

Abstract
Stenotrophomonas maltophilia is a ubiquitous bacterium and an emerging nosocomial pathogen. This bacterium is resistant to many antibiotics, associated with a number of infections, and a significant health risk, especially for immunocompromised patients. Given that Caenorhabditis elegans shares many conserved genetic pathways and pathway components with higher organisms, the study of its interaction with bacterial pathogens has biomedical implications. S. maltophilia has been isolated in association with nematodes from grassland soils, and it is likely that C. elegans encounters this bacterium in nature. We found that a local S. maltophilia isolate, JCMS, is more virulent than the other S. maltophilia isolates (R551-3 and K279a) tested. JCMS virulence correlates with intestinal distension and bacterial accumulation and requires the bacteria to be alive. Many of the conserved innate immune pathways that serve to protect C. elegans from various pathogenic bacteria also play a role in combating S. maltophilia JCMS. However, S. maltophilia JCMS is virulent to normally pathogen-resistant DAF-2/16 insulin-like signaling pathway mutants. Furthermore, several insulin-like signaling effector genes were not significantly differentially expressed between S. maltophilia JCMS and avirulent bacteria (Escherichia coli OP50). Taken together, these findings suggest that S. maltophilia JCMS evades the pathogen resistance conferred by the loss of DAF-2/16 pathway components. In summary, we have discovered a novel host-pathogen interaction between C. elegans and S. maltophilia and established a new animal model with which to study the mode of action of this emerging nosocomial pathogen.
INTRODUCTION
Stenotrophomonas maltophilia, a Gram-negative bacillus previously classified as both Pseudomonas and Xanthomonas (1), is an emerging nosocomial pathogen. From 1993 to 2004, S. maltophilia was found to be among the 11 organisms most frequently recovered from intensive care unit (ICU) patients in U.S. hospitals (2). A more recent study of patients with S. maltophilia bacteremia revealed that most cases were related to hospital admission, with some being associated with health care, such as outpatient intravenous antibiotics, treatment in a long-term-care facility, or chemotherapy (3). That study also found that intubation and ICU stay at the time of S. maltophilia bacteremia were associated with mortality (3). This is likely correlated with the propensity of S. maltophilia to adhere to plastics and form biofilms (4) and the opportunistic infection of patients with preexisting illnesses. In fact, S. maltophilia is the predominant bacterium recovered from the respiratory tract of patients with cystic fibrosis (5) and enhances the pathogenesis of Crohn's disease and ulcerative colitis (6, 7). Additionally, S. maltophilia can cause nosocomial pneumonia (8) and is associated with a number of infections, such as meningitis and endocarditis (reviewed in reference 9). Thus, S. maltophilia is a significant and medically important human pathogen. Furthermore, characterization of various S. maltophilia strains is imperative, as the genome sequences of environmental (including R551-3) and clinical (including K279a) S. maltophilia isolates contain heterogeneity that might help mediate adaptations to different environments (10).
Despite its medical importance, there are few model systems to investigate the mode of action of S. maltophilia (10–13). Furthermore, little is known about what is required for S. maltophilia virulence and if these requirements are bacterium and/or strain specific. Here, we develop Caenorhabditis elegans as a model for the study of S. maltophilia strain-specific responses. C. elegans has proven to be an excellent model for understanding development, neurobiology, behavior, and, more recently, innate immunity (14). Like other metazoans, C. elegans has evolved in the presence of microbes. The interaction between C. elegans and its associated microbes is multifaceted, as these nematodes feed on potentially pathogenic bacteria. Consequently, the C. elegans innate immune response has evolved both conserved and specific aspects to cope with the microbial world. Whereas the conserved Toll-like receptor (TLR) pathway plays a central role in innate immunity in other animals, the single C. elegans TLR gene, tol-1, is not involved in the response to several pathogens (15) and does not appear to control the expression of antimicrobial genes (16). Uniquely, tol-1 plays a role in the protection of pharyngeal tissue upon challenge with Salmonella enterica (17), suggesting a specific response that differs from the usual TLR signaling. Conversely, the functions of other innate immune pathways are conserved and studying the nematode immune response can be informative in understanding how higher organisms mount pathogen defenses (18, 19). For example, the highly conserved p38 mitogen-activated protein kinase (MAPK) pathway plays a major role in the response to human bacterial pathogens (20) such as Pseudomonas aeruginosa (21) and Staphylococcus aureus (22). The unfolded-protein response (UPR) ire-1–xbp-1 module is a downstream target of the p38 MAPK pathway in response to pore-forming toxin (PFT) (23), a virulence factor for a number of bacterial pathogens, including Bacillus thuringiensis (24). Other conserved pathways, such as the DBL-1/transforming growth factor β (TGF-β) pathway, play a role in the nematode response to a number of pathogens, including Drechmeria coniospora (25) and Serratia marcescens (26). The conserved insulin-like receptor DAF-2 negatively regulates the transcription factor DAF-16/ forkhead box O (FOXO), which induces the expression of numerous downstream effector genes. As a result, daf-2 mutants are long-lived on most bacteria tested to date, including Escherichia coli, Enterococcus faecalis, as well as several other human pathogens (15, 27). The DAF-2/16 pathway also regulates longevity, aging, and diapause in addition to its role in innate immunity (28–30). However, the regulation of longevity and innate immunity is distinct, involving pathway components that either play a role in both processes or have an exclusive role in longevity (31).
Each of the conserved innate immune pathways specifically regulates downstream effectors such as lysozymes, lectins, neuropeptide-like peptides (NLPs), and antimicrobial factors (reviewed in reference 20). For example, dbl-1 regulates caenacins but not the structurally related NLPs (25). Innate immunity effector genes such as members of the lysozyme family and the C-type lectin family are also pathogen specific (32), and innate immune pathway components can exhibit effector regulation independent of other pathway constituents (33). Finally, the p38 MAPK and DAF-2/16 pathways appear to function in parallel and positively regulate unique downstream genes (34). Thus, in addition to sharing conserved pathways with human innate immunity, C. elegans seemingly shares a conserved genetic architecture employing multiple pathways and corresponding effectors that function in parallel to combat various microbial assaults. Therefore, C. elegans is a valuable model system to study innate immune pathway function and specificity.
Previous work demonstrated that S. maltophilia K279a can kill C. elegans (13), and another strain, S. maltophilia G2, was shown to have nematicidal activity (35). S. maltophilia has also been detected in association with the nematode Pristionchus pacificus (36) as well as in the natural environment of C. elegans (B. Samuel, personal communication). Thus, it is likely that C. elegans encounters S. maltophilia in natural settings, suggesting that their interaction is evolutionarily significant. In this study, we report our discovery of an S. maltophilia strain, JCMS, that also kills C. elegans. Our results indicate that JCMS accumulates within C. elegans, does not appear to employ a toxin to confer virulence, and needs to be alive to be pathogenic. Our data also showed that the UPR, p38 MAPK, and DBL-1/TGF-β pathways are involved in a nonspecific innate immune response to bacteria. Furthermore, normally pathogen-resistant C. elegans mutants, such as daf-2, akt-1, and ins-7 mutants, were susceptible to S. maltophilia JCMS, suggesting that JCMS evades the downstream effects of these DAF-2/16 pathway components. These findings correlate with our observation that daf-2-regulated effector genes were not differentially expressed when nematodes were fed S. maltophilia JCMS compared to E. coli OP50. In summary, these results demonstrate the value of evaluating several conserved genetic pathways as a whole and provide evidence for specificity in the C. elegans innate immune response mounted against S. maltophilia.
MATERIALS AND METHODS
Nematode strains.
C. elegans strains containing the following alleles were obtained from the Caenorhabditis Genetics Center: LG (linkage group) I: daf-16(mu86), tol-1(nr2033), LG II: age-1(hx546), ire-1(v33), nsy-1(ag3), sma-6(wk7), LG III: daf-2(e1370), daf-2(e1368), sma-4(e729), sma-3(e491), tir-1(qd4), xbp-1(zc12), LG IV: htas-1 ins-7(ok1573), pmk-1(km25), sma-2(e502), LG V: akt-1(ok525), dbl-1(nk3), LG X: akt-2(ok393), pdk-1(sa680), sek-1(km4). Strain ZD350 agIs219 [T24B8.5p::GFP::unc-54-3′UTR + ttx-3p::GFP::unc-45-3′UTR] atf-7(qd137)III was provided by D. Pagano (MIT). N2 was used as the wild-type strain and was thawed yearly from a frozen stock for experimentation.
Bacterial strains and growth.
S. maltophilia JCMS was isolated by our laboratory from a culture of Mesorhabditis species nematodes found in soil samples from the Konza Prairie Biological Station in Manhattan, KS. Briefly, nematodes were isolated from soil cores, washed in sterile M9 buffer, and allowed to crawl on nematode growth medium (NGM) plates without any bacteria for 1 h. Nematodes were then moved to a plate seeded with E. coli OP50 for rearing. Bacteria that grew on the initial NGM plate were considered to be “nematode-associated bacteria.” Despite our efforts to ensure that JCMS was indeed associated with native soil nematodes, it is possible that this strain could have been present in the soil from which the nematodes were isolated or a laboratory contaminant. E. coli OP50 and OP50-GFP were obtained from the Caenorhabditis Genetics Center, S. maltophilia K279a was obtained from R. Ryan (University College Cork), S. maltophilia R551-3 was obtained from D. van der Lelie (Brookhaven National Laboratory), E. faecalis V583 was obtained from L. Hancock (Kansas State University), and P. aeruginosa PA14 was obtained from F. M. Ausubel (Harvard Medical School). Transformation of S. maltophilia strains was completed via the insertion of a mini-Tn7 expression cassette that expresses green fluorescent protein (GFP) (37), miniTn7KSGFP(pURR25), obtained from T. Ciche (Michigan State University), into the genome of each S. maltophilia strain. All bacterial strains were frozen at −80°C upon retrieval and were thawed regularly for use in experiments. S. maltophilia strains are naturally ampicillin (AMP) resistant and were streaked for colony isolation from a frozen stock onto Luria broth (LB) agar containing 100 μg/ml ampicillin to selectively prevent the growth of other bacterial contaminants. E. coli OP50 was streaked onto LB agar for colony isolation. For each bacterial strain, liquid LB was inoculated and shaken overnight at 32°C. Bacterial lawns used for survival were seeded onto NGM with the bacterial culture at log/lag phase and grown overnight at room temperature.
Nematode survival assays.
Nematodes were reared and synchronized as larval stage 4 (L4s) at 20°C on E. coli OP50 lawns. For survival analysis, 10 to 15 L4s were picked onto replicate lawns of bacteria and maintained at 25°C. The number of surviving nematodes was recorded daily, and death was determined by the lack of motion in response to prodding with a platinum wire pick. Nematodes were picked to new bacterial lawns for the first 5 to 6 days after the start of the experiment to separate them from their progeny. Dead nematodes were removed upon discovery. Sample sizes (number of nematodes) varied due to contamination and the removal of specimens that died via means other than the specified bacterial treatment, such as desiccation that occurs when nematodes leave the bacterial lawn and die at the plate edge. The infrequent presence of contamination was determined by observing bacterial lawn morphology, and contaminated replicates were discarded. Kaplan-Meier estimates of survival over time and determination of survival curve statistics using Cox proportional-hazard models were performed with R (R Foundation for Statistical Computing, Vienna, Austria). Survival curves can be statistically compared by using the log-rank or Cox proportional-hazard tests. Cox proportional-hazard models were used to test the effects of independent variables such as genotype and bacterium on the hazard, a dependent variable defined as the probability of dying at a given time (38). The model used for analysis is indicated in a footnote for each table, and the effect of the designated independent variable was considered significant if the P value was <0.05. Some models included a categorical variable that specified the date on which the experiment was completed. This categorical variable and the interaction between this variable and genotype or bacterial treatment were included in the model if they were found to be significant. Models were evaluated by testing for a nonzero slope and visualizing the Schoenfeld residuals (39). A nonzero slope is an indication of the violation of the proportional-hazard assumption, and models were fit to the data aiming to meet this assumption.
Bacterial load assays.
Bacterial load was determined by measuring CFU using methods modified from those reported in previous studies (40, 41). Synchronized L4s were fed non-GFP-expressing strains (except for E. coli due to the presence of ampicillin resistance) for 6 days on NGM plates. For each time point (30 min; 1 h; 6 h; 12 h; and days 1, 2, 4, and 6), three replicates of 7 to 10 nematodes were picked after exposure to E. coli OP50-GFP or S. maltophilia JCMS, R551-3, and K279a and fed non-GFP-expressing E. coli OP50 for 1 h to clear the intestinal lumen of nonadhering bacteria that we reasoned would be swept away during this feeding period. Only living nematodes were used, and the sample size varied due to the number of living individuals available at each time point. Nematodes were then placed onto unseeded plates of NGM containing 120 μg/ml doxycycline (NGM-doxycycline plates) for washing once with 25 mM levamisole-M9 (LM) buffer, twice with LM buffer with doxycycline (120 μg/ml), and twice with M9 buffer. Washed nematodes were then placed into a 1.7-ml microcentrifuge tube containing 50 μl of M9 buffer plus 1% Triton X-100 (Sigma-Aldrich) and homogenized by using a pestle motor. Crushed nematodes were diluted and plated onto LB agar containing 100 μg/ml ampicillin to select for the growth of adherent strains. Bacterial lawn CFU were quantified by plating five replicates of OP50, JCMS, R551-3, and K279a bacterial lawns onto LB agar containing 100 μg/ml ampicillin. For each replicate, 5 ml of M9 buffer was used to gather the bacterial lawn from an NGM plate (see “Bacterial strains and growth,” above).
Bacterial accumulation, distension, and pharyngeal pumping.
Synchronized L4s were fed GFP-containing bacteria for 11 days and maintained at 25°C. Prior to observation, nematodes were fed non-GFP-containing bacteria for 1 h of clearing. Nematodes were anesthetized (10 mM sodium azide) for observation daily at a ×1,000 magnification using a Zeiss Axioplan II microscope equipped with epifluorescence and differential interference contrast (DIC) optics. The GFP accumulation pattern (punctate or diffuse) was scored, and the degree of intestinal distension was quantified by using a micrometer. Only living nematodes were scored. The sample sizes varied depending upon the length of time that nematodes survived on the different bacteria. The GFP accumulation pattern throughout the nematode was observed, and distension was recorded for the anterior section of the intestine (most proximal to the pharynx) for a maximum of 30 worms per day for each bacterial treatment. To measure pharyngeal pumping rates, synchronized L4s were picked onto each treatment bacterium. Each treatment included three replicates of 10 to 15 nematodes. During the survival analysis, six nematodes from each treatment were randomly selected and observed daily. The number of pumps of the posterior bulb of the pharynx was counted for 30 s and extrapolated to 60-s intervals. The mean pumping rate is the average of the number of pharynx bulb pumps per minute observed on each day of the experiment.
Effect of bacterial viability and secretions on nematode killing.
Cultures of E. coli OP50 and S. maltophilia strains grown overnight were heat killed for 1 h at 92°C by using a Thermolyne DryBath. For antibiotic treatment (120 μg/ml doxycycline or 500 μg/ml ciprofloxacin), cultures grown overnight were shaken for 1 1/2 h at 32°C. The treated cultures were concentrated 20-fold as described previously (42) and used to seed NGM plates containing 100 μg/ml ampicillin to prevent the growth of E. coli OP50 bacteria transferred from plates used for nematode rearing. Prior to use, treated bacterial lawns were examined for colonies to determine whether any bacteria survived each treatment. To test whether S. maltophilia secretions might impact nematode viability, we performed a filter assay as previously described (43), using bacterial cultures grown on a 0.2-μM mixed-cellulose-ester filter (Millipore) and placed onto NGM plates at room temperature overnight. Prior to survival analysis, the filter containing the treatment strain was removed, and the plate was seeded with E. coli OP50 bacteria.
Germ line removal.
To assess whether the extended survival of age-1(hx546) mutants compared to that of wild type was dependent on the presence of the germ line, we used RNA interference (RNAi) to knock down cdc-25.1 as previously reported (44). Briefly, adult nematodes were picked onto RNAi plates (1 mM isopropyl-β-d-1-thiogalactopyranoside [IPTG] and 50 μg/ml ampicillin) seeded with E. coli HT115(DE3) bacteria expressing either double-strand RNA (dsRNA) or the empty vector (L4440) and allowed to lay eggs. Prior to seeding, each bacterial strain was shaken overnight at 32°C in LB containing ampicillin (50 μg/ml), and dsRNA expression was induced via shaking in 2 ml of fresh LB containing AMP in the presence of IPTG (1 mM) for 3 h. Treated adult nematodes were removed, and their eggs were allowed to develop into adults. Those without a proliferating germ line (Glp) were then picked onto NGM plates seeded with E. coli OP50 or S. maltophilia JCMS for survival analysis.
Reverse transcription-quantitative PCR.
Synchronized wild type and daf-2(e1368) mutant L4s were grown with E. coli OP50 or S. maltophilia JCMS at 25°C for 24 h, collected in M9 buffer, and lysed in TRIzol reagent (Life Technologies). RNA extraction and DNase treatment were completed by using the PureLink RNA minikit (Invitrogen) and on-column PureLink DNase treatment (Invitrogen). RNA quality was checked by visualizing 28S and 18S rRNA bands using gel electrophoresis and checking the ratios of absorbance at 260 nm/280 nm and at 260 nm/230 nm using a NanoDrop 2000 spectrophotometer. Intact RNA was used for cDNA synthesis using a SuperScript Vilo cDNA synthesis kit (Invitrogen). Reverse transcription-quantitative PCR (RT-qPCR) was completed by using 96-well plates and the CFX96 Touch real-time PCR detection system (Bio-Rad). Each amplification reaction was performed in triplicate, and three biological replicates were completed for each bacterium-nematode combination. Primer sequences for clec-85 (5′-CCTGTGCTACTCAATTTCCGC-3′ and 5′-CTGGAAGAAGCTCGGCTCAA-3′) and spp-1 (5′-GCCAATCCAGCTAACCCACT-3′ and 5′-AACGGCAACAGCATAGTCCA-3′) were designed by using NCBI Primer3. Primer sequences for csq-1 (5′-AACTGAGGTTCTGACCGAGAAG-3′ and 5′-TACTGGTCAAGCTCTGAGTCGTC-3′) were designed by using Geneious software (BioMatters Ltd.). All designed primer sequences were checked for specificity by using NCBI BLAST. Previously reported primer sequences for dod-22, K08D8.5, lys-7, and lys-1 were used (32). The efficiency of each primer pair was determined by using a standard curve on a pooled sample of cDNA. The efficiencies of the target and the reference gene csq-1 were determined to be approximately equal (45) and were assumed to be 100% during normalization and ΔCT quantification. The reference gene csq-1 was chosen due to its low level of variance between the bacterial treatments used. Differential expression was determined by comparing the 2−ΔCT values for biological replicates of the target gene in a daf-2 mutant background versus wild type on JCMS or OP50 and in wild-type nematodes grown on JCMS versus OP50 (46). Statistical significance (P < 0.05) was determined with Student's t test assuming equal variance.
Nucleotide sequence accession number.
The full-length S. maltophilia JCMS 16S rRNA gene sequence was deposited in GenBank with the accession number KF724885.
RESULTS
S. maltophilia JCMS kills C. elegans.
S. maltophilia strain JCMS was isolated in our laboratory (see Materials and Methods). We amplified the complete 16S rRNA gene, and sequence comparisons indicated that JCMS is more closely related to the clinical strain K279a than to the environmental isolate R551-3 (see Fig. S1 in the supplemental material). S. maltophilia strains R551-3, K279a, and JCMS are differentially virulent to C. elegans, with JCMS being the most severe (Fig. 1; see also Table S1 in the supplemental material), as seen by comparing the hazard ratios determined by the corresponding Cox proportional-hazard model (see Materials and Methods). Briefly, a hazard is the probability that an individual nematode dies at a given time. Therefore, in this case, the hazard ratio compared the relative hazards of two bacteria. Nematodes fed JCMS were ∼10 times more likely to die than those fed K279a and 4 times more likely to die than those fed R551-3 (see Table S1 in the supplemental material). Survival of nematodes grown on JCMS was not significantly different from that of nematodes grown on E. faecalis V583 and was significantly longer than that of nematodes grown on P. aeruginosa PA14 (Fig. 1; see also Table S1 in the supplemental material), both well-studied C. elegans and human pathogens. S. maltophilia K279a has been reported to kill C. elegans within 24 h (13). However, K279a was avirulent in our hands, as survival of nematodes grown on K279a was not significantly different from that with the standard C. elegans laboratory food E. coli OP50 (Fig. 1; see also Table S1 in the supplemental material). Similarly, and in contrast to data from a previous report (13), we observed that P. aeruginosa was significantly more virulent to C. elegans than was K279a (Fig. 1; see also Table S1 in the supplemental material). Although the source of this experimental discrepancy was unclear, this difference prompted us to test the effect of growing bacteria on different media. As previously demonstrated, P. aeruginosa PA14 was significantly more virulent to nematodes when grown on “fast-killing” peptone-glucose-sorbitol (PGS) medium (47), and E. coli OP50 was more virulent when grown on brain heart infusion (BHI) medium (40) (see Table S2 in the supplemental material). K279a was more virulent when grown on enriched BHI or PGS medium (see Table S2 in the supplemental material) but was not as virulent as previously reported (13). Intriguingly, nematodes were more likely to die from JCMS grown on NGM than from JCMS grown on PGS or BHI medium (see Table S2 in the supplemental material). While the medium-dependent difference in the tolerance of C. elegans to JCMS was interesting, the mechanism of nematode killing was not explored, as BHI and PGS media did not cause an increase in bacterial virulence, as was observed for the other S. maltophilia strains (see Table S2 in the supplemental material). In summary, JCMS is the most virulent S. maltophilia strain in this study, and neither K279a nor OP50 was virulent.
FIG 1.
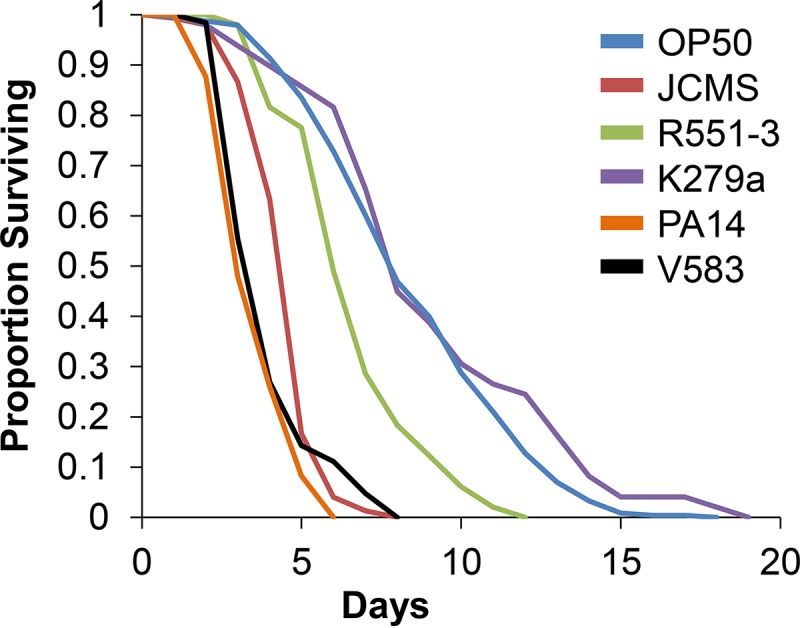
JCMS is the most virulent S. maltophilia strain. Shown is survival of wild-type nematodes fed S. maltophilia JCMS, S. maltophilia R551-3, S. maltophilia K279a, E. coli OP50, E. faecalis V583, or P. aeruginosa PA14. Results plotted are the proportions of surviving nematodes determined by using Kaplan-Meier estimates generated in R for at least three replicate samples of 10 to 15 nematodes. Sample sizes and P values from the application of Cox proportional-hazard models are included in Table S1 in the supplemental material. Survival of nematodes fed PA14, K279a, and R551-3 was significantly different (P < 0.05) from survival of nematodes fed JCMS. JCMS was more virulent than OP50 and K279a, and PA14 was more virulent than JCMS. Survival of nematodes fed JCMS was not significantly different from that of nematodes fed V583 (P = 0.0663).
S. maltophilia JCMS accumulates in the intestine.
Given the diverse levels of virulence displayed by the different S. maltophilia strains, we sought to determine whether the whole-nematode bacterial load was related to pathogenicity. In order to quantify the bacterial load, we measured CFU from crushed nematodes exposed to E. coli OP50-GFP (used instead of OP50 because it is ampicillin resistant) (see Materials and Methods) or S. maltophilia JCMS, K279a, or R551-3. Nematodes exposed to JCMS had 10-fold more CFU than any other strain tested at 30 min (Fig. 2A). The bacterial load in nematodes grown on JCMS increased exponentially after 12 h and leveled off after 4 days. Thus, the whole-nematode bacterial load correlates with the degree of pathogenic effect for all the bacterial strains tested. It was possible that the differences in bacterial loads could be caused by differences in the densities of bacteria in the lawns of the different strains; however, the number of CFU in each lawn was not correlated with S. maltophilia virulence (Fig. 2B). C. elegans feeds by the pharynx pumping bacteria from the mouth into the intestine. It is possible that the observed difference in the bacterial load was related to feeding behavior, as a rapid uptake of bacterial cells could cause an increase in the number of CFU. To address this question, we measured the pharyngeal pumping rates for nematodes fed each bacterial strain daily until death. Since pumping rates are known to decline with age (48), we included only data from the first 4 days. The pumping rate for nematodes fed JCMS was not substantially higher on any of the days observed (Fig. 2C). Thus, bacterial lawn density and nematode bacterial uptake do not correlate with pathogenicity or contribute to the observed strain-specific differences in bacterial loads.
FIG 2.

S. maltophilia JCMS persists in nematodes and causes intestinal distension. Shown are mean bacterial loads per nematode (A), mean bacterial lawn densities (B), pharynx pumping rates (C), and intestinal lumen distention (D) for synchronized wild-type nematodes fed E. coli OP50 or OP50-GFP or S. maltophilia JCMS, R551-3, or K279a. (A) Mean log CFU per nematode for nematodes cleared of nonadherent bacteria for 1 h on OP50 after feeding on the indicated bacterial strain. The inset shows an expanded view of the time points from the first day of feeding (n = 24 for day 6 of growth on JCMS and n = 30 for all other days and bacteria). (B) Mean log CFU for bacterial lawns (five replicates) of each indicated bacterial strain. Lawns of R551-3 had significantly more bacteria than did lawns of JCMS and OP50. OP50 lawns were significantly less dense than K279a and JCMS lawns. Statistical significance (P < 0.05) was determined with Student's t test, assuming equal variance. (C) Mean pumping rate (pharynx pumps per minute) for six nematodes picked at random per indicated bacterial treatment on days 1 to 4. (D) Adult worms were anesthetized for observation daily after exposure to each indicated bacterial strain. The width of the gut in the anterior region of each worm was scored for 6 to 11 days depending on nematode survival (n = 51 for growth on JCMS, n = 63 for growth on R551-3, n = 69 for growth on K279a, and n = 76 for growth on OP50). All error bars indicate standard errors of the means.
In order to further investigate the differences in the bacterial loads and pathogenicities of the S. maltophilia strains, we sought to determine where bacteria were localized within the nematode and whether localization was also correlated with pathogenicity. To better visualize and track bacteria within the C. elegans intestine, we introduced GFP into each S. maltophilia strain (JCMS, K279a, and R551-3). Integration of GFP did not cause a significant difference in nematode survival on these S. maltophilia strains (see Table S3 in the supplemental material). Bacterial pathogenicity was previously correlated with bacterial accumulation within the C. elegans intestine and distension of the intestinal lumen (40, 49). Thus, the extent of intestinal lumen distension (Fig. 2D) was measured when the pattern of GFP accumulation was observed (Fig. 3). We focused on the anterior portion of the intestine since it has been shown to be sensitive to the effects of pathogenic bacteria (50, 51). Intestinal distention was observed in nematodes fed each S. maltophilia strain but occurred earlier and to a greater extent for those fed the more pathogenic strains JCMS and R551-3 (Fig. 2D). Nematodes grown on E. coli OP50-GFP accumulated GFP-expressing bacteria within the intestinal lumen that had primarily a “punctate” pattern (Fig. 3B and C; see also Fig. S2A in the supplemental material). This pattern of accumulation appears to illuminate intact bacterial cells. Worms fed JCMS-GFP accumulated bacteria primarily in a “diffuse” pattern that appeared to involve higher numbers of tightly packed bacteria, some of which might have been lysed, resulting in the release of GFP into the intestinal lumen (Fig. 3D to F; see also Fig. S2B in the supplemental material). Efforts to characterize the exact cellular mechanism responsible for the diffuse GFP accumulation pattern have been inconclusive. However, the diffuse pattern of bacterial GFP accumulation was characteristic of all S. maltophilia strains and roughly correlated with the degree of virulence (see Fig. S2B in the supplemental material). Few GFP-labeled S. maltophilia cells were observed in tissues outside the intestinal lumen and not until nematodes were older and deteriorating, suggesting that S. maltophilia pathogenesis initially involved intestinal colonization, which became systemic after prolonged exposure. The extent of this effect, and the degree of intestinal distension, appeared to be generally consistent for all sections of the nematode (data not shown), signifying that the accumulation and distention patterns were representative.
FIG 3.

Accumulation of GFP-expressing bacteria in nematode intestines. Shown are micrographs of wild-type nematodes grown on either E. coli OP50-GFP (A to C) or S. maltophilia JCMS-GFP (D to F). The anterior is to the left in all panels. (A and D) Overlay of DIC and fluorescent images on day 6 at a ×10 magnification; bars, 100 μm. (B and E) DIC images of the anterior intestines of nematodes fed OP50-GFP (B) and JCMS-GFP (E) on day 4 at a ×1,000 magnification. Bars, 10 μm. Arrows on the DIC images indicate the intestinal wall. Both anterior intestines were distended, but JCMS-GFP-fed animals appear to contain more bacteria. (C and F) Fluorescence images of the same nematodes shown in panels B and E, respectively. Bars, 10 μm. The OP50-GFP-fed nematode shown in panel C displays the punctate pattern of accumulation of GFP-expressing bacteria, while the JCMS-GFP-fed nematode in panel F shows the diffuse pattern.
Intriguingly, whole-nematode bacterial loads increased more quickly than the GFP-labeled bacterial loads in the intestine and did not coordinate with intestinal distension. When the accumulation of GFP-expressing bacteria in all parts of the intestine was measured, between 7 and 17% of nematodes had a GFP signal on day 1, depending upon the strain (see Fig. S2C in the supplemental material). These data (see Fig. S2C in the supplemental material) correlate better with whole-nematode CFU (Fig. 2A), but differences remain. One possible explanation for these differences is that nematodes in the accumulation experiments were cleared of nonadherent bacteria via feeding on the same non-GFP-bearing strain, while in the bacterial load assay, nematodes were cleared of nonadherent bacteria on E. coli. To investigate this experimental difference, we compared accumulations of JCMS-GFP within nematodes after clearing on non-GFP-bearing OP50 or JCMS. JCMS-GFP initially accumulated to a greater extent when cleared on E. coli OP50 (see Fig. S2D in the supplemental material). Thus, JCMS is better at dislodging itself than OP50. This bacterium-specific difference in the ability to clear nonadherent bacteria may have contributed to the discrepancy between the GFP accumulation and bacterial load experiments. However, it should be noted that the measurements of nematode CFU and GFP accumulation within the nematode quantify two distinct aspects of S. maltophilia pathogenicity. A recent report demonstrated that the presence of a GFP signal and/or intestinal distension does not indicate that an infection has been established, suggesting that bacterial load and intestinal accumulation are not necessarily coupled (52). Thus, although GFP accumulation, distension, and bacterial load should intuitively be correlated, these phenomena likely represent different aspects of S. maltophilia pathogenicity.
Virulence of S. maltophilia JCMS is not mediated by a toxin and requires live bacteria.
The ability of S. maltophilia JCMS to colonize and accumulate within C. elegans (Fig. 2A) suggested that living bacteria were involved in S. maltophilia virulence. To test the requirement of living bacteria for virulence, we used heat to kill E. coli OP50 and each S. maltophilia strain. Nematodes fed heat-killed JCMS survived longer than did nematodes fed living bacteria, while survival of nematodes fed heat-killed R551-3 and K279a bacteria was not statistically different from survival of nematodes fed living bacteria of the same strains (Fig. 4A; see also Table S4 in the supplemental material). Surprisingly, heat-killed OP50 bacteria were more hazardous to nematodes than living bacteria. This effect is not likely due to a lack of nutrition, as these bacteria were concentrated in the same manner as the S. maltophilia strains. While the curves shown in Fig. 4A appear to indicate that heat-killed R551-3 and K279a bacteria were also more hazardous to nematodes, these differences were not statistically significant. These data suggest that a heat-stable compound might contribute to the virulence of the mildly pathogenic S. maltophilia strain R551-3. Additionally, JCMS-mediated killing appeared to be distinct from that of other S. maltophilia isolates in that virulence clearly required these bacteria to be alive. To confirm these data and determine if a heat-labile toxin could be involved in S. maltophilia JCMS virulence, we used the antibiotics doxycycline and ciprofloxacin to treat E. coli and each S. maltophilia strain. Both antibiotic treatments were optimized to remove all proliferating cells. As expected with antibiotic treatment (53), nematodes fed doxycycline-treated OP50 bacteria survived significantly longer than did nematodes fed living bacteria (Fig. 4B; see also Table S4 in the supplemental material). Nematodes fed JCMS and K279a bacteria treated with either antibiotic and R551-3 bacteria treated with ciprofloxacin also survived longer than did nematodes fed the respective living bacteria (Fig. 4B and C; see also Table S4 in the supplemental material). Surprisingly, survival of nematodes fed OP50 and R551-3 showed a treatment-specific interaction with each antibiotic, as survival of nematodes fed doxycycline-treated R551-3 or ciprofloxacin-treated OP50 bacteria was significantly shorter than survival of nematodes fed the respective living bacteria. This treatment-specific effect suggests that either antibiotic can induce a toxin that is detrimental to the nematode. However, taken together, these data indicate that proliferating bacteria and/or a factor associated with bacterial growth is required for S. maltophilia virulence. Moreover, since neither antibiotic treatment should denature proteins, we conclude that a protein toxin does not play a significant role in JCMS and likely also R551-3 pathogenicity. We also attempted to use UV treatment as another means to kill bacteria without denaturing proteins. However, we were unable to find a UV dose that completely killed all cells (data not shown).
FIG 4.

Virulence of S. maltophilia JCMS is not mediated by a toxin and requires living bacteria. (A to C) Survival of wild-type nematodes grown on untreated (UNT) or heat-killed (HK) bacteria (A) or of nematodes grown on bacteria treated with the antibiotic doxycycline (DT) (B) or ciprofloxacin (CT) (C). (D) Survival of wild-type nematodes grown on secretion (filtrate)-treated E. coli OP50 bacteria. Bacterial secretions were obtained from E. coli OP50, S. maltophilia JCMS, S. maltophilia R551-3, or S. maltophilia K279a. Results plotted are the proportions of surviving nematodes determined by using Kaplan-Meier estimates generated in R for at least three replicate samples of 10 to 15 nematodes. Sample sizes and P values from the application of Cox proportional-hazard models are included in Tables S4 and S5 in the supplemental material. Living JCMS bacteria were significantly (P < 0.05) more virulent than heat-killed and antibiotic-treated JCMS bacteria, while the virulence of OP50 bacteria treated with JCMS secretions (filtrate) was not significantly different from that of OP50 bacteria treated with OP50 secretions (P = 0.401).
We next used a filter assay to test for the contribution of S. maltophilia secretions to pathogenicity. We found that treatment of OP50 with secretions from any S. maltophilia strain did not cause a decrease in survival compared to treatment with OP50 secretions (Fig. 4D; see also Table S5 in the supplemental material). In fact, we observed a significant extension in survival time for nematodes fed OP50 bacteria treated with K279a secretions. However, it is possible that the filter bound the toxic secretion and/or that the molecule was too large to pass through the filter. While this issue could be addressed through the use of a nonfiltered bacterium-free supernatant obtained by centrifugation of bacterial cultures, we were unable to remove all viable S. maltophilia cells from the supernatant (data not shown). Therefore, we cannot rule out the role of a large and/or filter-bound toxin. However, taken together, these data strongly suggest that living bacteria, rather than a toxin, play a larger role in S. maltophilia nematode pathogenesis.
DAF-2/16 insulin-like signaling pathway component mutants are not resistant to S. maltophilia JCMS.
C. elegans daf-2 mutants display increased survival and increased pathogen resistance to practically all bacteria tested to date (14, 27, 31). To determine whether this was the case for S. maltophilia JCMS, we analyzed the survival of various daf-2 pathway mutants on JCMS and E. coli OP50 as a control. Compared to wild type, daf-2(e1368) and daf-2(e1370) mutants were long-lived on OP50, and neither mutant allele conferred extended survival on S. maltophilia JCMS (Fig. 5A and Table 1). Specifically, daf-2(e1370) mutants were more susceptible to S. maltophilia JCMS, while survival of daf-2(e1368) mutants was not significantly different from that of wild type. While we observed increased susceptibility for daf-2(e1370) mutants, these mutants also displayed pleiotropic effects (28) such as reduced brood size and abnormal development, which might have contributed to the observed phenotype for nematodes fed JCMS. Mutants of other DAF-2/16 pathway components, including the insulin-like ligand ins-7 and a serine/threonine kinase ortholog, akt-1, had extended survival times compared to that of wild type grown on OP50 but not on JCMS, with ins-7 mutants being more susceptible (Fig. 5B and Table 1). Intriguingly, mutants of another serine/threonine kinase ortholog, akt-2, were significantly longer-lived than wild type grown on JCMS, suggesting a role that is similar to that for growth on OP50. The differential roles of akt-1 and akt-2 in growth on JCMS may be linked to their differential regulation of antimicrobial gene expression (31). Contrary to data from previous reports (27, 31), we observed that age-1 mutants were not significantly longer-lived than wild type on OP50. This anomaly led us to ask whether previously reported effects of sterility on life span and pathogen resistance (54, 55) might be responsible. We used RNAi of cdc-25.1 to remove the germ line (44) in wild-type nematodes and age-1 mutants and found that age-1;cdc-25.1(RNAi) nematodes survived significantly longer than did cdc-25.1(RNAi) nematodes grown on OP50 (Fig. 5C and Table 1). There was not a significant difference between wild type and age-1 mutants fed the RNAi vector only, confirming that the knockdown of cdc-25.1 was the cause of the increased life span. Furthermore, there was not a significant difference in survival between age-1;cdc-25.1(RNAi) and cdc-25.1(RNAi) nematodes on JCMS. When the germ line-dependent effects of age-1 loss of function are taken into account, like most other DAF-2/16 pathway components, age-1 mutants were not resistant to JCMS. Finally, survival of forkhead box O (FOXO) homolog daf-16 mutants was not significantly different from survival of wild type fed JCMS or OP50, which is consistent with previously reported results (27, 34, 56). In summary, all tested components of the DAF-2/16 pathway except daf-16 were similarly involved in the response to OP50. We observed that most of the pathway mutants were not significantly different from wild type fed S. maltophilia JCMS, suggesting that DAF-2/16 signaling plays little or no role in the C. elegans defense response against these bacteria. Furthermore, the usually pathogen-resistant daf-2(e1370) and ins-7 mutants were slightly more susceptible to JCMS than were wild-type nematodes, indicating that their role in the response to JCMS is different than that for other bacterial pathogens.
FIG 5.

Survival of representative DAF-2/16 insulin-like signaling pathway mutants. (A) Survival of wild-type nematodes and daf-2(e1368) and daf-2(e1370) mutants grown on E. coli OP50 and S. maltophilia JCMS. (B) Survival of wild-type nematodes and ins-7(ok1573) and akt-1(ok525) mutants grown on E. coli OP50 and S. maltophilia JCMS. (C) Survival of adult nematodes without a proliferating germ line [cdc-25.1(RNAi)] and age-1;cdc-25.1(RNAi) mutants grown on E. coli OP50 and S. maltophilia JCMS. Results plotted are the proportions of surviving nematodes determined by using Kaplan-Meier estimates generated in R for at least three replicate samples of 10 to 15 nematodes. Sample sizes and P values from the application of Cox proportional-hazard models are included in Table 1. Survival of daf-2(e1370) and ins-7 mutants fed JCMS was significantly different from that of the wild type, while survival of the daf-2(e1368) and akt-1 mutants was not significantly different. Survival of the daf-2, ins-7, and akt-1 mutants on OP50 was significantly extended. Survival of age-1;cdc-25.1(RNAi) mutants was significantly longer than that of cdc-25.1(RNAi) nematodes grown on OP50 but not on JCMS. P values of <0.05 were considered significant.
TABLE 1.
C. elegans defense pathway mutant responses
| Genotype |
S. maltophilia JCMS |
E. coli OP50 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean survival time (days) | SE for survival time (days) | No. of nematodes tested | Hazard ratio | P valueb | Mean survival time (days) | SE for survival time (days) | No. of nematodes tested | Hazard ratio | P valueb | |
| Wild type | 4.69 | 0.088 | 150 | NAa | NA | 8.53 | 0.187 | 244 | NA | NA |
| cdc-25.1(RNAi) | 6.97 | 0.163 | 33 | NA | NA | 9.48 | 0.624 | 29 | NA | NA |
| DAF-2/16 | ||||||||||
| ins-7 | 4.1 | 0.139 | 58 | 1.609 | 0.0023 | 11.04 | 0.426 | 53 | 0.210 | 3.66E−15 |
| daf-2(e1368) | 4.88 | 0.187 | 50 | 0.794 | 0.161 | 10.84 | 0.515 | 50 | 0.108 | 2.22E−16 |
| daf-2(e1370) | 3.26 | 0.167 | 74 | 2.163 | 8.6E−08 | 9.59 | 0.585 | 56 | 0.193 | 2.96E−13 |
| akt-1 | 4.54 | 0.121 | 59 | 1.19 | 0.262 | 12.58 | 0.169 | 60 | 0.0741 | <2E−16 |
| akt-2 | 5.36 | 0.164 | 59 | 0.552 | 0.00016 | 12.13 | 0.278 | 47 | 0.195 | <2E−16 |
| age-1 | 5.28 | 0.2 | 210 | 0.594 | 0.00112 | 8.6 | 0.194 | 278 | 0.835 | 0.164 |
| age-1;cdc-25.1(RNAi) | 7.3 | 0.163 | 33 | 1.229 | 0.393* | 11.77 | 1.119 | 31 | 0.304 | 0.0014c* |
| daf-16 | 4.38 | 0.128 | 50 | 1.22 | 0.153 | 8.19 | 0.309 | 58 | 1.22 | 0.175 |
| UPR | ||||||||||
| xbp-1 | 3.11 | 0.17 | 57 | 2.747 | 1.9E−10 | 5.98 | 0.478 | 58 | 1.68 | 0.00042 |
| ire-1 | 3.14 | 0.13 | 50 | 5.806 | <2E−16 | 5.74 | 0.298 | 54 | 2.88 | 2.26E−11 |
| p38 MAPK | ||||||||||
| nsy-1 | 2.7 | 0.076 | 60 | 31.296 | <2E−16 | 2.43 | 0.208 | 60 | 13.9 | <2E−16 |
| tir-1 | 2.7 | 0.089 | 57 | 11.317 | <2E−16 | 3.56 | 0.194 | 56 | 8.03 | <2E−16 |
| sek-1 | 1.13 | 0.044 | 60 | 227.79 | <2E−16 | 3.28 | 0.228 | 57 | 7.18 | <2E−16 |
| pmk-1 | 2.58 | 0.072 | 60 | 16.492 | <2E−16 | 8.3 | 0.404 | 60 | 0.948 | 0.711 |
| atf-7 | 3.02 | 0.091 | 57 | 6.717 | <2E−16 | 6.43 | 0.306 | 82 | 1.82 | 4.07E−06 |
| DBL-1/TGF-β | ||||||||||
| dbl-1 | 3.0 | 0.138 | 59 | 3.841 | <2E−16 | 9.8 | 0.374 | 59 | 0.543 | 6.98E−05 |
| sma-6 | 3.54 | 0.125 | 48 | 3.297 | 1.2E−11 | 9.01 | 0.452 | 46 | 0.647 | 8.63E−03 |
| sma-2 | 3.39 | 0.157 | 51 | 2.953 | 1.4E−10 | 5.5 | 0.216 | 59 | 3.67 | <2E−16 |
| sma-3 | 3.76 | 0.137 | 49 | 2.388 | 2.5E−07 | 6.52 | 0.377 | 50 | 1.51 | 8.74E−03 |
| sma-4 | 3.65 | 0.128 | 46 | 3.073 | 4.1E−10 | 6.68 | 0.457 | 42 | 1.52 | 1.35E−02 |
| TLR | ||||||||||
| tol-1 | 5.07 | 0.144 | 60 | 0.729 | 0.041 | 8.38 | 0.341 | 60 | 0.878 | 0.377 |
NA, not applicable.
P values of <0.05 were considered significant and are given for the survival predictor of treatment (mutant genotype) for Cox proportional-hazard models in R. *, P value for age-1;cdc-25.1(RNAi) mutants versus the cdc-25.1(RNAi) mutant.
The date of experimentation was observed to have a significant effect and was included in this model.
The degree to which mutations of DAF-2/16 pathway components affect survival was inferred from the value of the mutant-to-wild-type hazard ratio (from the corresponding Cox proportional-hazard model) (see Materials and Methods). Hazard ratio values near 1 reveal that the mutant hazard is not different from that of wild type and suggest that the mutated gene is not involved in the response to the given bacterium, while values that deviate from 1 suggest involvement. Values of >1 indicate that the mutants have a shorter life span than that of wild-type nematodes, and values of <1 indicate that the mutants have a longer life span than that of wild type. We did not consider mutant hazard ratios that were not significantly different from that of the wild type in these analyses of differential degrees of mutant effects. The hazard ratios for DAF-2/16 pathway mutants fed OP50 were all less than, and deviated the most, from 1 (Table 1), suggesting that this pathway is key to the nematode response to OP50. We observed significantly extended survival on JCMS for only akt-2 mutants, which are about one-half as likely to die as wild-type nematodes. In contrast, akt-2 mutants are nearly one-fifth as likely to die as wild-type nematodes fed OP50, indicating a smaller role in survival of nematodes fed JCMS. In fact, the hazard ratios of all DAF-2/16 pathway mutants deviated further from 1 on OP50 than on JCMS, suggesting that this pathway plays a greater role in survival of nematodes fed OP50. Furthermore, the hazard ratios of DAF-2/16 pathway mutants fed JCMS deviated the least from 1 compared to the other defense pathway mutants, suggesting that this pathway as a whole is more expendable.
Conserved role for other C. elegans defense pathways in combating S. maltophilia JCMS.
Nematodes with mutations affecting the UPR, p38 MAPK, and DBL-1/TGF-β pathway components ire-1, pmk-1, and dbl-1 had increased susceptibility on JCMS (Fig. 6 and Table 1). These results suggested an involvement of these C. elegans defense pathways and were bolstered by our analysis of mutants affecting multiple genes within each pathway (Fig. 6 and Table 1; see also Fig. S3 and S4 in the supplemental material). Mutants of the p38 MAPK pathway signaling components sek-1, nsy-1, tir-1, and atf-7 were each susceptible to OP50 and JCMS, but pmk-1 mutants were susceptible to JCMS only (Fig. 6A and Table 1; see also Fig. S3 in the supplemental material). These results agreed with previously reported data in that pmk-1 mutants were susceptible to pathogenic P. aeruginosa and that the absence of pmk-1 in nematodes fed E. coli is irrelevant (34). The hazard ratios for p38 MAPK mutants were higher than those for other pathway mutants for growth on both JCMS and OP50 (Table 1). In addition, the hazard ratio for a given mutant fed JCMS was higher than that for OP50 for these genes. For example, the hazard ratios for sek-1 and nsy-1 for growth on JCMS were 228 and 31.3, while for growth on OP50, they were 7.18 and 13.9 (Table 1). Thus, the loss of the p38 MAPK pathway is the most detrimental for survival on both bacteria, with the loss being more severe for growth on JCMS.
FIG 6.

p38 MAPK, DBL-1/TGF-β, and UPR defense pathway mutants. (A) Survival of wild-type nematodes and pmk-1(km25), sek-1(km4), and nsy-1(ag3) mutants grown on E. coli OP50 and S. maltophilia JCMS. (B) Survival of wild-type nematodes and dbl-1(nk3) and sma-6(wk7) mutants grown on E. coli OP50 and S. maltophilia JCMS. (C) Survival of wild-type nematodes and xbp-1(zc12) and ire-1(v33) mutants grown on E. coli OP50 and S. maltophilia JCMS. Results plotted are the proportions of surviving nematodes determined by using Kaplan-Meier estimates generated in R for at least three replicate samples of 10 to 15 nematodes. Sample sizes and P values from the application of Cox proportional-hazard models are included in Table 1. All pathway mutants had significantly decreased survival during growth on JCMS. Survival for pmk-1 mutants was not significantly different from that of wild type, and the dbl-1 and sma-6 mutants had significantly extended survival during growth on OP50. All other immune pathway mutants were significantly susceptible to OP50. P values of <0.05 were considered significant.
Mutations in the DBL-1/TGF-β pathway components that form the SMAD complex (sma-2, sma-3, and sma-4) also increased susceptibility to both OP50 and JCMS (Table 1; see also Fig. S4 in the supplemental material). Thus, the SMAD complex components have similar roles in growth on JCMS and OP50. However, mutants of the upstream components dbl-1 and sma-6 were specifically susceptible to JCMS and long-lived when grown on OP50 (Fig. 6B and Table 1). The hazard ratios of these mutants were closer to 1 for growth on OP50 than on JCMS. Thus, it appears that these components are necessary for the response to JCMS and, perhaps, less important for the response to OP50. In fact, with the exception of sma-2, the hazard ratios for DBL-1/TGF-β pathway mutants deviated more from 1 for growth on JCMS. Comparatively, the deviations from 1 for this pathway are not as great as those for the p38 MAPK pathway. Thus, the loss of the DBL-1/TGFβ pathway as a whole is only mildly detrimental to nematodes fed JCMS.
UPR pathway components were also found to play similar roles in the response to JCMS and OP50. Both ire-1 and xbp-1 mutants had significantly shorter life spans than those of wild-type nematodes during growth on both JCMS and OP50 (Fig. 6C and Table 1). Similar to the p38 MAPK and DBL-1/TGFβ pathways, the hazard ratios for UPR pathway mutants were higher for growth on JCMS. In addition, the hazard ratio of ire-1 mutants was higher than that of xbp-1 mutants grown on JCMS and OP50, suggesting a greater role for this serine/threonine protein kinase in both bacteria. Finally, survival of the Toll-like receptor (TLR) tol-1 mutant was not significantly different from that of wild type grown on OP50 but was slightly increased with growth on JCMS (Table 1). The resistance of tol-1 mutants to JCMS was only marginally significant, with the hazard ratio indicating little involvement.
Differential regulation of immune effector genes in nematodes grown on S. maltophilia JCMS.
We used RT-qPCR to investigate the expression of several innate immune effector genes in nematodes fed S. maltophilia JCMS and E. coli OP50. We chose to focus on clec-85, lys-1, lys-7, dod-22, K08D8.5, and spp-1 due to their demonstrated regulation by the p38 MAPK, DBL-1/TGF-β, and/or DAF-2/16 signaling pathway (32, 57). These genes were also of interest because they are expressed in the intestine and pharynx, sites of pathogen contact (32). Our analysis of DAF-2/16 pathway mutant survival suggested that these genes do not play a role in the C. elegans innate immune response to S. maltophilia JCMS. Accordingly, we sought to determine the dependence of each effector gene on DAF-2/16 signaling by comparing expression levels in wild type and daf-2 mutants. Of the putative DAF-2/16-regulated genes chosen, only clec-85, lys-7, and dod-22 were significantly differentially expressed between daf-2 and wild type for growth on JCMS (Fig. 7A). Consistent with data from previous work (32, 57), clec-85 and lys-7 were upregulated and dod-22 was downregulated in daf-2 mutants grown on OP50. However, the expression of spp-1 and K08D8.5 in the daf-2 background was not significantly different from that of wild type fed either bacterium, and these genes were not evaluated further since their expression trends did not agree with data from previous studies (32, 57). Additionally, as expected for a daf-2-independent gene, the expression of lys-1 in the daf-2 background was not significantly different from that for wild type with either bacterium. The expressions of lys-1, lys-7, dod-22, and clec-85 are known to be upregulated when C. elegans comes into contact with pathogenic bacteria (26, 32). Thus, the expression of these immune effector genes is typically expected to be upregulated during growth on pathogenic bacteria such as JCMS versus a less pathogenic or nonpathogenic bacterium like OP50. However, since clec-85, dod-22, and lys-7 are regulated by DAF-2/16 signaling and DAF-2/16 signaling was not primarily involved in the JCMS response, we hypothesized that these effectors would not be differentially expressed. As expected, only lys-1 was significantly upregulated during growth on JCMS versus OP50 (Fig. 7B). These results support a role for lys-1 and the upstream pathways that regulate lys-1 in the C. elegans defense response to JCMS, while DAF-2/16-dependent effector genes have little or no role in the response to these bacteria.
FIG 7.

daf-2-regulated genes are not regulated in nematodes grown on S. maltophilia JCMS. RNA was extracted from synchronized wild-type nematodes and daf-2(e1368) mutants that were grown on E. coli OP50 or S. maltophilia JCMS for 24 h. Differential expression was determined by comparing biological replicates of the target gene in daf-2(e1368) mutants versus wild-type (WT) nematodes (control) grown on OP50 or JCMS (A) and wild-type nematodes grown on JCMS versus OP50 (control) (B). Fold changes are shown in reference to expression levels in the control sample (wild type grown on JCMS and/or OP50). Statistical significance (P < 0.05) was determined with Student's t test, assuming equal variance. The effector genes dod-22, lys-7, and clec-85 are significantly regulated by daf-2 on JCMS. Only clec-85 was significantly regulated by daf-2 when nematodes were grown on OP50. lys-1 was marginally significantly (P = 0.058) upregulated during growth on JCMS versus OP50 and was not significantly regulated by daf-2 during growth on either bacterium.
DISCUSSION
S. maltophilia JCMS is a C. elegans bacterial pathogen.
We isolated a strain of the emerging nosocomial pathogen S. maltophilia that kills C. elegans. Like E. faecalis, P. aeruginosa, and S. marcescens (20, 40), S. maltophilia JCMS accumulates in the gut and causes intestinal distention. In contrast to P. aeruginosa (58) and/or B. thuringiensis (24), JCMS virulence requires the presence of proliferating bacteria and does not involve a toxin. Furthermore, JCMS causes a diffuse GFP accumulation pattern that may represent a bacterial lysate and/or the presence of an intestinal biofilm. Biofilm formation is thought to be a survival mechanism of clinically relevant organisms such as E. faecalis (reviewed in reference 59). E. faecalis also establishes a proliferating infection within the C. elegans intestine (40), and survival on S. maltophilia JCMS was indistinguishable from survival on E. faecalis V583 (Fig. 1 and Table 1). Also, like S. maltophilia JCMS, E. faecalis does not kill nematodes when treated with antibiotics (40). These data suggest that the modes of action utilized by S. maltophilia JCMS and E. faecalis could be similar, possibly involving opportunistic proliferation and biofilm formation.
Comparison of the effects of S. maltophilia JCMS, R551-3, and K279a on C. elegans revealed several characteristics that likely underlie their differences in virulence. For example, differences in the onset of bacterial accumulation suggest distinct modes of action. Additionally, unlike JCMS, R551-3 virulence did not always require that these bacteria be alive, as heat killing does not change nematode survival, while antibiotic treatment produces differential effects (Fig. 4; see also Table S5 in the supplemental material). Curiously, our data suggest that unlike the other S. maltophilia strains that we tested, R551-3 produces a heat-resistant, ciprofloxacin-sensitive toxin that is induced by doxycycline. If true, the mode of action of R551-3 might be similar to that of P. aeruginosa, which, when heat treated, is as virulent as living bacteria on fast-killing medium (47) and employs heat-stable diffusible toxins called phenazines (58). However, unlike for R551-3, antibiotic treatment of P. aeruginosa renders these bacteria less pathogenic to nematodes (47). Thus, it appears that P. aeruginosa and S. maltophilia R551-3 do not act similarly in all respects.
The DAF-2/16 pathway plays a relatively small role in defense against S. maltophilia JCMS.
Binding of the insulin/insulin growth factor (IGF) receptor ortholog DAF-2 negatively regulates the transcription factor DAF-16/FOXO. When the functions of DAF-2 or other members of the DAF-2/16 pathway are disrupted, DAF-16 is free to enter the nucleus to promote the expression of numerous genes, including those involved in stress and defense responses (57). Here, we show that S. maltophilia JCMS evades the downstream effects induced by disrupting the functions of members of the DAF-2/16 pathway. Thus, in C. elegans, not all of the conserved innate immune pathways function to defend against all bacterial pathogens. Specifically, it appears that JCMS evades downstream effects that include the activation of daf-16-regulated general stress effectors such as sod-3 and antimicrobial genes such as lys-7 (57). Such genes are considered to be innate immune effectors, as they are induced by bacterial pathogens such as P. aeruginosa, S. marcescens, and S. aureus (32). Thus, our data suggest that the activation of these genes is insufficient protection against JCMS and that JCMS causes cellular stress that is distinct from that caused by other bacteria that require the action of the DAF-2/16 pathway. Our observation that daf-2-regulated effector genes were not differentially expressed between JCMS and avirulent E. coli strain OP50 (Fig. 7) also supports this notion. Taken together, these results support a response to JCMS that is independent of the DAF-2/16 signaling pathway.
However, survival of ins-7 and akt-2 mutants on S. maltophilia JCMS revealed the possibility of a small, but perhaps significant, role for these genes. Specifically, our data suggest that ins-7 functions to protect nematodes from JCMS. However, the role of ins-7 appears to be relatively minor, as the hazard ratio was the lowest among those for the significantly susceptible innate immune pathway mutants fed JCMS. On the other hand, the removal of akt-2 in nematodes fed JCMS appears to be beneficial. This result may be attributed to the dual role of akt-2 in pathogen resistance and longevity (31). As age-1 also has a dual role (31), it is possible that this resistance is dependent on the germ line, as we demonstrated for age-1 mutants. In fact, akt-2 and age-1 mutants with an intact germ line had similar survival phenotypes and hazard ratios (Table 1). However, like ins-7 mutants, the mutant-to-wild-type hazard ratio is fairly low compared to the hazard ratios of other mutants (Table 1). Therefore, if C. elegans akt-2 has a role in growth on S. maltophilia JCMS, this role is nearly negligible. Still, the resistance of akt-1 mutants to JCMS is curious and suggests that akt-1 and akt-2 might have different functions in the response to these bacteria. This is not unprecedented, as akt-1 and akt-2 have been shown to differentially regulate innate immune effectors such as thn-2 on E. coli and spp-1 on E. coli and P. aeruginosa (31). Thus, akt-1 and akt-2 might play different roles in the response to JCMS by regulating different effector genes.
Conserved defense pathways are involved in the C. elegans response to S. maltophilia JCMS.
We performed a survey of the known C. elegans bacterial defense pathways and found that several conserved pathways were involved in the response to S. maltophilia JCMS and E. coli OP50, while the Toll-like receptor gene tol-1 was not. Specifically, we found that tol-1 was minimally involved in the response to JCMS and, as previously shown for other nematode-bacterium interactions (15), was not involved in the response to OP50. On the other hand, the role of the UPR, p38 MAPK, and TGF-β-like pathways is largely conserved, as these pathways are involved in the response to other pathogenic bacteria (21–23, 25, 26), JCMS and OP50. The differential expression of the p38 MAPK- and TGF-β-regulated gene lys-1 (32) in nematodes fed JCMS and OP50 further highlights the necessity of these pathways in C. elegans defense. Furthermore, lys-1 was upregulated in nematodes fed JCMS (Fig. 7B), as observed previously for other bacterial pathogens (26, 32), suggesting that it functions as an antimicrobial gene. We observed that the loss of the more universally acting p38 MAPK pathway (60; reviewed in reference 14) was the most detrimental, followed by the UPR and TGF-β pathways. Our data also revealed that the loss of genes encoding signaling proteins, such as nsy-1, was very detrimental. This suggests that signaling through these gene products plays a central role in multiple pathways and/or a number of biological processes.
However, a closer inspection of the hazard ratios of S. maltophilia JCMS and E. coli OP50 suggests that individual components within the p38 MAPK and TGF-β pathways might function in a bacterium-specific manner. For example, PMK-1 is the terminal kinase in the p38 MAPK pathway and is important for the switch of the leucine zipper transcription factor ATF-7 from a repressor to an activator in response to P. aeruginosa (61). Our data suggest that PMK-1 mediates this switch in response to JCMS, and thus, nematode defense against these bacteria requires both atf-7- and pmk-1-dependent genes. On the other hand, we have evidence that the response to OP50 is atf-7 dependent and pmk-1 independent. For the TGF-β pathway, it appears that sma-6 and dbl-1 activities during growth on OP50 are somehow disadvantageous for wild-type nematodes but are required for the response to JCMS. Furthermore, the role for TGF-β-like pathway components during growth on JCMS seems similar to their role in S. marcescens infection (26).
Therefore, in wild-type nematodes, all the p38 MAPK and TGF-β pathway components likely play a defense role in the response to JCMS, while only some components are required for the response to OP50, suggesting a bacterium- or pathogen-specific action of individual pathway components. Intriguingly, we did not observe a bacterium-specific action for the tested UPR pathway components, which is likely due to a more generalized role in nematode stress responses (62). Together, these data support an interaction between C. elegans and bacteria involving conserved mechanisms that include genes that are specific to the bacterial environment. While the significance of bacterium-specific action for components within individual pathways is not clear, it can be revealed only by evaluating the functions of multiple pathway components and suggests that further investigation of the roles of individual components in the response to different bacteria is warranted.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by a grant from the National Science Foundation (NSF EF 0723862) to Michael A. Herman and by the Kansas State University Division of Biology. Most nematode strains were provided by the Caenorhabditis elegans Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40OD010440).
We thank Joseph Coolon for identification of S. maltophilia JCMS, Lynn Hancock for help with the generation of S. maltophilia GFP-expressing strain derivatives, and past and present members of the Herman laboratory for stimulating discussions.
Funding Statement
This work was funded by a grant from the National Science Foundation (NSF EF 0723862) to Michael A. Herman and by the Kansas State University Division of Biology.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00711-15.
REFERENCES
- 1.Denton M, Kerr KG. 1998. Microbiological and clinical aspects of infection associated with Stenotrophomonas maltophilia. Clin Microbiol Rev 11:57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, Diekema DJ, Quinn JP, Doern GV. 2007. Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J Clin Microbiol 45:3352–3359. doi: 10.1128/JCM.01284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garazi M, Singer C, Tai J, Ginocchio CC. 2012. Bloodstream infections caused by Stenotrophomonas maltophilia: a seven-year review. J Hosp Infect 81:114–118. doi: 10.1016/j.jhin.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 4.De Oliveira-Garcia D, Dall'Agnol M, Rosales M, Azzuz ACGS, Alcántara N, Martinez MB, Girón JA. 2003. Fimbriae and adherence of Stenotrophomonas maltophilia to epithelial cells and to abiotic surfaces. Cell Microbiol 5:625–636. doi: 10.1046/j.1462-5822.2003.00306.x. [DOI] [PubMed] [Google Scholar]
- 5.Steinkamp G, Wiedemann B, Rietschel E, Krahl A, Gielen J, Bärmeier H, Ratjen F. 2005. Prospective evaluation of emerging bacteria in cystic fibrosis. J Cyst Fibros 4:41–48. doi: 10.1016/j.jcf.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Parent K, Mitchell P. 1978. Cell wall-defective variants of Pseudomonas-like (group Va) bacteria in Crohn's disease. Gastroenterology 75:368–372. [PubMed] [Google Scholar]
- 7.Parent K, Mitchell PD. 1976. Bacterial variants: etiologic agent in Crohn's disease? Gastroenterology 71:365–368. [PubMed] [Google Scholar]
- 8.A'Court C, Garrard CS. 1992. Nosocomial pneumonia in the intensive care unit: mechanisms and significance. Thorax 47:465–473. doi: 10.1136/thx.47.6.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooke JS. 2012. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev 25:2–41. doi: 10.1128/CMR.00019-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryan RP, Monchy S, Cardinale M, Taghavi S, Crossman L, Avison MB, Berg G, van der Lelie D, Dow JM. 2009. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat Rev Microbiol 7:514–525. doi: 10.1038/nrmicro2163. [DOI] [PubMed] [Google Scholar]
- 11.Looney WJ, Narita M, Mühlemann K. 2009. Stenotrophomonas maltophilia: an emerging opportunist human pathogen. Lancet Infect Dis 9:312–323. doi: 10.1016/S1473-3099(09)70083-0. [DOI] [PubMed] [Google Scholar]
- 12.Steinert M. 2011. Pathogen-host interactions in Dictyostelium, Legionella, Mycobacterium and other pathogens. Semin Cell Dev Biol 22:70–76. doi: 10.1016/j.semcdb.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Fouhy Y, Scanlon K, Schouest K, Spillane C, Crossman L, Avison MB, Ryan RP, Dow JM. 2007. Diffusible signal factor-dependent cell-cell signaling and virulence in the nosocomial pathogen Stenotrophomonas maltophilia. J Bacteriol 189:4964–4968. doi: 10.1128/JB.00310-07. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Irazoqui JE, Ausubel FM. 2010. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Caenorhabditis elegans as a model to study tissues involved in host immunity and microbial pathogenesis. Clin Exp Immunol 160:48–57. doi: 10.1111/j.1365-2249.2010.04122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pujol N, Link EM, Liu LX, Kurz CL, Alloing G, Tan MW, Ray KP, Solari R, Johnson CD, Ewbank JJ. 2001. A reverse genetic analysis of components of the Toll signaling pathway in Caenorhabditis elegans. Curr Biol 11:809–821. doi: 10.1016/S0960-9822(01)00241-X. [DOI] [PubMed] [Google Scholar]
- 16.Couillault C, Pujol N, Reboul J, Sabatier L, Guichou J-F, Kohara Y, Ewbank JJ. 2004. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol 5:488–494. doi: 10.1038/ni1060. [DOI] [PubMed] [Google Scholar]
- 17.Tenor JL, Aballay A. 2008. A conserved Toll-like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep 9:103–109. doi: 10.1038/sj.embor.7401104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irazoqui JE, Urbach JM, Ausubel FM. 2010. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol 10:47–58. doi: 10.1038/nri2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan MW, Shapira M. 2011. Genetic and molecular analysis of nematode-microbe interactions. Cell Microbiol 13:497–507. doi: 10.1111/j.1462-5822.2011.01570.x. [DOI] [PubMed] [Google Scholar]
- 20.Marsh EK, May RC. 2012. Caenorhabditis elegans, a model organism for investigating immunity. Appl Environ Microbiol 78:2075–2081. doi: 10.1128/AEM.07486-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan M-W, Ausubel FM. 2002. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 22.Sifri CD, Begun J, Ausubel FM, Calderwood SB. 2003. Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect Immun 71:2208–2217. doi: 10.1128/IAI.71.4.2208-2217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bischof LJ, Kao C-Y, Los FCO, Gonzalez MR, Shen Z, Briggs SP, van der Goot FG, Aroian RV. 2008. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog 4:e1000176. doi: 10.1371/journal.ppat.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bravo A, Gill SS, Soberón M. 2007. Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control. Toxicon 49:423–435. doi: 10.1016/j.toxicon.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zugasti O, Ewbank JJ. 2009. Neuroimmune regulation of antimicrobial peptide expression by a noncanonical TGF-beta signaling pathway in Caenorhabditis elegans epidermis. Nat Immunol 10:249–256. doi: 10.1038/ni.1700. [DOI] [PubMed] [Google Scholar]
- 26.Mallo GV, Kurz CL, Couillault C, Pujol N, Granjeaud S, Kohara Y, Ewbank JJ. 2002. Inducible antibacterial defense system in C. elegans. Curr Biol 12:1209–1214. doi: 10.1016/S0960-9822(02)00928-4. [DOI] [PubMed] [Google Scholar]
- 27.Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM. 2003. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300:1921. doi: 10.1126/science.1080147. [DOI] [PubMed] [Google Scholar]
- 28.Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, Larsen PL, Riddle DL. 1998. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150:129–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dillin A, Crawford DK, Kenyon C. 2002. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 298:830–834. doi: 10.1126/science.1074240. [DOI] [PubMed] [Google Scholar]
- 30.Huang X, Zhang H, Zhang H. 2011. The zinc-finger protein SEA-2 regulates larval developmental timing and adult lifespan in C. elegans. Development 138:2059–2068. doi: 10.1242/dev.057109. [DOI] [PubMed] [Google Scholar]
- 31.Evans EA, Chen WC, Tan M-W. 2008. The DAF-2 insulin-like signaling pathway independently regulates aging and immunity in C. elegans. Aging Cell 7:879–893. doi: 10.1111/j.1474-9726.2008.00435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alper S, McBride SJ, Lackford B, Freedman JH, Schwartz DA. 2007. Specificity and complexity of the Caenorhabditis elegans innate immune response. Mol Cell Biol 27:5544–5553. doi: 10.1128/MCB.02070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans EA, Kawli T, Tan M-W. 2008. Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog 44:e1000175. doi: 10.1371/journal.ppat.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. 2006. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2:e183. doi: 10.1371/journal.pgen.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang X, Liu J, Ding J, He Q, Xiong R, Zhang K. 2009. The investigation of nematocidal activity in Stenotrophomonas maltophilia G2 and characterization of a novel virulence serine protease. Can J Microbiol 55:934–942. doi: 10.1139/W09-045. [DOI] [PubMed] [Google Scholar]
- 36.Rae R, Riebesell M, Dinkelacker I, Wang Q, Herrmann M, Weller AM, Dieterich C, Sommer RJ. 2008. Isolation of naturally associated bacteria of necromenic Pristionchus nematodes and fitness consequences. J Exp Biol 211:1927–1936. doi: 10.1242/jeb.014944. [DOI] [PubMed] [Google Scholar]
- 37.Ciche TA, Groffredi SK. 2007. General methods to investigate microbial symbioses, p 394–419. In Reddy CA, Beveridge TJ, Breznak JA, Marzluf GA, Schmidt TM, Snyder LR (ed), Methods for general and molecular microbiology, 3rd ed ASM Press, Washington, DC. [Google Scholar]
- 38.Goel MK, Khanna P, Kishore J. 2010. Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res 1:274–278. doi: 10.4103/0974-7788.76794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.UCLA Statistical Consulting Group. 13 October 2014. Supplemental notes to applied survival analysis. UCLA Statistical Consulting Group, Los Angeles, CA: http://www.ats.ucla.edu/stat/examples/asa/test_proportionality.htm. [Google Scholar]
- 40.Garsin DA, Sifri CD, Mylonakis E, Qin X, Singh KV, Murray BE, Calderwood SB, Ausubel FM. 2001. A simple model host for identifying Gram-positive virulence factors. Proc Natl Acad Sci U S A 98:10892–10897. doi: 10.1073/pnas.191378698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Portal-Celhay C, Bradley ER, Blaser MJ. 2012. Control of intestinal bacterial proliferation in regulation of lifespan in Caenorhabditis elegans. BMC Microbiol 12:49. doi: 10.1186/1471-2180-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruber JAN, Tang SY, Halliwell B. 2007. Evidence for a trade-off between survival and fitness caused by resveratrol treatment of Caenorhabditis elegans. Ann N Y Acad Sci 1100:530–542. doi: 10.1196/annals.1395.059. [DOI] [PubMed] [Google Scholar]
- 43.Twumasi-Boateng K, Shapira M. 2012. Dissociation of immune responses from pathogen colonization supports pattern recognition in C. elegans. PLoS One 7:e35400. doi: 10.1371/journal.pone.0035400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shapira M, Hamlin BJ, Rong J, Chen K, Ronen M, Tan M-W. 2006. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc Natl Acad Sci U S A 103:14086–14091. doi: 10.1073/pnas.0603424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Applied Biosystems. 1997. User bulletin #2: ABI Prism 7700 sequence detection 889 system. Applied Biosystems, Waltham, MA. [Google Scholar]
- 46.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 47.Tan MW, Mahajan-Miklos S, Ausubel FM. 1999. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A 96:715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang C, Xiong C, Kornfeld K. 2004. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditis elegans. Proc Natl Acad Sci U S A 101:8084–8089. doi: 10.1073/pnas.0400848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sifri CD, Begun J, Ausubel FM. 2005. The worm has turned—microbial virulence modeled in Caenorhabditis elegans. Trends Microbiol 13:119–127. doi: 10.1016/j.tim.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Spanier B, Starke M, Higel F, Scherer S, Fuchs TM. 2010. Yersinia enterocolitica infection and tcaA-dependent killing of Caenorhabditis elegans. Appl Environ Microbiol 76:6277–6285. doi: 10.1128/AEM.01274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM. 2010. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog 6:e1000982. doi: 10.1371/journal.ppat.1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsiao J-Y, Chen C-Y, Yang M-J, Ho H-C. 2013. Live and dead GFP-tagged bacteria showed indistinguishable fluorescence in Caenorhabditis elegans gut. J Microbiol 51:367–372. doi: 10.1007/s12275-013-2589-8. [DOI] [PubMed] [Google Scholar]
- 53.Garigan D, Hsu A-L, Fraser AG, Kamath RS, Ahringer J, Kenyon C. 2002. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics 161:1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsin H, Kenyon C. 1999. Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399:362–366. doi: 10.1038/20694. [DOI] [PubMed] [Google Scholar]
- 55.Miyata S, Begun J, Troemel ER, Ausubel FM. 2008. DAF-16-dependent suppression of immunity during reproduction in Caenorhabditis elegans. Genetics 178:903–918. doi: 10.1534/genetics.107.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kerry S, TeKippe M, Gaddis NC, Aballay A. 2006. GATA transcription factor required for immunity to bacterial and fungal pathogens. PLoS One 1:e77. doi: 10.1371/journal.pone.0000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. 2003. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 58.Mahajan-Miklos S, Tan M, Rahme LG, Ausubel FM. 1999. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96:47–56. [DOI] [PubMed] [Google Scholar]
- 59.Donlan RM, Costerton JW. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shivers RP, Kooistra T, Chu SW, Pagano DJ, Kim DH. 2009. Tissue-specific activities of an immune signaling module regulate physiological responses to pathogenic and nutritional bacteria in C. elegans. Cell Host Microbe 6:321–330. doi: 10.1016/j.chom.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shivers RP, Pagano DJ, Kooistra T, Richardson CE, Reddy KC, Whitney JK, Kamanzi O, Matsumoto K, Hisamoto N, Kim DH. 2010. Phosphorylation of the conserved transcription factor ATF-7 by PMK-1 p38 MAPK regulates innate immunity in Caenorhabditis elegans. PLoS Genet 6:e1000892. doi: 10.1371/journal.pgen.1000892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richardson CE, Kooistra T, Kim DH. 2010. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463:1092–1095. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.