signaling pathways in the host cell blocking the binding between cAMP and PKA.
Importantly, the histological results corroborated the main role of BPE005 in the modulation of the fibrotic phenotype during B. abortus infection, thus confirming the relevance of the in vitro findings.
All together, these results indicate that upon infection of LX-2 cells, B. abortus triggers a profibrotic response characterized by inhibition of MMP-9 secretion, inducing concomitant collagen deposition and TGF-β1 secretion in a way that involved a functional T4SS and its BPE005 effector protein through a mechanism(s) that involved the cAMP and PKA signaling pathway.
ACKNOWLEDGMENTS
We thank Scott Friedman for LX-2 cells and also Horacio Salomón and the staff of the Instituto de Investigaciones Biomédicas en Retrovirus y Sida (INBIRS) for their assistance with biosafety level 3 laboratory uses.
Funding Statement
This work was supported by grants PICT2010-0023, PICT 2011-1501, PICT 2011-1200, PICT 2012-2252, and PICT 2011 01485 from Agencia Nacional of Promoción Científica y Tecnológica (ANPCYT, Argentina) and by grants UBACYT 20020090200012 and 20020120100128 from Universidad de Buenos Aires. P.C.A.B. and C.K.H. are recipients of a fellowship from CONICET. A.I.P.V. is a recipient of a fellowship from ANPCyT. G.H.G., D.J.C., S.V., and M.V.D. are members of the Research Career of CONICET. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Madkour MM. 2001. Gastrointestinal brucellosis, p 150–158. In Madkour MM. (ed), Madkour's brucellosis, 2nd ed Springer-Verlag, Berlin, Germany. [Google Scholar]
- 2.Pappas G, Akritidis N, Bosilkovski M, Tsianos E. 2005. Brucellosis. N Engl J Med 352:2325–2336. doi: 10.1056/NEJMra050570. [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL. 2003. Liver fibrosis—from bench to bedside. J Hepatol 38(Suppl 1):S38–S53. [DOI] [PubMed] [Google Scholar]
- 4.Bataller R, Brenner DA. 2005. Liver fibrosis. J Clin Invest 115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Meyer C, Muller A, Herweck F, Li Q, Mullenbach R, Mertens PR, Dooley S, Weng HL. 2011. IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-beta-independent Smad signaling. J Immunol 187:2814–2823. doi: 10.4049/jimmunol.1003260. [DOI] [PubMed] [Google Scholar]
- 6.Bissell DM, Wang SS, Jarnagin WR, Roll FJ. 1995. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 96:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos RM, Norton P, Degli Esposti S, Zern MA. 1998. TGF-beta isoforms in alcoholic liver disease. J Gastroenterol 33:383–389. doi: 10.1007/s005350050100. [DOI] [PubMed] [Google Scholar]
- 8.Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y, Inoue K. 2002. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 35:49–61. doi: 10.1053/jhep.2002.30083. [DOI] [PubMed] [Google Scholar]
- 9.Xu GF, Li PT, Wang XY, Jia X, Tian DL, Jiang LD, Yang JX. 2004. Dynamic changes in the expression of matrix metalloproteinases and their inhibitors, TIMPs, during hepatic fibrosis induced by alcohol in rats. World J Gastroenterol 10:3621–3627. doi: 10.3748/wjg.v10.i24.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rengel Y, Ospelt C, Gay S. 2007. Proteinases in the joint: clinical relevance of proteinases in joint destruction. Arthritis Res Ther 9:221. doi: 10.1186/ar2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arthur MJ. 2002. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology 122:1525–1528. doi: 10.1053/gast.2002.33367. [DOI] [PubMed] [Google Scholar]
- 12.Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, Sands E, Suliman I, Trim N, Knorr A, Arthur MJ, Benyon RC, Iredale JP. 2004. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 126:1795–1808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Parés A, Caballería J, Bruguera M, Torres M, Rodés J. 1986. Histological course of alcoholic hepatitis. Influence of abstinence, sex and extent of hepatic damage. J Hepatol 2:33–42. [DOI] [PubMed] [Google Scholar]
- 14.Inagaki M, Wang Z, Carr BI. 1994. Transforming growth factor beta 1 selectively increases gene expression of the serine/threonine kinase receptor 1 (SKR1) in human hepatoma cell lines. Cell Struct Funct 19:305–313. doi: 10.1247/csf.19.305. [DOI] [PubMed] [Google Scholar]
- 15.Breitkopf K, Lahme B, Tag CG, Gressner AM. 2001. Expression and matrix deposition of latent transforming growth factor beta binding proteins in normal and fibrotic rat liver and transdifferentiating hepatic stellate cells in culture. Hepatology 33:387–396. doi: 10.1053/jhep.2001.21996. [DOI] [PubMed] [Google Scholar]
- 16.Arriola Benitez PC, Scian R, Comerci DJ, Serantes DR, Vanzulli S, Fossati CA, Giambartolomei GH, Delpino MV. 2013. Brucella abortus induces collagen deposition and MMP-9 down-modulation in hepatic stellate cells via TGF-beta1 production. Am J Pathol 183:1918–1927. doi: 10.1016/j.ajpath.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Zechner EL, Lang S, Schildbach JF. 2012. Assembly and mechanisms of bacterial type IV secretion machines. Philos Trans R Soc Lond B Biol Sci 367:1073–1087. doi: 10.1098/rstb.2011.0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franco IS, Shuman HA, Charpentier X. 2009. The perplexing functions and surprising origins of Legionella pneumophila type IV secretion effectors. Cell Microbiol 11:1435–1443. doi: 10.1111/j.1462-5822.2009.01351.x. [DOI] [PubMed] [Google Scholar]
- 19.Ninio S, Roy CR. 2007. Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol 15:372–380. doi: 10.1016/j.tim.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Rolán HG, Tsolis RM. 2008. Inactivation of the type IV secretion system reduces the Th1 polarization of the immune response to Brucella abortus infection. Infect Immun 76:3207–3213. doi: 10.1128/IAI.00203-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roux CM, Rolan HG, Santos RL, Beremand PD, Thomas TL, Adams LG, Tsolis RM. 2007. Brucella requires a functional type IV secretion system to elicit innate immune responses in mice. Cell Microbiol 9:1851–1869. doi: 10.1111/j.1462-5822.2007.00922.x. [DOI] [PubMed] [Google Scholar]
- 22.Gomes MT, Campos PC, Oliveira FS, Corsetti PP, Bortoluci KR, Cunha LD, Zamboni DS, Oliveira SC. 2013. Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J Immunol 190:3629–3638. doi: 10.4049/jimmunol.1202817. [DOI] [PubMed] [Google Scholar]
- 23.Marchesini MI, Herrmann CK, Salcedo SP, Gorvel JP, Comerci DJ. 2011. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell Microbiol 13:1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comerci DJ, Martinez-Lorenzo MJ, Sieira R, Gorvel JP, Ugalde RA. 2001. Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cell Microbiol 3:159–168. doi: 10.1046/j.1462-5822.2001.00102.x. [DOI] [PubMed] [Google Scholar]
- 25.Scian R, Barrionuevo P, Giambartolomei GH, De Simone EA, Vanzulli SI, Fossati CA, Baldi PC, Delpino MV. 2011. Potential role of fibroblast-like synoviocytes in joint damage induced by Brucella abortus infection through production and induction of matrix metalloproteinases. Infect Immun 79:3619–3632. doi: 10.1128/IAI.05408-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hibbs MS, Hasty KA, Seyer JM, Kang AH, Mainardi CL. 1985. Biochemical and immunological characterization of the secreted forms of human neutrophil gelatinase. J Biol Chem 260:2493–2500. [PubMed] [Google Scholar]
- 27.Scian R, Barrionuevo P, Giambartolomei GH, Fossati CA, Baldi PC, Delpino MV. 2011. Granulocyte-macrophage colony-stimulating factor- and tumor necrosis factor alpha-mediated matrix metalloproteinase production by human osteoblasts and monocytes after infection with Brucella abortus. Infect Immun 79:192–202. doi: 10.1128/IAI.00934-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tullberg-Reinert H, Jundt G. 1999. In situ measurement of collagen synthesis by human bone cells with a Sirius red-based colorimetric microassay: effects of transforming growth factor beta2 and ascorbic acid 2-phosphate. Histochem Cell Biol 112:271–276. doi: 10.1007/s004180050447. [DOI] [PubMed] [Google Scholar]
- 29.Xu R, Zhang Z, Wang FS. 2012. Liver fibrosis: mechanisms of immune-mediated liver injury. Cell Mol Immunol 9:296–301. doi: 10.1038/cmi.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delpino MV, Fossati CA, Baldi PC. 2009. Proinflammatory response of human osteoblastic cell lines and osteoblast-monocyte interaction upon infection with Brucella spp. Infect Immun 77:984–995. doi: 10.1128/IAI.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delpino MV, Barrionuevo P, Scian R, Fossati CA, Baldi PC. 2010. Brucella-infected hepatocytes mediate potentially tissue-damaging immune responses. J Hepatol 53:145–154. doi: 10.1016/j.jhep.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 32.Rittig MG, Kaufmann A, Robins A, Shaw B, Sprenger H, Gemsa D, Foulongne V, Rouot B, Dornand J. 2003. Smooth and rough lipopolysaccharide phenotypes of Brucella induce different intracellular trafficking and cytokine/chemokine release in human monocytes. J Leukoc Biol 74:1045–1055. doi: 10.1189/jlb.0103015. [DOI] [PubMed] [Google Scholar]
- 33.Flamm SL. 2012. Granulomatous liver disease. Clin Liver Dis 16:387–396. doi: 10.1016/j.cld.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Leask A, Abraham DJ. 2004. TGF-beta signaling and the fibrotic response. FASEB J 18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 35.Vaday GG, Schor H, Rahat MA, Lahat N, Lider O. 2001. Transforming growth factor-beta suppresses tumor necrosis factor alpha-induced matrix metalloproteinase-9 expression in monocytes. J Leukoc Biol 69:613–621. [PubMed] [Google Scholar]
- 36.Zambon AC, Zhang L, Minovitsky S, Kanter JR, Prabhakar S, Salomonis N, Vranizan K, Dubchak I, Conklin BR, Insel PA. 2005. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc Natl Acad Sci U S A 102:8561–8566. doi: 10.1073/pnas.0503363102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen P. 2002. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov 1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 38.Westwick JK, Cox AD, Der CJ, Cobb MH, Hibi M, Karin M, Brenner DA. 1994. Oncogenic Ras activates c-Jun via a separate pathway from the activation of extracellular signal-regulated kinases. Proc Natl Acad Sci U S A 91:6030–6034. doi: 10.1073/pnas.91.13.6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akritidis N, Tzivras M, Delladetsima I, Stefanaki S, Moutsopoulos HM, Pappas G. 2007. The liver in brucellosis. Clin Gastroenterol Hepatol 5:1109–1112. doi: 10.1016/j.cgh.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 40.Meneghin A, Hogaboam CM. 2007. Infectious disease, the innate immune response, and fibrosis. J Clin Invest 117:530–538. doi: 10.1172/JCI30595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monack DM, Mueller A, Falkow S. 2004. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol 2:747–765. doi: 10.1038/nrmicro955. [DOI] [PubMed] [Google Scholar]
- 42.Adams DO. 1976. The granulomatous inflammatory response. A review. Am J Pathol 84:164–192. [PMC free article] [PubMed] [Google Scholar]
- 43.Elkington PT, O'Kane CM, Friedland JS. 2005. The paradox of matrix metalloproteinases in infectious disease. Clin Exp Immunol 142:12–20. doi: 10.1111/j.1365-2249.2005.02840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. 2003. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med 198:545–556. doi: 10.1084/jem.20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myeni S, Child R, Ng TW, Kupko JJ III, Wehrly TD, Porcella SF, Knodler LA, Celli J. 2013. Brucella modulates secretory trafficking via multiple type IV secretion effector proteins. PLoS Pathog 9:e1003556. doi: 10.1371/journal.ppat.1003556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME, Messer K, Canbay A, Hoffman HM, Feldstein AE. 2014. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 92:1069–1082. doi: 10.1007/s00109-014-1170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SY, Kim HJ, Lee WJ, Joo SH, Jeon SJ, Kim JW, Kim HS, Han SH, Lee J, Park SH, Cheong JH, Kim WK, Ko KH, Shin CY. 2008. Differential regulation of matrix metalloproteinase-9 and tissue plasminogen activator activity by the cyclic-AMP system in lipopolysaccharide-stimulated rat primary astrocytes. Neurochem Res 33:2324–2334. doi: 10.1007/s11064-008-9737-2. [DOI] [PubMed] [Google Scholar]
- 48.Lee CW, Lin CC, Lin WN, Liang KC, Luo SF, Wu CB, Wang SW, Yang CM. 2007. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 292:L799–L812. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Sun SQ, Hassid A, Ostrom RS. 2006. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol 70:1992–2003. doi: 10.1124/mol.106.028951. [DOI] [PubMed] [Google Scholar]
- 50.Schiller M, Dennler S, Anderegg U, Kokot A, Simon JC, Luger TA, Mauviel A, Bohm M. 2010. Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J Biol Chem 285:409–421. doi: 10.1074/jbc.M109.038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. 2005. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci U S A 102:437–442. doi: 10.1073/pnas.0408704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mallat A, Gallois C, Tao J, Habib A, Maclouf J, Mavier P, Preaux AM, Lotersztajn S. 1998. Platelet-derived growth factor-BB and thrombin generate positive and negative signals for human hepatic stellate cell proliferation. Role of a prostaglandin/cyclic AMP pathway and cross-talk with endothelin receptors. J Biol Chem 273:27300–27305. [DOI] [PubMed] [Google Scholar]
- 53.Ikeda N, Murata S, Maruyama T, Tamura T, Nozaki R, Kawasaki T, Fukunaga K, Oda T, Sasaki R, Homma M, Ohkohchi N. 2012. Platelet-derived adenosine 5′-triphosphate suppresses activation of human hepatic stellate cell: in vitro study. Hepatol Res 42:91–102. doi: 10.1111/j.1872-034X.2011.00893.x. [DOI] [PubMed] [Google Scholar]
- 54.Berman B, Duncan MR. 1989. Pentoxifylline inhibits normal human dermal fibroblast in vitro proliferation, collagen, glycosaminoglycan, and fibronectin production, and increases collagenase activity. J Investig Dermatol 92:605–610. doi: 10.1111/1523-1747.ep12712140. [DOI] [PubMed] [Google Scholar]
- 55.Berman B, Wietzerbin J, Sanceau J, Merlin G, Duncan MR. 1992. Pentoxifylline inhibits certain constitutive and tumor necrosis factor-alpha-induced activities of human normal dermal fibroblasts. J Investig Dermatol 98:706–712. doi: 10.1111/1523-1747.ep12499916. [DOI] [PubMed] [Google Scholar]
- 56.Préaux AM, Mallat A, Rosenbaum J, Zafrani ES, Mavier P. 1997. Pentoxifylline inhibits growth and collagen synthesis of cultured human hepatic myofibroblast-like cells. Hepatology 26:315–322. [DOI] [PubMed] [Google Scholar]
- 57.Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, Grotendorst GR. 1999. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J 13:1774–1786. [PubMed] [Google Scholar]
- 58.Ignotz RA, Massague J. 1986. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 261:4337–4345. [PubMed] [Google Scholar]
- 59.Raghow R, Postlethwaite AE, Keski-Oja J, Moses HL, Kang AH. 1987. Transforming growth factor-beta increases steady-state levels of type I procollagen and fibronectin messenger RNAs posttranscriptionally in cultured human dermal fibroblasts. J Clin Invest 79:1285–1288. doi: 10.1172/JCI112950. [DOI] [PMC free article] [PubMed] [Google Scholar]