

Abstract
Streptococcus pneumoniae is the world's foremost human pathogen. Acquisition of the first row transition metal ion zinc is essential for pneumococcal colonization and disease. Zinc is acquired via the ATP-binding cassette transporter AdcCB and two zinc-binding proteins, AdcA and AdcAII. We have previously shown that AdcAII is reliant upon the polyhistidine triad (Pht) proteins to aid in zinc recruitment. Pht proteins generally contain five histidine (His) triad motifs that are believed to facilitate zinc binding and therefore play a significant role in pneumococcal metal ion homeostasis. However, the importance and potential redundancy of these motifs have not been addressed. We examined the effects of mutating each of the five His triad motifs of PhtD. The combination of in vitro growth assays, active zinc uptake, and PhtD expression studies show that the His triad closest to the protein's amino terminus is the most important for zinc acquisition. Intriguingly, in vivo competitive infection studies investigating the amino- and carboxyl-terminal His triad mutants indicate that the motifs have similar importance in colonization. Collectively, our new insights into the contributions of the individual His triad motifs of PhtD, and by extension the other Pht proteins, highlight the crucial role of the first His triad site in zinc acquisition. This study also suggests that the Pht proteins likely play a role beyond zinc acquisition in pneumococcal virulence.
INTRODUCTION
Streptococcus pneumoniae is responsible for a broad range of major human diseases, including pneumonia, sepsis, meningitis, and otitis media, which together contribute to significant global morbidity and mortality (1). The ability of the bacterium to import zinc (Zn2+) ions from the extracellular environment is critical for its pathogenicity (2–5). Pneumococcal Zn2+ acquisition occurs via the cluster A-I substrate binding proteins (SBPs), AdcA and AdcAII, which deliver the metal ion to AdcCB, the ATP-binding cassette transporter, for cellular import (2–4).
The polyhistidine triad (Pht) family of proteins have also been shown to contribute to pneumococcal Zn2+ acquisition (4–6). S. pneumoniae encodes four related Pht proteins, PhtA, PhtB, PhtD, and PhtE, which are thought to be predominantly anchored in the pneumococcal cell wall (7, 8). The exposure of the Pht proteins on the cell surface has led to significant investigation into their potential use in next-generation pneumococcal vaccines, with clinical trials up to phase II having been completed (9–13). Despite this, their contribution to Zn2+ acquisition has only recently been revealed. A role for the Pht proteins in scavenging transition metal ions was initially reported by Rioux et al. (14), and it was subsequently suggested that PhtD and AdcAII could interact to transfer Zn2+ from the former to the latter (8, 15). Recently, we showed that AdcAII is largely dependent on the Pht proteins to acquire Zn2+ under Zn2+-limiting conditions. In contrast, AdcA is capable of contributing to Zn2+ acquisition independent of the Pht proteins, likely via its histidine-rich loop and C-terminal ZinT domain, features that are absent from AdcAII (4). Consistent with the high level of similarity between PhtA, PhtB, PhtD, and PhtE, each of the four Pht proteins has been shown to contribute to AdcAII-mediated Zn2+ acquisition (5). Furthermore, deletion of the pht genes in an ΔadcAII mutant reduced pneumococcal invasion relative to the parental ΔadcAII strain, indicating that the Pht proteins hold roles in addition to AdcAII-mediated Zn2+ acquisition (5). Currently, it is not known whether these other functions are related to zinc homeostasis.
The defining feature of the Pht proteins is the presence of multiple copies of the histidine triad motif (HxxHxH). PhtD, the most highly conserved Pht protein across pneumococcal strains, contains five His triad (HT) motifs, and each motif is thought to bind one Zn2+ ion (8). Loisel et al. produced a truncated derivative of PhtD containing only one HT motif and determined the affinity of the fragment for Zn2+ to be 131 ± 10 nM, which is weaker than that of AdcA (2.4 ± 0.1 nM) and other Zn2+-binding cluster A-I SBPs (4, 8, 16). This is consistent with the model for Zn2+ transfer to the higher-affinity site of a Zn2+-binding SBP for transport into the bacterial cell. The structure of this PhtD fragment has also been analyzed by nuclear magnetic resonance spectroscopy, with the single His triad site shown to bind Zn2+ in an assumed tetrahedral conformation by His83 (Nε2), His86 (Nε2), His88 (Nδ1), and Glu63 (15). This structure shows similarities to that previously derived by X-ray crystallography for a truncated PhtA fragment, which contained a different HT motif (17). In both cases, the motifs were observed to comprise part of a three-stranded β-sheet, and this suggests that a similar secondary structure could occur in the full-length Pht proteins (15, 17). However, it is important to note that these observations were made from truncated Pht fragments; their relevance to the full-length proteins remains to be confirmed. Further, there is a paucity of information on the flexibility and movement of SBPs and whether they could colocalize with the Pht proteins to effect Zn2+ transfer.
To date, there has been no investigation into the role of the HT motifs in the context of intact pneumococci or on their potential contribution to the process of Zn2+ uptake or to the other proposed functions of the proteins. Since these motifs are the defining feature of this protein family, with each of the Pht proteins sharing significant sequence similarity and contributing to Zn2+ acquisition, we have addressed these questions by examining the phenotypes of five PhtD mutant strains, each lacking one of the five HT motifs.
MATERIALS AND METHODS
Strains and growth media.
Defined, nonpolar deletion replacement or unmarked deletion mutants of phtA, phtB, phtD, phtE, or adcA in S. pneumoniae D39 have been generated previously (4, 18) (Table 1). To modify the HT motifs of PhtD, forward and reverse primers were designed complementary to the sequences encoding the motifs and were paired with primers flanking phtD to perform PCR to amplify the gene in two parts. The primers were designed with mutations to substitute phenylalanine residues in place of each histidine residue of the relevant triad (Table 2). The two PCR products were then fused by overlap extension PCR and used to transform the ΔadcA ΔphtABE strain, resulting in the mutant strains designated HT1 to HT5, respectively. Each of these strains produced a mutant variant of PhtD wherein each of the His residues of one of the five HT motifs had been substituted by phenylalanine residues. Mutagenesis of phtD in the HT strains was confirmed by DNA sequencing. Opaque phase variants were used in all experiments and a complete list of strains used in this study is in Table 1. Bacteria were routinely grown at 37°C in 5% CO2 on Columbia agar supplemented with 5% (vol/vol) horse blood or, for challenge of mice, in serum broth (10% heat-inactivated horse serum in nutrient broth). Alternatively, cation-defined medium (CDM; prepared as described previously [4, 19, 20]) was used. To assess the effect of Zn2+ starvation, the Zn(II)-chelating agent N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) was supplemented at an equimolar concentration to Zn2+, as determined in CDM by inductively coupled plasma-mass spectrometry (ICP-MS) to yield CDM-TPEN. To examine growth kinetics, cells were harvested from Columbia agar plates supplemented with 5% (vol/vol) horse blood incubated for 18 h at 37°C in 5% CO2 and inoculated in CDM(-TPEN) to an optical density at 600 nm (OD600) of 0.01. For all other analyses, cells were harvested similarly, but CDM-TPEN cultures were inoculated at an OD600 of 0.05.
TABLE 1.
Strains used in this study
| S. pneumoniae strain | Description or characteristic(s) | Source or reference |
|---|---|---|
| D39 | Serotype 2 parent strain | NCTC7466 |
| ΔadcA mutant | D39 with adcA deleted | 4 |
| ΔadcA ΔphtABE mutant | adcA, phtA, phtB, and phtE deleted | 5 |
| HT1 | ΔadcA ΔphtABE mutant with HT site 1 mutated | This study |
| HT2 | ΔadcA ΔphtABE mutant with HT site 2 mutated | This study |
| HT3 | ΔadcA ΔphtABE mutant with HT site 3 mutated | This study |
| HT4 | ΔadcA ΔphtABE mutant with HT site 4 mutated | This study |
| HT5 | ΔadcA ΔphtABE mutant with HT site 5 mutated | This study |
| ΔadcA ΔphtABDE mutant | adcA, phtA, phtB, phtD, and phtE deleted | 5 |
TABLE 2.
Oligonucleotides used in this study
| Primera | Sequence (5′−3′)b |
|---|---|
| 16s_F (qRT-PCR) | CATGCAAGTAGAACGCTGAA |
| 16s_R (qRT-PCR) | TGTCATGCAACATCCACTCT |
| phtD_F (qRT-PCR) | GTATTAGACAAAATGCTGTGGAG |
| phtD_R (qRT-PCR) | CTGTATAGGAGTCGGTTGACTTTC |
| HT1_F | ATGTGACCTCTTTTGGAGACTTCTATTTTTACTATAATGGCAAGGTTC |
| HT1_R | GAACCTTGCCATTATAGTAAAAATAGAAGTCTCCAAAAGAGGTCACAT |
| HT2_F | ATCGTTCCTTTCGGCGACTTTTACTTTTACATTCCTAAGAGTGATTTG |
| HT2_R | CAAATCACTCTTAGGAATGTAAAAGTAAAAGTCCCCGAAAGGAACGAT |
| HT3_F | GCTGTACCGTTCGGAGACTTTTATTTTTTTATTCCTTATTCACAACTG |
| HT3_R | CAGTTGTGAATAAGGAATAAAAAAATAAAAGTCTCCGAACGGTACAGC |
| HT4_F | CCTATGTAACTCCATTTATGACCTTTAGCTTCTGGATTAAAAAAGATAG |
| HT4_R | CTATCTTTTTTAATCCAGAAGCTAAAGGTCATAAATGGAGTTACATAGG |
| HT5_F | AGTTTAATCATACCTTTTTATGACTTTTACTTTAACATCAAATTTGAGT |
| HT5_R | ACTCAAATTTGATGTTAAAGTAAAAGTCATAAAAAGGTATGATTAAACT |
The direction of the primer is indicated by the primer designation suffix (F, forward; R, reverse).
Nucleotides targeting histidine residues for mutation to phenylalanine are indicated in boldface.
Growth analysis.
For growth analyses, S. pneumoniae D39 and mutant strains were grown in CDM with 1 μM MnSO4 until they reached an OD600 of 0.3. They were then subcultured into 200 μl CDM or CDM-TPEN to a final OD600 of 0.01. The bacteria were incubated at 37°C in a CO2-enriched atmosphere, and growth was monitored by measurement of the OD600 at 30-min intervals.
Flow cytometry.
Flow cytometry was performed essentially as described previously (11). In brief, cells were grown in CDM-TPEN until they reached an OD600 of 0.3 and incubated for 1 h at 37°C with murine anti-PhtD antisera in phosphate-buffered saline (PBS) (1:100 [vol/vol]) generated previously (11), followed by Alexa Fluor 488-rabbit anti-mouse IgG(H+L) (Thermo Fisher Scientific) for 30 min at 4°C. The cells were washed three times in 1 ml of PBS between each step. Fluorescence measurements from 10,000 events were collected using a BD FACSCanto flow cytometer (BD Biosciences). Data were analyzed by using the software package FlowJo (Tree Star). The results are a single representative of two independent experiments.
Whole-cell metal ion accumulation.
S. pneumoniae strains were grown in CDM-TPEN to an OD600 of 0.3, and their metal ion content was determined essentially as described previously (21). In brief, the bacteria were washed three times with PBS containing 5 mM EDTA and then twice with PBS. Bacterial pellets were desiccated by heating at 95°C overnight. The dry cell weight was measured, and the pellets were resuspended in 35% HNO3. Metal ion content was measured on an Agilent 7500cx inductively coupled plasma-mass spectrometer.
Measuring intracellular zinc using FluoZin-3.
S. pneumoniae strains were grown in CDM to an OD600 of 0.3. The cells were then washed three times in PBS, incubated with 5 μM cell permeant FluoZin-3 AM, probenecid, and PowerLoad (Thermo Fisher Scientific), and further mixed for 30 min at room temperature, similar to a method described previously (22). After washing in PBS to remove extracellular FluoZin-3 AM, the fluorescence was measured (excitation, 494 nm; emission, 516 nm). ZnSO4 was added to a final concentration of 10 μM, and the fluorescence was examined after 20 min. The data, corrected to cells not supplemented with ZnSO4, represent the mean of at least three independent experiments. Statistical significance was assessed by one-way analysis of variance (ANOVA) using Dunnett's posttest, comparing each strain to the ΔadcA ΔphtABE strain.
Quantitative immunoblot and qRT-PCR analyses.
Wild-type and mutant S. pneumoniae strains were grown under the same conditions as for ICP-MS. After reaching an OD600 of 0.3, samples were taken for protein analysis and RNA extraction. For protein extraction, cells were incubated with 0.1% sodium deoxycholate (Sigma-Aldrich) at 37°C for 60 min to induce lysis. Protein concentrations were determined (DC Bio-Rad protein assay; Bio-Rad), and 20 μg of total protein was loaded into each lane of a 4 to 12% NuPage Bis-Tris gel. After electrophoretic separation by SDS-PAGE, the proteins were transferred to a nitrocellulose membrane using an iBlot system (Thermo Fisher Scientific). Blots were incubated with previously generated murine antisera against PhtD (11). This was followed by incubation with anti-mouse IRDye 800 and analysis using an Odyssey infrared imaging system (LI-COR). Band intensities were measured using the manufacturer's application software. The results are representative of three independent experiments. For RNA extraction and quantitative reverse transcription-PCR (qRT-PCR) analysis, 500 μl of culture was mixed with 1 ml of RNA protect (Qiagen). RNA was extracted and purified using an RNeasy Protect Bacteria minikit (Qiagen) after enzymatic lysis using lysozyme and mutanolysin, according to the manufacturer's instructions. The total RNA samples were treated with DNase I (Roche) and qRT-PCR was carried out using a SuperScript III One-Step RT-PCR kit (Thermo Fisher Scientific) on an LC480 real-time cycler (Roche). Transcription levels of genes analyzed were normalized to those obtained for 16S rRNA. Primer sequences are available in Table 2. The results were representative of three independent experiments.
Assessment of bacterial fitness.
Outbred 5- to 6-week-old female CD1 (Swiss) mice were used in all animal experiments. Mice were anesthetized by intraperitoneal injection of pentobarbital sodium (Nembutal; Rhone-Merieux) at a dose of 66 μg per g of body weight, followed by intranasal administration of 50 μl of bacterial suspension containing ∼107 CFU. For competition experiments, 5 × 106 CFU of each of the two relevant strains was mixed prior to intranasal administration. The challenge dose was confirmed retrospectively by serial dilution and plating on blood agar, and for competition experiments, this was followed by patching of colonies onto blood agar plates with or without antibiotic to allow discrimination of the two strains.
At 24 h (absolute counts) and 48 h (absolute counts and competition) postchallenge, mice were euthanized by CO2 asphyxiation. Blood was collected by syringe from the posterior vena cava. The pleural cavity was lavaged with 1 ml of sterile PBS introduced through the diaphragm. Pulmonary vasculature was perfused by infusion of sterile PBS through the heart. Lungs were subsequently excised. The trachea was then exposed, and 1 ml of PBS was inserted into the tracheal end of the upper respiratory tract and collected from the nose (nasal wash). Lastly, the nasopharynx/upper palate was excised (nasal tissue). Tissues were homogenized, and all samples were serially diluted and plated on blood agar. For competition experiments, colonies were subsequently patched onto blood plates with or without the relevant antibiotic to allow discrimination of the two mutant strains and determination of output ratios. Competitive indices (the ratio of one mutant strain to the other relative to the input ratio) were then calculated and compared to a theoretical value of 1 (which would indicate no difference in fitness between strains) by one-sample t tests.
All procedures performed here were conducted with a view to minimizing the discomfort of the animals and used the minimum numbers to generate reproducible and statistically significant data. All experiments were approved by the University of Adelaide Animal Ethics Committee (Animal Welfare Assurance number A5491-01; project approval number S-2013-053) and were performed in strict adherence to guidelines dictated by the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
RESULTS
Mutation of the HT sites results in compromised growth under Zn2+-limiting conditions.
Zinc recruitment under Zn2+-replete conditions occurs via AdcA and AdcAII in the pneumococcus. AdcAII has a more significant role during Zn2+ deficiency due to its high level of upregulation under such conditions. However, AdcAII relies on the Pht proteins for full functionality. To examine the role of the HT motifs in the Pht proteins, mutants lacking one of the five HT motifs in PhtD were constructed in a previously generated ΔadcA ΔphtABE background strain. In these mutant strains, Zn2+ acquisition was entirely dependent upon AdcAII and the four remaining functional HT motifs of PhtD. In this way, the contribution of the individual HT sites could be examined. The PhtD HT motifs have been designated HT1 to HT5, with HT1 at the amino terminus predicted to be closest to the cell membrane and HT5 at the carboxyl terminus closest to the extracellular environment (14, 23, 24). In the HT mutants, each of the histidine residues (HxxHxH) have been mutated to phenylalanine, removing their Zn2+ binding capacity while maintaining a similar steric bulk. The HT mutant strains were assessed for growth in vitro in comparison to the wild-type strain D39, the ΔadcA ΔphtABE mutant strain (direct parent strain), and the ΔadcA ΔphtABDE mutant strain (lacking all Pht proteins). Growth of all mutant strains remained essentially unchanged compared to that of the wild type in CDM, i.e., Zn2+-replete conditions (Fig. 1A). To examine the effect of Zn2+ limitation on growth of these strains, the medium was supplemented with the Zn2+-chelating agent TPEN at an equimolar concentration to the Zn2+ present in the media. In general, these stress conditions affected growth of all strains, including the wild type, but to vastly different extents (Fig. 1B). Most strikingly, growth was largely inhibited in the HT1 mutant, a phenotype similar to that seen for the ΔadcA ΔphtABDE mutant, indicating that mutation of HT1 abolished the functionality of PhtD in Zn2+ recruitment. The HT2 and HT3 mutants also showed compromised growth, but not to the same extent as the HT1 mutant. In contrast, the HT4 and HT5 mutants had growth phenotypes that were essentially identical to that of the parental strain (ΔadcA ΔphtABE). Taken together, these results indicate a potential hierarchy in the HT sites for their importance in growth under Zn2+ limited conditions, with the relative contribution of each of the sites to Zn2+ acquisition generally decreasing from the amino terminus to the carboxyl terminus of the protein (HT sites 1 to 5).
FIG 1.

In vitro growth comparison of the S. pneumoniae HT mutant strains. Bacteria were grown in CDM (A) or CDM-TPEN (B) (i.e., under Zn2+-restricted conditions) at 37°C, and growth was monitored by OD600 measurements every 30 min. Data are representative mean OD600 measurements ± the standard errors of the mean (SEM) from three independent biological experiments. WT, wild type.
The HT mutants express PhtD at the cell surface.
To ascertain whether the differences in the growth kinetics of the HT site mutants under Zn2+-limiting conditions were due to expression issues and/or mislocalization of PhtD, we examined the strains for surface-localized PhtD by flow cytometry. Cells were incubated with an anti-PhtD antiserum, followed by a fluorescently labeled secondary antibody, to determine the presence of cell surface PhtD, as previously described (11). Consistent with expectations, the Pht-null mutant (ΔadcA ΔphtABDE) showed a single population representing negative events, similar to that seen for the unlabeled wild-type sample (Fig. 2). The labeled wild-type and ΔadcA strains showed a similar pattern to each other, with a broad distribution of fluorescence intensities seen, indicating that PhtD was expressed but to substantially different levels. The combined deletion of adcA, phtA, phtB, and phtE (ΔadcA ΔphtABE) resulted in a dramatic shift, with most cells showing ∼10-fold-higher fluorescence levels than the ΔadcA strain. Subsequent mutation of the HT sites resulted in only minor changes compared to the parental strain, indicating that mutation of the HT sites did not have a detrimental impact on PhtD localization. However, the flow cytometric analyses of the HT1 mutant, and to some extent the HT2 mutant, indicate that PhtD may be expressed at even higher levels in these mutant strains than in the parent strain (ΔadcA ΔphtABE).
FIG 2.
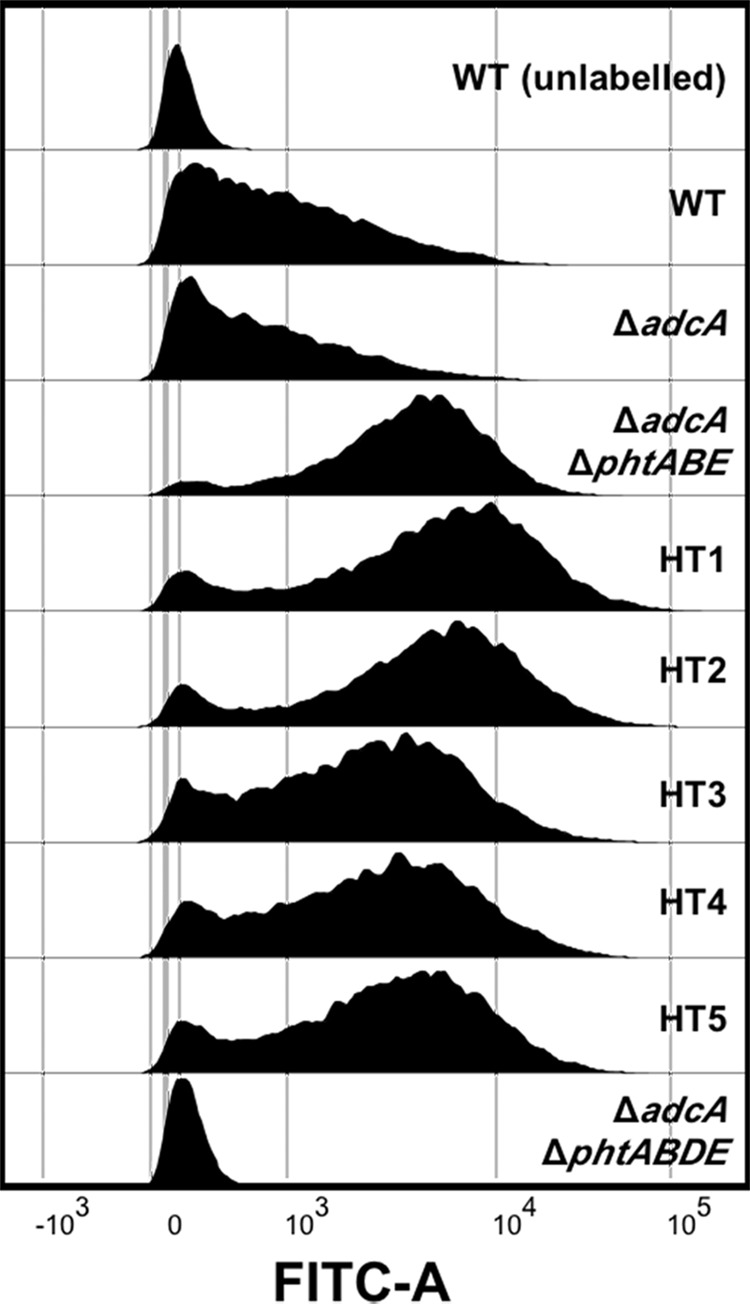
Flow cytometric measurement of the presence of PhtD at the bacterial surface of the HT mutants and control strains. A representative histogram of fluorescence profiles for each strain is shown. An unlabeled wild-type strain (WT) and a phtD deletion strain (ΔadcA ΔphtABDE) were included to represent the profiles of negative cells.
Mutation of PhtD HT1 results in increased PhtD expression.
To gain greater insight into potential differences in the abundance of PhtD, as revealed by our flow cytometric analyses, we examined the transcript levels of phtD, which is part of the Zn2+-responsive AdcR regulon (25). Deletion of adcA resulted in a minor increase in phtD transcription (∼3-fold) and the additional deletion of phtA, phtB, and phtE (ΔadcA ΔphtABE) resulted in an ∼4.4-fold increase in expression, compared to the wild type (Fig. 3A). Mutation of the PhtD HTs in the ΔadcA ΔphtABE strain revealed highly intriguing differences. Compared to the ΔadcA ΔphtABE parental strain, the HT1 mutant showed a significant 10-fold (P < 0.0001) upregulation of phtD. Despite a modest increase in phtD expression in the HT2, HT3, HT4, and HT5 mutants (2.5- to 3.1-fold), this was not significantly different from the ΔadcA ΔphtABE strain. Examination of PhtD protein expression by quantitative immunoblot analysis showed a pattern consistent with the mRNA levels. However, the overall extent of the differences between strains was more modest (Fig. 3B and C). Minor, nonsignificant differences in PhtD expression were observed for the ΔadcA and ΔadcA ΔphtABE strains compared to the wild type. However, the HT1 mutant displayed a significant 2-fold (P < 0.0001) increase in expression of PhtD relative to its ΔadcA ΔphtABE parent strain. In contrast, the HT2 mutant exhibited a statistically insignificant increase in PhtD expression (1.3-fold), with the expression of PhtD in the HT3, HT4 and HT5 mutants unchanged compared to that in the ΔadcA ΔphtABE strain. Taken together, since phtD is upregulated by the Zn2+-dependent transcription regulator AdcR in response to Zn2+ deficiency, these data strongly suggest that mutation of HT site 1 resulted in a significantly lower level of Zn2+ within the pneumococcus.
FIG 3.
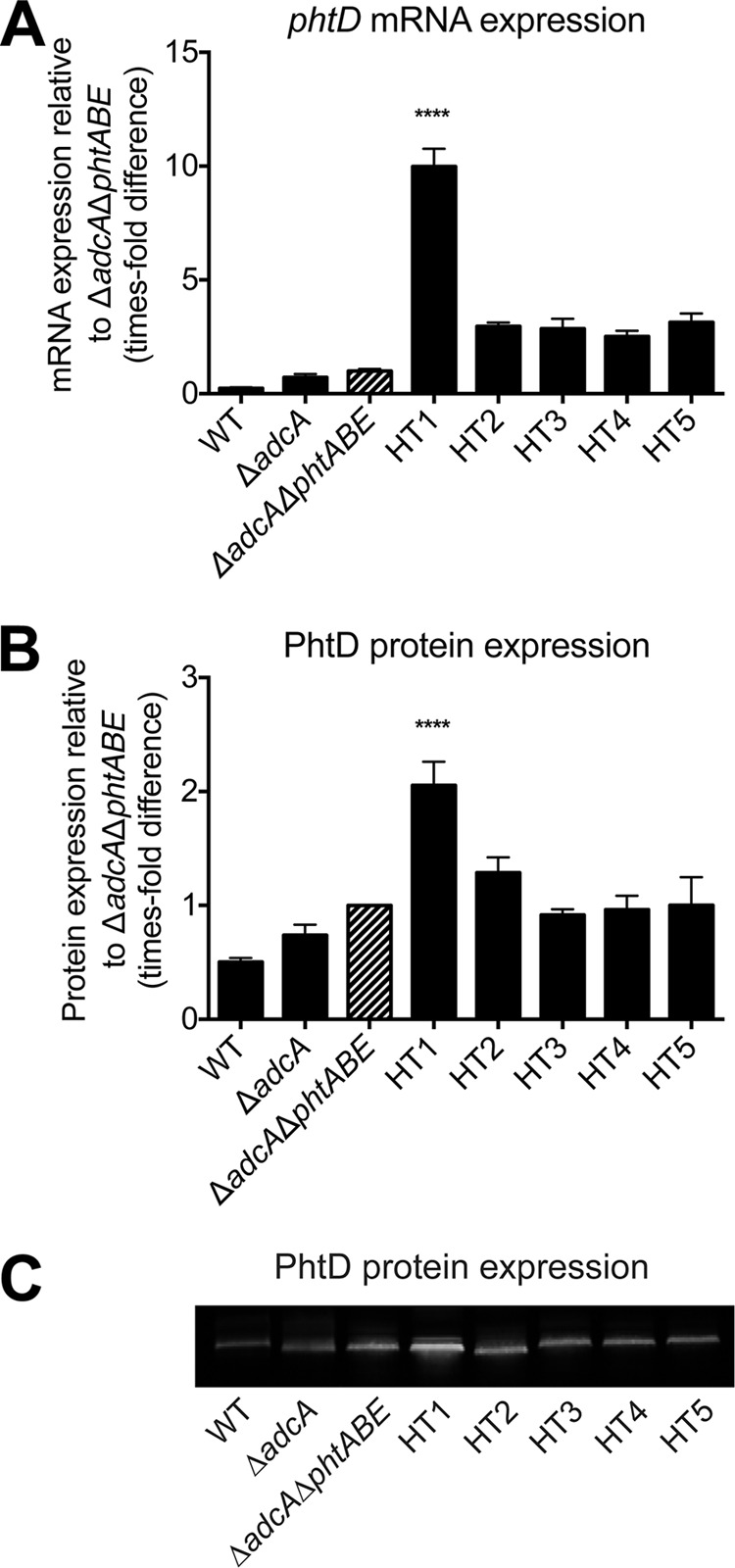
Analysis of PhtD expression levels in HT mutant strains grown in CDM-TPEN. mRNA (A) and protein (B) expression levels are shown, normalized to the levels in the ΔadcA ΔphtABE strain. For mRNA, measurements were made relative to 16S rRNA. Strains were compared to the ΔadcA ΔphtABE mutant (striped bars) by one-way ANOVA using Dunnett's posttest, and statistically significant differences, where present, are indicated (****, P < 0.0001). The results (± the SEM) are representative of three independent experiments. WT, wild type. (C) Representative Western blot for the determination of PhtD protein expression.
Metal ion accumulation of HT deficient strains.
To further examine the effect of mutating the HT motifs of PhtD, the cellular metal content of the HT mutant, wild-type, and control strains was assessed following growth in CDM-TPEN to an OD600 of 0.3. Consistent with our previous observations, deletion of adcA in conjunction with phtA, phtB, and phtE (ΔadcA ΔphtABE) resulted in a significant reduction in cellular Zn2+ abundance relative to that for the wild type (P < 0.001) as determined by ICP-MS (Fig. 4A) (5). Although only minor, the additional deletion of phtD (ΔadcA ΔphtABDE) resulted in a further reduction in Zn2+ accumulation (15%; P < 0.05). Due to the minimal difference in Zn2+ accumulation between the ΔadcA ΔphtABE and ΔadcA ΔphtABDE strains, the effect of mutating the individual HT sites on total cellular Zn2+ accumulation was not able to be resolved via ICP-MS. To ascertain whether the HT mutant strains had differences in the ability to acquire Zn2+, we performed a Zn2+ uptake assay using the intracellular Zn2+-responsive fluorophore FluoZin-3 AM. Analysis of the strains revealed similar levels of Zn2+ uptake in the wild-type, ΔadcA, and ΔadcA ΔphtABE strains after the addition of ZnSO4 (Fig. 4B). Thus, the combined action of AdcAII and PhtD is sufficient for wild-type levels of Zn2+ uptake. In the absence of AdcA and all four Pht proteins, the ΔadcA ΔphtABDE strain exhibited a significant decrease in Zn2+ uptake (23%; P < 0.01) relative to the level for the wild type, with the Zn2+ acquisition entirely dependent on AdcAII. The uptake of Zn2+ was also significantly reduced in the HT1, HT2, HT3, and HT4 strains (∼25%), but consistent with the expression and growth data, mutation of the HT5 site had no effect on Zn2+ accumulation. Overall, these data show that although total cellular Zn2+ accumulation was not found to be significantly different in any of the HT mutant strains, active Zn2+ uptake was impaired when any of the first four HT sites of PhtD were mutated. In contrast, the HT5 site, located at the carboxyl terminus of PhtD, appears to be the least important site for Zn2+ acquisition.
FIG 4.
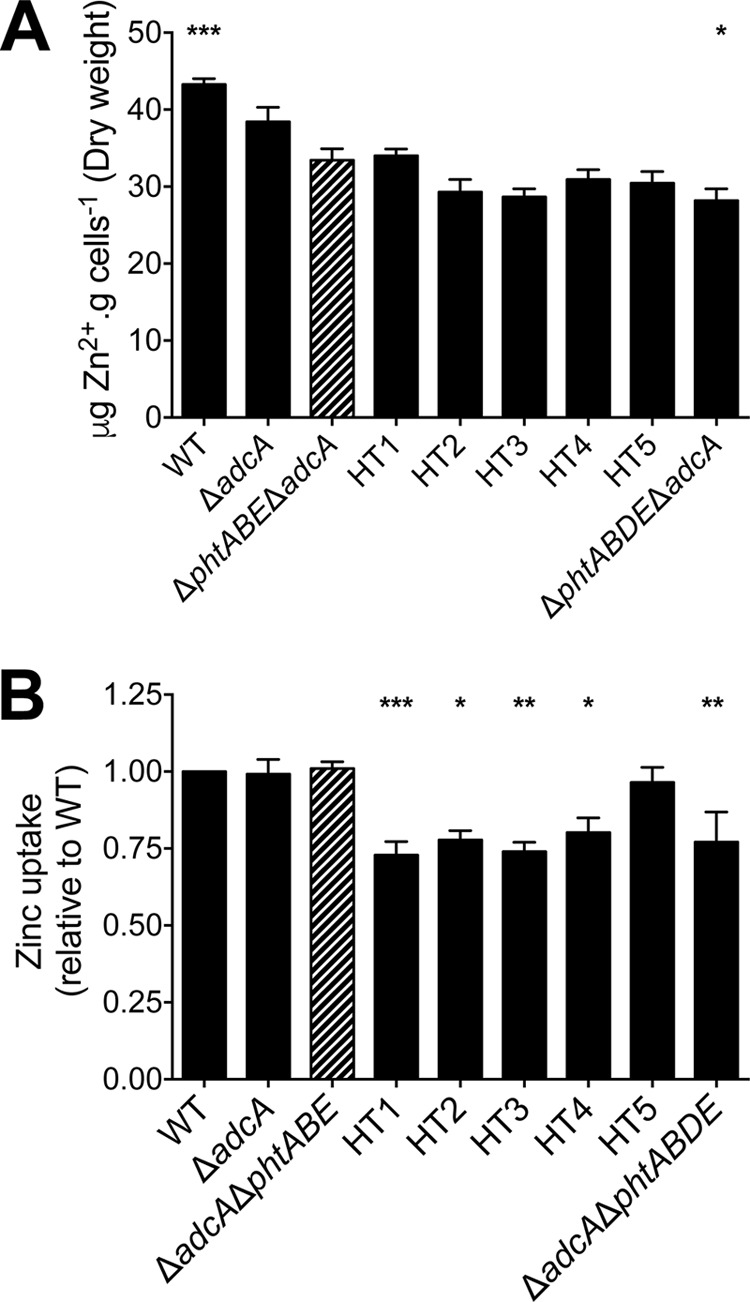
(A) Whole-cell Zn2+ accumulation of control and HT mutant strains as determined by ICP-MS. (B) FluoZin-3 AM Zn2+ uptake assay examining control and HT mutant strains. Zinc uptake for each of the strains is shown relative to that for the wild type. In both assays, all strains were compared to the ΔadcA ΔphtABE mutant (striped bars) by one-way ANOVA using Dunnett's posttest, and statistical significance is indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Data are means (± the SEM) from a minimum of three biological replicates.
Role of PhtD HT sites in vivo.
To examine whether the HT1 site also played a critical role during pneumococcal infection, we assessed the ability of the HT1 mutant strain to colonize and persist in various niches in a murine host. Colonization by the HT1 strain was compared to that of the S. pneumoniae D39 wild-type, ΔadcA, ΔadcA ΔphtABE, ΔadcA ΔphtABDE and HT5 mutant strains at 24 and 48 h. At 24 h the wild-type strain, and to a lesser degree the ΔadcA strain, was able to colonize various niches to a greater extent than the ΔadcA ΔphtABE strain, with a significantly higher level of colonization observed for the wild-type strain in the nasopharynx (Fig. 5A). To assess the impact of the HT mutations in PhtD, we first established the difference in colonization between the PhtD-only strain (ΔadcA ΔphtABE) and the strain lacking all Pht proteins (ΔadcA ΔphtABDE). Statistically significant differences (one-way ANOVA) between these two strains were only observed in the blood (P < 0.01) and pleural lavage fluid (P < 0.01) at 24 h postchallenge (Fig. 5A). Further analysis of these niches at 24 h revealed that results for the HT1 mutant strain were significantly different (P < 0.05) from results for the PhtD-only containing strain (ΔadcA ΔphtABE) in the blood but not in the pleural lavage fluid. Although not statistically significant, colonization of the HT5 mutant was also attenuated relative to that of the PhtD-only containing strain, and no statistically significant difference was observed between the HT1 and HT5 mutant strains. The lungs and pleural lavage fluid of the majority of mice infected with the HT1 or HT5 strain showed levels of bacterial burden intermediate between the parental strain (ΔadcA ΔphtABE) and the complete pht deletion strain (ΔadcA ΔphtABDE). As a consequence, the absolute quantitation of bacterial colonization did not allow for discrimination of the in vivo fitness between the HT mutants. To examine their colonization potential in greater detail, we then conducted an in vivo competition assay, in which mice were challenged with the HT1 or HT5 mutant strain and an equal dose of the parent strain (ΔadcA ΔphtABE). Since a number of mice infected with the HT mutant strains did not show detectable levels of bacteria in the blood 24 h postchallenge (Fig. 5A), we only examined the competitive index at 48 h. Both the HT1 and HT5 mutant strains were significantly compromised in their ability to infect the lungs, pleural cavity, and blood compared to the ΔadcA ΔphtABE strain (one-sample t test; P < 0.0001) (Fig. 6). The low competitive index of the HT5 mutant strain, relative to that of the ΔadcA ΔphtABE strain, was unanticipated given the in vitro characteristics of the strain, wherein the HT5 mutant was essentially comparable to the ΔadcA ΔphtABE strain in terms of Zn2+ uptake. Altogether, despite the differences between the HT mutants and the parent strain, no significant differences were seen in the competitive indices between the individual HT mutants. The observation that this finding does not directly correlate with the in vitro data suggests that the role of the individual HT motifs is more complex than first anticipated and worthy of further investigation.
FIG 5.

Burden of S. pneumoniae infection was assessed by determination of the bacterial load (CFU ml−1) recovered from infected mice at 24 h (A) and 48 h (B) after challenge. The number of pneumococci in each niche at each time point is plotted (one point represents one niche from one mouse). Solid lines denote the median of each group; dashed lines denote the limit of detection. Statistically significant differences from the ΔadcA ΔphtABE mutant (circles) are indicated, as assessed by one-way ANOVA (*, P < 0.05; **, P < 0.01; ****, P < 0.0001).
FIG 6.

In vivo competition between the ΔadcA ΔphtABE mutant and HT1 or HT5. Eight mice were used per group. The results are shown as the median competitive indices (the ratio of the HT mutant to the ΔadcA ΔphtABE mutant corrected for the input ratio). The competitive index was compared to a theoretical mean of 1 (dashed line) by one-sample t tests, and the statistical significance is indicated (****, P < 0.0001).
DISCUSSION
Zinc acquisition in the pneumococcus is central to its ability to colonize and cause disease. As surface-associated proteins, the Pht family are proposed to hold key roles in aiding delivery of Zn2+ to AdcAII for transport by AdcCB under Zn2+-limiting conditions, although the molecular details of the Zn2+ transfer process remain unclear. As the most highly conserved Pht protein, PhtD has been extensively studied and holds potential as a novel protein-based vaccine candidate. However, the roles of the individual HT motifs have not been thoroughly investigated. Here we have shown that, in vitro, the HT1 site is most important for growth in Zn2+-restricted media, where the phenotypic impact was similar to that observed for a mutant lacking all Pht proteins. Overall, our in vitro data indicate a hierarchy of importance, where the HT site(s) at the amino terminus, i.e., closest to the cell membrane, has the most significant role in Zn2+ acquisition, whereas the HT5 site at the carboxyl terminus, which is closest to the extracellular environment, has the smallest contribution. Similar to other cluster A-I SBPs, AdcAII is anticipated to have an affinity for Zn2+ of <20 nM, facilitating this transfer and the delivery of Zn2+ to the AdcCB transporter. Notably, the amino termini of the different Pht proteins are also the most highly conserved portion of the protein, but the significance of this remains to be determined.
Intriguingly, while the hierarchy of importance of the HT sites holds true in vitro, the situation in vivo appears to be more complex. Although the HT1 site played a greater role in Zn2+ acquisition in vitro, the HT1 and HT5 strains were each similarly less competitive than the ΔadcA ΔphtABE strain, in a murine model of disease. A role for Pht proteins other than in AdcAII-mediated zinc recruitment was also demonstrated by an ΔadcAII ΔphtABDE deletion mutant, which was significantly impaired in its ability to cause invasive disease compared to an ΔadcAII strain (5). PhtD, together with the other Pht proteins, has previously been suggested to contribute to pneumococcal adherence to respiratory epithelial cells and aid in invasion (26) as well as contribute to the evasion of complement (27). Whether or not HT5, predicted to be the most surface-exposed HT motif, significantly contributes to any of these processes and thus disease progression requires further investigation. Alternatively, a functional HT5 motif may provide a competitive advantage for the acquisition of Zn2+ at the host-pathogen interface without significantly contributing to pneumococcal Zn2+ uptake.
In summary, the data presented here demonstrate that the PhtD HT sites hold an important role in Zn2+ acquisition and pneumococcal disease. Despite the significant advances made in uncovering the role of Pht proteins in pneumococcal Zn2+ acquisition, the precise pathway and molecular mechanism of Zn2+ movement across the capsule and cell wall barrier via the Pht proteins to AdcAII anchored to the outer face of the membrane require further analysis. Questions still remain as to the role of each of the HT sites in the translocation of Zn2+ and whether the Zn2+ is passed to AdcAII directly or indirectly from the Pht proteins. Further characterization of the overall role of the carboxyl-terminal HT sites in vivo will also be required to elucidate the manner in which they contribute to disease pathogenesis.
ACKNOWLEDGMENT
J.C.P. is an NHMRC Senior Principal Research Fellow.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, O'Brien KL, Campbell H, Black RE. 2013. Global burden of childhood pneumonia and diarrhoea. Lancet 381:1405–1416. doi: 10.1016/S0140-6736(13)60222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayle L, Chimalapati S, Schoehn G, Brown J, Vernet T, Durmort C. 2011. Zinc uptake by Streptococcus pneumoniae depends on both AdcA and AdcAII and is essential for normal bacterial morphology and virulence. Mol Microbiol 82:904–916. doi: 10.1111/j.1365-2958.2011.07862.x. [DOI] [PubMed] [Google Scholar]
- 3.Lewis VG, Ween MP, McDevitt CA. 2012. The role of ATP-binding cassette transporters in bacterial pathogenicity. Protoplasma 249:919–942. doi: 10.1007/s00709-011-0360-8. [DOI] [PubMed] [Google Scholar]
- 4.Plumptre CD, Eijkelkamp BA, Morey JR, Behr F, Counago RM, Ogunniyi AD, Kobe B, O'Mara ML, Paton JC, McDevitt CA. 2014. AdcA and AdcAII employ distinct zinc acquisition mechanisms and contribute additively to zinc homeostasis in Streptococcus pneumoniae. Mol Microbiol 91:834–851. doi: 10.1111/mmi.12504. [DOI] [PubMed] [Google Scholar]
- 5.Plumptre CD, Hughes CE, Harvey RM, Eijkelkamp BA, McDevitt CA, Paton JC. 2014. Overlapping functionality of the Pht proteins in zinc homeostasis of Streptococcus pneumoniae. Infect Immun 82:4315–4324. doi: 10.1128/IAI.02155-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plumptre CD, Ogunniyi AD, Paton JC. 2012. Polyhistidine triad proteins of pathogenic streptococci. Trends Microbiol 20:485–493. doi: 10.1016/j.tim.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Plumptre CD, Ogunniyi AD, Paton JC. 2013. Surface association of Pht proteins of Streptococcus pneumoniae. Infect Immun 81:3644–3651. doi: 10.1128/IAI.00562-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loisel E, Chimalapati S, Bougault C, Imberty A, Gallet B, Di Guilmi AM, Brown J, Vernet T, Durmort C. 2011. Biochemical characterization of the histidine triad protein PhtD as a cell surface zinc-binding protein of pneumococcus. Biochemistry 50:3551–3558. doi: 10.1021/bi200012f. [DOI] [PubMed] [Google Scholar]
- 9.Godfroid F, Hermand P, Verlant V, Denoel P, Poolman JT. 2011. Preclinical evaluation of the Pht proteins as potential cross-protective pneumococcal vaccine antigens. Infect Immun 79:238–245. doi: 10.1128/IAI.00378-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prymula R, Pazdiora P, Traskine M, Ruggeberg JU, Borys D. 2014. Safety and immunogenicity of an investigational vaccine containing two common pneumococcal proteins in toddlers: a phase II randomized clinical trial. Vaccine 32:3025–3034. doi: 10.1016/j.vaccine.2014.03.066. [DOI] [PubMed] [Google Scholar]
- 11.Plumptre CD, Ogunniyi AD, Paton JC. 2013. Vaccination against Streptococcus pneumoniae using truncated derivatives of polyhistidine triad protein D. PLoS One 8:e78916. doi: 10.1371/journal.pone.0078916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira DM, Jambo KC, Gordon SB. 2011. Experimental human pneumococcal carriage models for vaccine research. Trends Microbiol 19:464–470. doi: 10.1016/j.tim.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Pauksens K, Nilsson AC, Caubet M, Pascal TG, Van Belle P, Poolman JT, Vandepapeliere PG, Verlant V, Vink PE. 2014. Randomized controlled study of the safety and immunogenicity of pneumococcal vaccine formulations containing PhtD and detoxified pneumolysin with alum or adjuvant system AS02V in elderly adults. Clin Vaccine Immunol 21:651–660. doi: 10.1128/CVI.00807-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rioux S, Neyt C, Di Paolo E, Turpin L, Charland N, Labbe S, Mortier MC, Mitchell TJ, Feron C, Martin D, Poolman JT. 2011. Transcriptional regulation, occurrence and putative role of the Pht family of Streptococcus pneumoniae. Microbiology 157:336–348. doi: 10.1099/mic.0.042184-0. [DOI] [PubMed] [Google Scholar]
- 15.Bersch B, Bougault C, Roux L, Favier A, Vernet T, Durmort C. 2013. New insights into histidine triad proteins: solution structure of a Streptococcus pneumoniae PhtD domain and zinc transfer to AdcAII. PLoS One 8:e81168. doi: 10.1371/journal.pone.0081168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ilari A, Alaleona F, Tria G, Petrarca P, Battistoni A, Zamparelli C, Verzili D, Falconi M, Chiancone E. 2014. The Salmonella enterica ZinT structure, zinc affinity and interaction with the high-affinity uptake protein ZnuA provide insight into the management of periplasmic zinc. Biochim Biophys Acta 1840:535–544. doi: 10.1016/j.bbagen.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Riboldi-Tunnicliffe A, Isaacs NW, Mitchell TJ. 2005. 1.2 Angstroms crystal structure of the S. pneumoniae PhtA histidine triad domain a novel zinc binding fold. FEBS Lett 579:5353–5360. doi: 10.1016/j.febslet.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 18.Ogunniyi AD, Grabowicz M, Mahdi LK, Cook J, Gordon DL, Sadlon TA, Paton JC. 2009. Pneumococcal histidine triad proteins are regulated by the Zn2+-dependent repressor AdcR and inhibit complement deposition through the recruitment of complement factor H. FASEB J 23:731–738. doi: 10.1096/fj.08-119537. [DOI] [PubMed] [Google Scholar]
- 19.McDevitt CA, Ogunniyi AD, Valkov E, Lawrence MC, Kobe B, McEwan AG, Paton JC. 2011. A molecular mechanism for bacterial susceptibility to zinc. PLoS Pathog 7:e1002357. doi: 10.1371/journal.ppat.1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Begg SL, Eijkelkamp BA, Luo Z, Counago RM, Morey JR, Maher MJ, Ong CL, McEwan AG, Kobe B, O'Mara ML, Paton JC, McDevitt CA. 2015. Dysregulation of transition metal ion homeostasis is the molecular basis for cadmium toxicity in Streptococcus pneumoniae. Nat Commun 6:6418. doi: 10.1038/ncomms7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eijkelkamp BA, Morey JR, Ween MP, Ong CL, McEwan AG, Paton JC, McDevitt CA. 2014. Extracellular zinc competitively inhibits manganese uptake and compromises oxidative stress management in Streptococcus pneumoniae. PLoS One 9:e89427. doi: 10.1371/journal.pone.0089427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clementi EA, Marks LR, Roche-Hakansson H, Hakansson AP. 2014. Monitoring changes in membrane polarity, membrane integrity, and intracellular ion concentrations in Streptococcus pneumoniae using fluorescent dyes. J Vis Exp 2014:e51008. doi: 10.3791/51008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamou JE, Heinrichs JH, Erwin AL, Walsh W, Gayle T, Dormitzer M, Dagan R, Brewah YA, Barren P, Lathigra R, Langermann S, Koenig S, Johnson S. 2001. Identification and characterization of a novel family of pneumococcal proteins that are protective against sepsis. Infect Immun 69:949–958. doi: 10.1128/IAI.69.2.949-958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beghetto E, Gargano N, Ricci S, Garufi G, Peppoloni S, Montagnani F, Oggioni M, Pozzi G, Felici F. 2006. Discovery of novel Streptococcus pneumoniae antigens by screening a whole-genome lambda-display library. FEMS Microbiol Lett 262:14–21. doi: 10.1111/j.1574-6968.2006.00360.x. [DOI] [PubMed] [Google Scholar]
- 25.Shafeeq S, Kloosterman TG, Kuipers OP. 2011. Transcriptional response of Streptococcus pneumoniae to Zn2+ limitation and the repressor/activator function of AdcR. Metallomics 3:609–618. doi: 10.1039/c1mt00030f. [DOI] [PubMed] [Google Scholar]
- 26.Kallio A, Sepponen K, Hermand P, Denoel P, Godfroid F, Melin M. 2014. Role of Pht proteins in attachment of Streptococcus pneumoniae to respiratory epithelial cells. Infect Immun 82:1683–1691. doi: 10.1128/IAI.00699-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melin M, Di Paolo E, Tikkanen L, Jarva H, Neyt C, Kayhty H, Meri S, Poolman J, Vakevainen M. 2010. Interaction of pneumococcal histidine triad proteins with human complement. Infect Immun 78:2089–2098. doi: 10.1128/IAI.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]