

Abstract
We previously associated a missense mutation of the tc0668 gene of serial in vitro-passaged Chlamydia muridarum, a murine model of human urogenital C. trachomatis, with severely attenuated disease development in the upper genital tract of female mice. Since these mutants also contained a TC0237 Q117E missense mutation that enhances their in vitro infectivity, an effort was made here to isolate and characterize a tc0668 single mutant to determine its individual contribution to urogenital pathogenicity. Detailed genetic analysis of C. muridarum passages revealed a truncated variant with a G216* nonsense mutation of the 408-amino-acid TC0668 protein that does not produce a detectable product. Intracellular growth and infectivity of C. muridarum in vitro remain unaffected in the absence of TC0668. Intravaginal inoculation of the TC0668 null mutant into C3H/HeJ mice results in a typical course of lower genital tract infection but, unlike a pathogenic isogenic control, is unable to elicit significant chronic inflammation of the oviduct and fails to induce hydrosalpinx. Thus, TC0668 is demonstrated as an important chromosome-encoded urogenital pathogenicity factor of C. muridarum and the first with these characteristics to be discovered for a Chlamydia pathogen.
INTRODUCTION
Chlamydia muridarum is a Gram-negative obligate intracellular pathogen that was isolated from a steady-state respiratory infection of laboratory mice in the early 1940s (1, 2). Like other chlamydial organisms, C. muridarum has a biphasic life cycle that alternates between infectious elementary body (EB) and replicating reticulate body (RB) morphologies. The genome of C. muridarum is reductively evolved, containing a 1.07-Mb circular chromosome and single 7.5-kb extrachromosomal plasmid (3). In that C. muridarum and other chlamydial pathogens have fewer than 1,000 genes and ∼900 encoded proteins, roughly half the number encoded by environmental chlamydial organisms that parasitize simple single-celled eukaryotes (4), it is not known which of the many cryptic genetic factors allow them to thrive within and harm complex vertebrates.
In the laboratory, C. muridarum is used as a model of urogenital disease resulting from sexually transmitted Chlamydia trachomatis in women. The basic biology and genomes of these two pathogens are highly conserved. The urogenital biovar of C. trachomatis is responsible for the most reported cases of bacterial infection in the United States (5) and is a pervasive global health problem (6). In women, ascending infection from the lower to upper genital tracts, separated by the cervical barrier, can lead to loss of the ciliated epithelium and irreversible fibrotic remodeling of the fallopian tubes after primary infection is resolved (7). If left untreated, often because of asymptomatic infection (8), afflicted women can experience severe chronic sequelae, such as tubal blockage, hydrosalpinx, spontaneous abortion, ectopic pregnancy, and tubal factor infertility (9, 10). Genital inoculation of female mice with C. muridarum results in analogous established lower genital tract infection, ascension to the upper genital tract, high rates of hydrosalpinx, and long-lasting infertility (11–16). As a result, hydrosalpinx serves as a surrogate marker of chronic reproductive tract dysfunction in mice.
The genetic factors of Chlamydia that contribute to the development of upper genital tract sequelae are only now being elucidated due to recent technical advancements. Development of a C. trachomatis plasmid manipulation system and a transformation system for C. muridarum helped to reveal that plasmid-encoded Pgp3 and Pgp5 are crucial for C. muridarum induction of upper genital tract disease (17, 18). Deficiency in Pgp3 or Pgp5 results in reduced ascending infection to and chronic inflammatory infiltration in the upper genital tract. Chromosomal mutants of C. muridarum have previously been associated with reduced urogenital pathogenicity, but no individual lesions could be attributed to attenuation (19–21). Without a proven chromosomal genetic manipulation system, the contribution of other cryptic chromosomal factors to pathogenicity remains elusive and difficult to explore.
We recently reported that serial in vitro passage of C. muridarum with host cell adherence and entry selection pressure reduced its pathogenicity in the upper genital tract of intravaginally inoculated C3H/HeJ mice (22). Although a Q117E missense mutation of the chromosome-encoded protein TC0237 definitively enhanced in vitro infectivity in response to this pressure, a G322R missense mutation of TC0668, an unrelated chromosome-encoded hypothetical protein, remained loosely associated with in vivo attenuation. In this study, we sought to verify whether TC0668 is indeed an upper genital tract pathogenicity factor of C. muridarum. Comprehensive genome population analysis of C. muridarum passages leading to isolation of a TC0668 null mutant has allowed this hypothesis to be tested here.
MATERIALS AND METHODS
Chlamydia strains and culture.
Parental nonclonal C. muridarum organisms were initially acquired from the laboratory of Robert Brunham (University of Manitoba, Winnipeg, Manitoba, Canada) in 1999. C. muridarum EBs grown in HeLa cells (human cervical carcinoma epithelial cells; ATCC CCL-2) were density gradient purified as described previously (23). C. trachomatis serovar D was acquired from the laboratory of Harlan Caldwell (Rocky Mountain Laboratories, NIAID, NIH, Hamilton, MT) and maintained using the same protocol as for C. muridarum.
Prior to in vitro infection, host cells were suspended in Dulbecco's minimal essential medium (DMEM; Gibco BRL, Rockville, MD) containing 10% heat-inactivated fetal bovine serum (FBS; Gibco BRL) (D10 medium), seeded in 6-, 24-, or 96-circular-well tissue culture plates, and grown to confluence. In vitro infections were consistent with assisted or unassisted techniques described previously (22) with slight modifications for individual experiments in this study. Serial in vitro passage of C. muridarum strain Nigg3 was continued from passage 28 (G28) to passage 40 (G40) using the same alternating assisted/unassisted-passage scheme described for generating G28.
Whole-genome sequencing.
Whole-genome DNA sequencing (DNA-seq) was performed by using next-generation sequencing (NGS) as described previously (22). Briefly, genomic DNA was isolated from density gradient-purified EBs with a Quick-gDNA MiniPrep kit (Zymo Research, Irvine, CA), fragmented, bar coded with a TruSeq DNA sample preparation kit (Illumina, Inc.), and sequenced with a 50-bp single-end module on an Illumina HiSeq 2000 platform. FASTQ files of demultiplexed reads were exported with CASAVA (Illumina) prior to bioinformatics analysis.
Bioinformatics analysis.
DNA-seq data were pipelined through and processed with the BioProfile MATLAB (MathWorks, Natick, MA) class, an in-house object-oriented NGS analysis software tool (patent pending). First, parental C. muridarum (G0) reads were mapped to the Chlamydia muridarum strain Nigg reference chromosome (GenBank accession number NC_002620.2) and pMoPn plasmid (NC_002182.1) using the Burrows-Wheeler Aligner MEM algorithm (24) with default parameters. The resulting sequence alignment map (SAM) file was converted to binary alignment map (BAM) format and sorted by the private MATLAB executable bamaccessmex command. Insertions and deletions (indels) were realigned using the RealignerTargetCreator and IndelRealigner methods of the Genome Analysis Toolkit (25–27) with default parameters. A BioProfile object was created from the resulting reads by the BioProfile object constructor method with default parameters. The unbiased nondegenerate consensus sequence of the parental C. muridarum chromosome was calculated, exported to FASTA format, submitted to GenBank for archiving, and annotated by the NCBI Prokaryotic Genome Annotation Pipeline (28). An automated genome BLAST search of the parental C. muridarum chromosome placed it outside the strain Nigg, Nigg2, and Weiss clades, and it was thus designated Chlamydia muridarum strain Nigg3. The resident plasmid of Nigg3 is identical to the published pMoPn plasmid sequence and not deposited to a public database.
DNA-seq data sets, including the parental sample, were then pipelined through and processed by the BioProfile MATLAB class as described above, with the exception of mapping reads to the C. muridarum strain Nigg3 chromosome (GenBank accession number CP009760.1) in place of the Nigg reference chromosome. Nucleotide variants were detected and functionally characterized by the BioProfile “curate variants” method using GenBank annotations for the Nigg3 chromosome and pMoPn plasmid. Only variants satisfying minimum thresholds for mean Phred base quality score (inverse log of probability of error; Q = −10 × log10Perror) of Q30 (Perror = 0.1%, or 99.9% precision), mapping quality of Q10 (90% precision), positive-strand bias between 20 and 80%, and a fraction of depth of 1% or higher were characterized and reported. Sequencing error variants were redacted after being logically excluded by their common fraction of depth in both passaged-population and plaque clone sequencing samples, the latter of which have only consensus variants. The remaining variants lie within the open read frames (ORFs) of hypothetical proteins DNC_01195 (Nigg TC0237 or TC_RS01195), DNC_02045 (Nigg TC0412 or TC_RS02045), and DNC_03395 (Nigg TC0668 or TC_RS03395). For consistency with the literature and standard practices in the field, these genes are referred to herein by their old Nigg locus tag (i.e., TC0237).
Variants affecting the tc0412 gene were individually replicated as mutated nucleotide sequences and sorted according to ascending gene position. This set of mutant sequences was processed by the ORF Figure program in MATLAB (accessible from the MathWorks File Exchange under file identification number [ID] 48955) for in silico translation of ORFs with 50 codons or more in all three forward frames, alignment to the annotated tc0412 nucleotide sequence as a reference, and figure drawing according to predicted ORF position, frame, and reference homology (blue for match; red for mismatch). Predictions, referred to as proteoforms, were labeled according to common proteoform patterns and mutation position within the group of patterns (29).
Plaque isolation, genotyping, and sizing.
Plaque-forming assays were used to isolate clonal organisms from purified EBs of Nigg3 passages G0, G28, and G40 and from passage G13 and G16 source material as described previously (22). Isolates were amplified in 96-well tissue culture plates under assisted-infection conditions, boiled in SDS, and diluted in Tris-EDTA (TE) buffer. The coding regions of tc0237, tc0412, and tc0668 were PCR amplified from template DNA using a KOD Hot Start master mix (EMD Millipore, Billerica, MA) according to the manufacturer's instructions. Primer sequences, associated ORF nucleotide positions, and ORF directions are listed in Table S1 in the supplemental material. PCR amplicons from passage G0, G28, and G40 plaques were then Sanger sequenced (ABI 3130XL [Thermo Fisher Scientific, Waltham, MA] operated by the University of Texas Health Science Center at San Antonio, San Antonio [UTHSCSA] Nucleic Acids Core Facility; ABI 3730XL operated by Beckman Coulter Genomics, Inc., Danvers, MA) to full coverage (two to three reads for each ORF), and sequence reads were assembled using the MATLAB sequence assembly application (accessible from the MathWorks File Exchange under file ID 46071). Passage G13 and G16 plaques were genotyped using a single Sanger sequence read with full coverage of known single nucleotide polymorphisms (SNPs) (Beckman Coulter Genomics, Inc.). EBs of select clones were density gradient purified following secondary plaque isolation, genotyping to verify desired mutations, and minimal HeLa propagation under assisted-infection conditions. Plaque clones were labeled by joining their Nigg3 passage, primary plaque number, and secondary plaque number, if applicable.
A plaque-forming assay was also used to compare relative plaque sizes of C. muridarum organisms after 6 days of incubation. Monolayers were stained through their agarose overlay with a 0.06% (wt/vol) solution of neutral red in phosphate-buffered saline (PBS) for 1 h at room temperature (RT) and documented with a flatbed scanner. Images were imported into MATLAB, and plaque diameters were measured in pixels using the pixel distance measurement tool.
Mutant allele frequency estimation.
The population frequencies of tc0237 and tc0668 nucleotide mutations were estimated using Sanger sequencing and subsequent signal analysis in MATLAB. Genomic DNA was purified from Nigg3 passaged whole-cell lysate with a Quick-gDNA MiniPrep kit (Zymo Research) according to the manufacturer's instructions. The coding regions of tc0237 and tc0668 were PCR amplified with KOD Hot Start master mix (EMD Millipore) and Sanger sequenced (ABI 3130XL operated by the UTHSCSA Nucleic Acids Core Facility) with a single read to cover known SNPs. Sequence traces were imported into MATLAB and plotted using the traceplot function, and wild-type and mutant nucleotide base relative fluorescence units (RFU) were recorded for known tc0237 and tc0668 single nucleotide polymorphisms (SNPs). Mutant allele frequency was calculated as the mutation SNP RFU count divided by the wild-type RFU count.
Western blotting.
CT389 and TC0668 protein were detected by Western blotting. Approximately 1 × 107 C. trachomatis serovar D or C. muridarum Nigg3 purified EBs were heat denatured in SDS and β-mercaptoethanol for 10 min. Denatured protein samples were size separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) containing 4% acrylamide stacking gel and 12% acrylamide separation gel layers. The SDS-PAGE gel was then laterally transferred to a nitrocellulose membrane. For protein staining, the membrane was blocked with 5% powdered bovine milk in PBS, probed for CT389/TC0668 with a polyclonal anti-glutathione S-transferase (GST)-tagged CT389 antibody (anti-GST-CT389) raised in mice, and probed again by goat anti-mouse secondary antibody conjugated with horseradish peroxidase (HRP) (Jackson ImmunoResearch). Blotted protein was visualized by 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Sigma-Aldrich) chemiluminescence captured on X-ray film.
Centrifugation dependence assay.
Cell culture infectivity of C. muridarum organisms was measured by comparing their dependence on centrifugation for in vitro infection. Purified C. muridarum EBs were serially diluted in DMEM before being inoculated onto confluent HeLa monolayers grown on glass coverslips in 24-well tissue culture plates in duplicate. The assisted-infection conditions described previously (22) were modified by removing DEAE-dextran pretreatment and including 1 h of preincubation before centrifugation in order to equal a total of 2 h of incubation required for unassisted infections. Monolayers were fixed and processed for immunofluorescence assay (IFA) detection of C. muridarum inclusion-forming units (IFU) following 24 h of growth. Centrifuge dependence was calculated by dividing the titer, measured in IFU, of the assisted infection by that of the unassisted infection derived from the same inoculum material.
Intracellular growth assay.
Intracellular growth and development of Nigg3 clones were characterized by quantitating the number of progeny organisms per input organism during logarithmic intracellular growth. Duplicate 24-well tissue culture plates with confluent HeLa monolayers were inoculated in quadruplicate with purified C. muridarum EBs diluted in sucrose-phosphate-glutamic acid (SPG) medium at a multiplicity of infection (MOI) of 0.1 under assisted-infection conditions. Following 24 h of incubation, one plate was snap-frozen at −80°C for 30 min, thawed, lysed via sonication, and retitrated for progeny inclusion-forming units (IFU) by indirect immunofluorescence assay (IFA) in a 96-well tissue culture plate. The other plate was fixed, permeabilized, and titrated for input IFU also by IFA. The number of progeny organisms that replicated per input organism was calculated by dividing the amount of progeny IFU by its paired input amount of IFU from the same inoculum.
Mouse infection and monitoring.
The animal experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol (05053-34) was approved by the Committee on the Ethics of Laboratory Animal Experiments of the University of Texas Health Science Center at San Antonio.
Female 6-week-old C3H/HeJ mice were acquired from The Jackson Laboratory (Bar Harbor, ME) and allowed to acclimate to the new facility for 1 week. Five days prior to infection, mice were subcutaneously injected on the abdomen with 2.5 mg of colloidal depot medroxyprogesterone (Depo-Provera; Pharmacia and Upjohn LLC, Kalamazoo, MI) suspended in sterile phosphate-buffered saline (PBS). Mice were inoculated with 2 × 105 inclusion forming units (IFU) of purified C. muridarum elementary bodies (EBs) diluted in 10 μl of SPG buffer into the vaginal cavity. Mice infected with the same C. muridarum organism were housed at five mice per cage. Lower genital tract infection was monitored using cervicovaginal swabbing and titration. Swabs were clipped into 0.5 ml of sterile SPG medium containing sterile glass beads, vortexed for 2 min, and titrated via IFA as described above. A gentle preswab was performed immediately prior to C. muridarum infection to clear the vaginal cavity of debris without damaging the tissue. Cervicovaginal swabs were collected and titrated for 3 days postinfection (dpi), 7 dpi, and weekly thereafter up to 49 dpi. Mice were anesthetized and euthanized by cervical dislocation at 56 dpi for observation of pathology.
Pathology and inflammation assessments.
Upon euthanasia, mouse genital tracts were excised, and gross pathology of hydrosalpinx was observed and documented by high-resolution digital photography. Hydrosalpinx was scored according to an ordinal scale where 0 indicates no hydrosalpinx, 1 indicates hydrosalpinx that is only observable under magnification, 2 indicates visible hydrosalpinx smaller than the size of the oviduct, 3 indicates hydrosalpinx roughly equal to the size of the oviduct, and 4 indicates hydrosalpinx larger than the oviduct. Bilateral hydrosalpinx severity was calculated for each mouse as the summed scores of the left and right oviducts. Hydrosalpinx incidence was calculated as the number of mice with a bilateral score of 1 or higher divided by the total number of mice in the group.
Chronic inflammation or infiltration of mononuclear cells into the tissue surrounding the oviduct of excised genital tracts was evaluated histologically. Following gross pathology assessment, whole genital tracts were fixed in 10% neutral formalin, stored in 70% ethanol in water, embedded in paraffin, and serially sectioned longitudinally across the plane of the whole genital tract at 5-μm intervals on an AccuCut SRM 200 microtome (Sakura, Torrance, CA). Three nearly equidistant sections at roughly 5 μm, 30 μm, and 55 μm into the lateral surface of the oviduct were subjected to hematoxylin and eosin (H&E) staining, sealed on glass tissue slides, and observed under a microscope for infiltration of mononuclear inflammatory cells into tissue surrounding the oviduct. These infiltrates were scored on an ordinal scale where 0 represents no cellular foci, 1 indicates a single focus, 2 indicates two to four loci, 3 indicates more than four foci, and 4 indicates confluent infiltration. The median of the three scores served as a single value for each oviduct unilateral inflammation score, and both unilateral scores for each mouse were combined to form a bilateral score. Bilateral oviduct dilation scores were acquired from the same three sections of each oviduct in the same manner as the inflammation scores but using the criteria for scoring hydrosalpinx described above.
Statistical analyses.
Statistical tests were performed using GraphPad Prism, version 6 (GraphPad Software Inc., La Jolla, CA). In vitro assays were analyzed by one-way analysis of variance (ANOVA) with a Holm-Sidak multiple-comparison test. Hydrosalpinx incidence from mutant clones versus the background organism was tested using a two-tailed Fisher's exact test. Scored measures (hydrosalpinx severity, oviduct dilation, and oviduct inflammation) were analyzed by Kruskal-Wallis nonparametric ANOVA and Dunn's multiple-comparison test. Oviduct inflammation and dilation scores were correlated by nonparametric Spearman rank correlation. Deming linear regression was used to generalize the relationship between oviduct dilation and chronic inflammation scores since it does not assume one variable is a function of the other. All multiple comparison tests were performed between each mutant clone and the background control. Reported P values are exact with the exception of the Spearman rank correlation, which is approximate.
Nucleotide sequence accession numbers.
All NGS sequence reads were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioProject accession number PRJNA227769. Where available, BioSample, SRA experiment/sample/run, and GenBank accession numbers for passaged C. muridarum populations (G0, G28, and G40) and derived clones (G13.32.1, G13.11.1, G40.50.2, and G28.51.1) are listed in Table S2 in the supplemental material.
RESULTS
A TC0668 nonsense mutant developed during passage of C. muridarum Nigg3.
Serial in vitro passage of C. muridarum strain Nigg3 in HeLa cells was continued from passage 28 (G28) to 40 (G40) in order to establish long-term trends and fix stable mutations. As before, alternating unassisted (no DEAE or centrifugation) and assisted (host cell pretreatment with DEAE-dextran and inoculum centrifugation) passages were implemented to select for organisms with enhanced in vitro infectivity and amplify the selected genomic population, respectively. Whole-genome Illumina HiSeq 2000 reads from the parental (G0), intermediate (G28), and final (G40) passage populations were analyzed with the BioProfile MATLAB class to detect and functionally characterize high-quality, low-frequency chromosomal and plasmid nucleotide variants. The previously reported TC0237 Q117E mutation (encoded by the nucleotide change G277313C) continued as a consensus variant into G40, while the TC0668 G322R variant (G797661T) rose from 25% to 36% population frequency between G28 and G40 (Table 1). Another unobserved chromosomal variant, G797979A, mutated the glycine (GGA) codon at position 216 to a stop codon (TGA) (G216*) of TC0668. The G216* variant followed a pattern of spontaneous development and steady growth similar to that of G322R, albeit at a lower population frequency of 5% in G28 to 10% in G40. The tc0668 gene contains 408 codons; thus, the G216* nonsense mutation is predicted to truncate the protein product at roughly its midpoint. No other plasmid or chromosomal variants were observed to have arisen in passaged populations.
TABLE 1.
Nucleotide variants after serial passage of C. muridarum Nigg3d
| Reference sequence | Position (nt)a | Variant | Nigg3 population frequency (%) |
Location | Variant type | Gene nucleotide variant | Gene codon variante | Gene protein variantc | ||
|---|---|---|---|---|---|---|---|---|---|---|
| G0 | G28 | G40 | ||||||||
| G | 277313 | C | 0.00 | 100.00 | 99.58 | TC_0237 | Conservative | G349C | CAA117GAA | Q117E |
| AATGGTAAGT | 472758 | -------- | 2.87 | 2.60 | 5.84 | Intergenicb | ||||
| G | 472761 | A | 6.25 | 0.08 | 0.07 | TC_0412 | Silent | G3A | ATG1ATA | M1M |
| T | 472829 | G | 6.94 | 0.00 | 0.00 | TC_0412 | Nonsense | T71G | TTA24TGA | L24* |
| - | 472842 | T | 10.44 | 47.97 | 11.19 | TC_0412 | Frameshift | -84T | ACT28ACT | C29L fs 47* |
| - | 472884 | T | 2.90 | 0.00 | 0.00 | TC_0412 | Frameshift | -126T | TGT42TGT | S43F fs 47* |
| - | 472923 | C | 7.37 | 2.93 | 6.32 | TC_0412 | Frameshift | -165C | TAT55TAC | S56* fs 56* |
| C | 472963 | T | 16.78 | 0.45 | 1.46 | TC_0412 | Nonsense | C205T | CAG69TAG | Q69* |
| G | 473020 | T | 7.59 | 25.89 | 34.22 | TC_0412 | Nonsense | G262T | GAG88TAG | E 88* |
| GGATATCA | 473072 | -------- | 4.63 | 0.00 | 0.08 | TC_0412 | Frameshift | GGATATCA314------- | TGG105TGC | W105C fs 118* |
| h | 473193 | T | 6.50 | 8.51 | 26.65 | TC_0412 | Frameshift | -435T | TCT145TCT | S146* fs 146* |
| C | 473587 | T | 2.36 | 0.00 | 0.00 | TC_0412 | Nonsense | C829T | CAA277TAA | Q277* |
| - | 473691 | T | 1.27 | 2.36 | 1.09 | TC_0412 | Frameshift | -933T | AGT311AGT | I312Y fs 323* |
| T | 473759 | - | 1.20 | 0.00 | 0.00 | TC_0412 | Frameshift | T1001- | ATT334ATA | T335Q fs 338* |
| G | 797661 | T | 0.00 | 6.36 | 12.02 | TC_0668 | Nonsense | G646T | GGA216TGA | G216* |
| G | 797979 | A | 0.00 | 25.87 | 35.42 | TC_0668 | Missense | G964A | GGA322AGA | G322R |
nt, nucleotide.
A 10-bp deletion extends into tc0412.
Variant types are in relation to the protein sequence. An asterisk (*) represents a single stop codon. Frameshifted (fs) sequences are indicated, and the subsequent positions listed indicate the first occurrences of a stop codon.
Hyphens indicate nucleotide deletions.
Mutated nucleotides are shown in boldface type.
The tc0412 gene was under strong negative selection prior to passage.
The parental C. muridarum Nigg3 population (G0) is heterogeneous due to 13 unique low-depth variants overlapping or within the chromosomal tc0412 gene (Table 1). Consistent with numerous mutations identified by different research groups in both tc0412 and its C. trachomatis homologs (19, 20, 30–33), the 13 genetic lesions are predicted to delete or disrupt its start codon, frameshift translation by a small insertion or deletion (indel), or terminate translation early by nonsense mutation. In order to visualize the functional consequences, mutant tc0412 nucleotide sequences were modeled and translated in silico in all three forward frames and mapped to the annotated protein sequence (Table 2). Distinct patterns of primary and alternative ORFs, collectively referred to as a proteoform, of each mutant emerge from the predictions. Putative TC0412 proteoforms were therefore grouped and labeled according to common structures (B1 to G1), with the exception of two proteoforms with hypothetical designations Z1 and Z2, which could not be validated outside NGS detection during the course of this study. Given the preponderance of inactivating mutations in C. muridarum Nigg3 and other laboratory-maintained Chlamydia organisms, it is likely that tc0412 is under strong negative selection in vitro.
TABLE 2.
Predicted proteoforms of tc0412 nucleotide variantsa

The wild-type sequence, or proteoform A1, is the annotated tc0412 sequence from the C. muridarum Nigg reference chromosome with no disruptions. ORFs of 50 codons or longer were detected and locally aligned to the A1 proteoform to determine their homology to the wild-type protein sequence. Red represents no homology to the reference sequence, and blue represents a perfect match. The three frames are spatially separated from top to bottom within each box. Frameshifted sequences sometimes contain alternative ORFs in the second frame because the alternative start codon was originally in frame with the annotated start codon. Mutations detected by NGS but not verified by clone genotyping are assigned to the hypothetical class Z. Hyphens indicate the deleted nucleotides.
TC0668 mutations developed independent of TC0237 Q117E.
By Nigg3 passage 28, the TC0237 Q117E variant was ubiquitous throughout the population while TC0668 G216* and G322R were not. This observation implies that tc0668 mutation occurred in organisms with a prior tc0237 mutation. In order to determine the passage at which these variants arose, mutant allele frequencies were estimated from Sanger sequencing traces of tc0237 and tc0668 amplicons of early-passage genomes. This targeted signal processing bypassed more complex NGS analysis while still allowing for identification of shifts in allele abundances. Contrary to expectation, TC0668 G216* and G322R polymorphisms reached confidently detectable abundance by Nigg3 passage 6, but TC0237 Q117E signal was absent until passage 11 (Fig. 1A). The tc0668 variants displayed a steady rise regardless of whether passage was assisted or unassisted. In contrast, the TC0237 Q117E SNP consistently rose at a much greater rate during unassisted than assisted passages, supporting prior evidence that it is an in vitro infectivity adaptation. The strong selective pressure of TC0237 Q117E under these conditions allowed a selective sweep that largely spanned only two unassisted passages, 13 and 15. During this, sweep mutant alleles of tc0668 had a negative relationship to the TC0237 Q117E mutation, suggesting that tc0668 and tc0237 mutations were in linkage equilibrium or two independent events.
FIG 1.
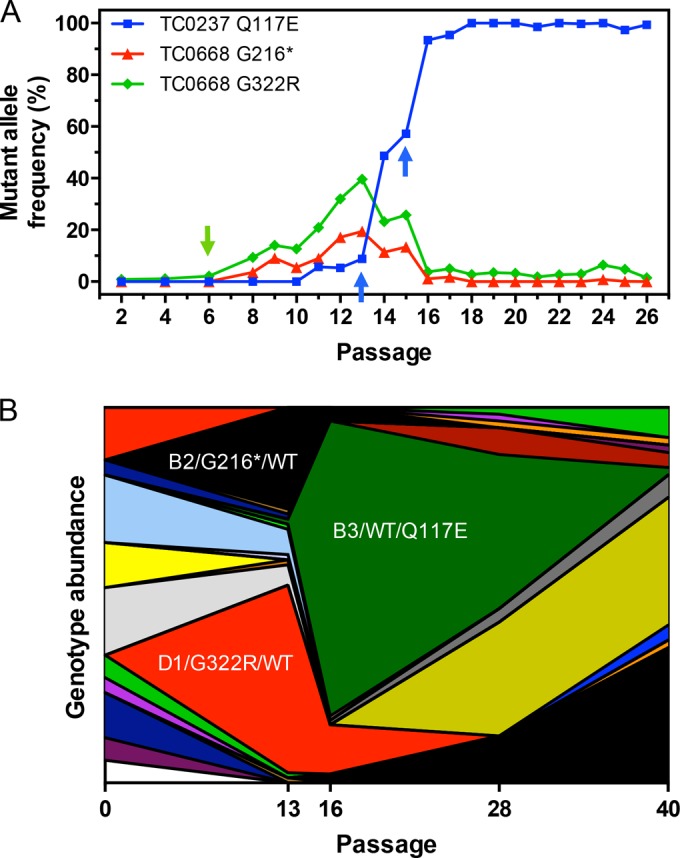
Population dynamics of mutations arising from C. muridarum Nigg3 passage. (A) Mutant allele frequencies of tc0237 and tc0668 variants across early C. muridarum Nigg3 passage were calculated from the ratio of mutant to wild-type allele relative fluorescence signals in Sanger sequencing chromatograms. Source DNA was purified from passaged populations and PCR amplified prior to sequencing. The green arrow indicates the first appearance of tc0668 mutation (passage 6), and blue arrows indicate selective sweep of TC0237 Q117E (unassisted passages 13 and 15). (B) Combined TC0412, TC0237, and TC0668 genotypes of Nigg3 plaque clones as determined by Sanger sequencing. Each continuous area represents a unique genotype encompassing all three genes. For genotype B2/G216*/WT (black), B2 signifies the proteoform of TC0412, G216* represents the nonsense mutation of TC0668, and wild type (WT) indicates no mutation in TC0237. The D1/G322R/WT and B3/WT/Q117E genotypes are highlighted in red and dark green, respectively. Abundances are in relation to the number of plaques isolated from each Nigg3 passage shown, with a total of 326 clones across the five passages sampled. Discontinuity in unique genotypes is largely due to selective sweep of TC0237 Q117E.
In order to verify the independent nature of tc0237 and tc0668 mutations, a total of 362 plaque clones were isolated from Nigg3 passages G0, G13, G16, G28, and G40, and their tc0412, tc0237, and tc0668 genes were genotyped by high-throughput Sanger sequencing (see Table S3 in the supplemental material). The G13 and G16 data sets represent the beginning and end of the TC0237 Q117E selective sweep and allow greater resolution of genetic linkage prior to and after the sudden event. Only one of the G13 plaque clones (G13.80; 1.1%) exhibited the TC0237 Q117E polymorphism alongside wild-type tc0668 and the B3 proteoform of TC0412 (early gene frameshift). By G16, this unique genotype encompassed a majority (80%) of the C. muridarum population (Fig. 1B). As inferred from mutant allele frequency estimation, the TC0668 G216* and G322R polymorphisms are independent of TC0237 Q117E and each other in G13 and G16. The G322R lesion is associated with the D1 proteoform (middle gene frameshift) of TC0412 and the G216* mutation with B2 (start codon deletion) in G13, suggesting that they developed on these tc0412 genetic backgrounds.
Significant recombination between the G16 and G40 passages was evident as tc0668 mutations dispersed across other TC0412 proteoforms and combined with TC0237 Q117E. All clonal isolates (n =326) had a major disruption to the tc0412 ORF matching a single nucleotide variant detected by NGS, but no discernible trends of selection of specific proteoforms were observed from G0 to G40. The negative selection of tc0412 therefore saturated the C. muridarum stock population prior to passage. That only a single tc0412 disruption was found in each isolate, while tc0237 and tc0668 alleles were exchanged at a high rate, implies that chromosomal recombination breakpoints occur more frequently across large genomic intervals. In total these findings demonstrate that tc0412, tc0237, and tc0668 mutations arose independently of one another with no evidence to suggest functional linkage.
The TC0668 G216* mutant is a null mutant.
The competitive nature of the TC0668 G216* mutant during in vitro passage suggests that the wild-type protein is dispensable for in vitro fitness of C. muridarum. A set of four isogenic plaque clone organisms with the TC0412 B2 genetic background (G13.32.1), TC0668 G216* (G13.11.1), TC0237 Q117E (G40.50.2), and TC0668 G216* with TC0237 Q117E (G28.51.1), were therefore characterized to dissect the contribution of individual and combined lesions on C. muridarum TC0668 protein expression. C. muridarum EBs were subjected to Western blotting using a polyclonal antibody raised against the C. trachomatis serovar D homolog of TC0668, CT389. Whereas ∼47-kDa CT389 and TC0668 proteins were observed in wild-type C. trachomatis and C. muridarum EBs, respectively, no trace of TC0668 could be discerned at either the expected or lower molecular weights of C. muridarum TC0668 G216* mutants (Fig. 2). Thus, the G216* variant is a spontaneous TC0668 null mutant.
FIG 2.
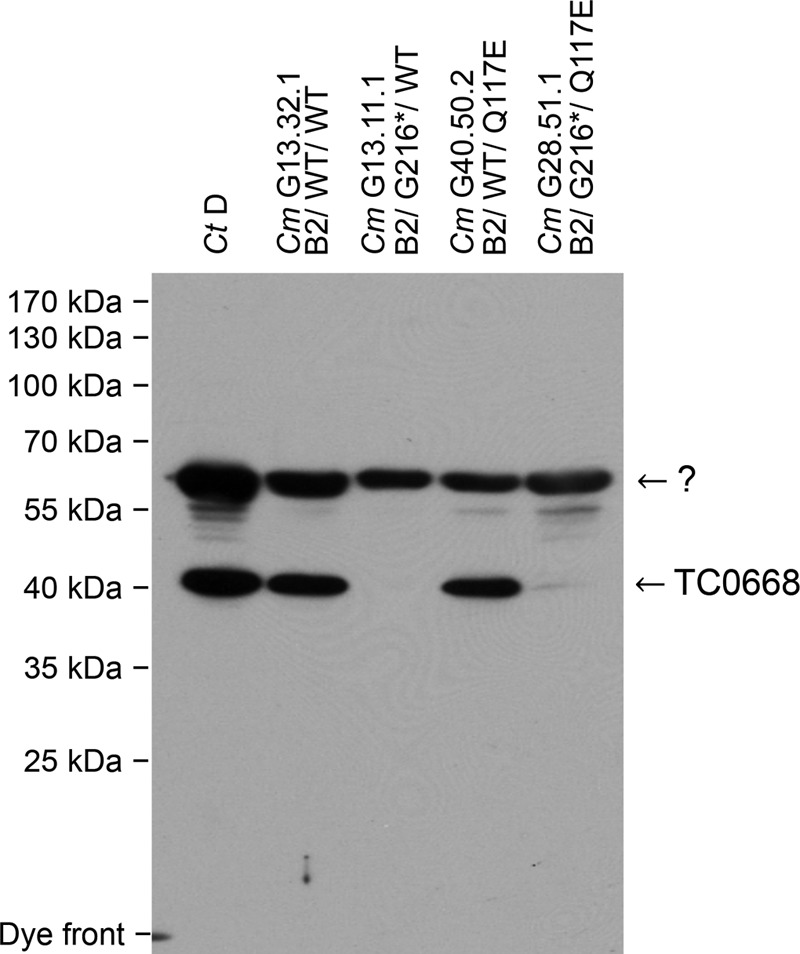
Western blot detection of TC0668 protein in C. muridarum Nigg3 mutant clones. The CT389 and TC0668 proteins from 1 × 107 heat-denatured C. trachomatis serovar D (Ct D) and C. muridarum (Cm) Nigg3 EBs were detected by a mouse polyclonal anti-CT389 antibody. Nigg3 EB organisms are labeled by their clone designations followed by their TC0412/TC0668/TC0237 proteoform/genotypes as indicated on top of the figure, including C. muridarum variant clones G13.31.1 with a genotype of B2/WT/WT, G13.11.1 with a genotype of B2/G216*/WT, G40.50.2 with a genotype of B2/WT/Q117E, and G28.51.1 with a genotype of B2/G216*/Q117E (see Table S2 in the supplemental material for details). TC0668 protein (∼47 kDa) is abundant in clones with the wild-type tc0668 sequence but not detectable in clones with the G216* mutation. An unknown (?) band of approximately ∼65 kDa is likely a secondary target of the polyclonal antibody and serves as a loading reference.
TC0668 null mutants display normal in vitro fitness.
It was previously shown that TC0237 Q117E mutation enhances infectivity of C. muridarum and that addition of TC0668 G322R to TC0237 Q117E has no obvious effect on infectivity. Using the same centrifugation dependence assay as before, the TC0668 null mutant (G13.11.1) was shown to be equally reliant on centrifugation for infecting cultured HeLa cells as the background control (G13.32.1) with a consistent 40-fold enhancement (Fig. 3A). TC0237 Q117E mutants (G40.50.2 and G28.51.1), on the other hand, are much less dependent on centrifugation as evidenced by a 14-fold increase in titers between noncentrifuged and centrifuged infections. Once inside the cell, all mutants tested grew equally well, suggesting that neither TC0668 nor TC0237 is essential for normal intracellular growth and development (Fig. 3B). The relative sizes of cell monolayer plaques formed by TC0237 Q117E mutants (G40.50.2 and G28.51.1) were significantly smaller than those of the background control (G13.32.1), but the TC0668 null mutant (G13.11.1) displayed normal plaque size (Fig. 4A and B). Although TC0237 is involved in the host cell invasion process, TC0668 has no apparent contribution to basal fitness of C. muridarum.
FIG 3.

In vitro fitness phenotypes of C. muridarum Nigg3 mutant clones. (A) Clone dependence on inoculum centrifugation for infection. Fold increase of HeLa cell infection was calculated by dividing the titers of the centrifuged by those of the noncentrifuged samples derived from the same source material (P < 0.0001, by one-way ANOVA; **** P < 0.0001, by a Holm-Sidak multiple-comparison test against the control organism). Bars are representative of means of two to three experiments with standard errors of the means. (B) Intracellular growth and development of clones during logarithmic growth. The number of progeny IFU per input IFU was calculated from duplicate infections using the same source material; one infection was directly titrated, while another was harvested at 24 h postinfection (hpi) and titrated (P = 0.75, by one-way ANOVA). Bars represent means from four experiments with standard errors of the means.
FIG 4.

Plaque sizes of C muridarum Nigg3 mutant clones. (A) Plaques formed by clones after 6 days of growth in confluent McCoy B monolayers. (B) Relative sizes of plaques in scanned-plate image pixels formed by the mutant clones (P < 0.0001 by one-way ANOVA; ****, P < 0.0001 by a Holm-Sidak multiple-comparison test against the control organism). Individual values are shown as gray points, and means with standard errors of the means are shown as black lines.
TC0668 null mutants are severely attenuated in vivo.
Prior experiments indicated that TC0668 mutation is associated with in vivo attenuation of C. muridarum in the murine genital tract infection model. In order to dissect the contribution of TC0668 to infection dynamics and development of hydrosalpinx, C3H/HeJ mice were intravaginally inoculated with the TC0668 null mutant (G13.11.1) and its isogenic controls. Cervicovaginal swabbing and titration during the course of infection indicated that mice infected with C. muridarum containing the TC0237 Q117E mutation had a marginally stronger shedding course beginning around 35 dpi, whereas the TC0668 null mutant (G13.11.1) had lower genital tract dynamics equal to those of its TC0412 B2 background control (G13.32.1) (Fig. 5A and B). Thus, TC0668 has no apparent effect on the lower genital tract fitness of C. muridarum.
FIG 5.
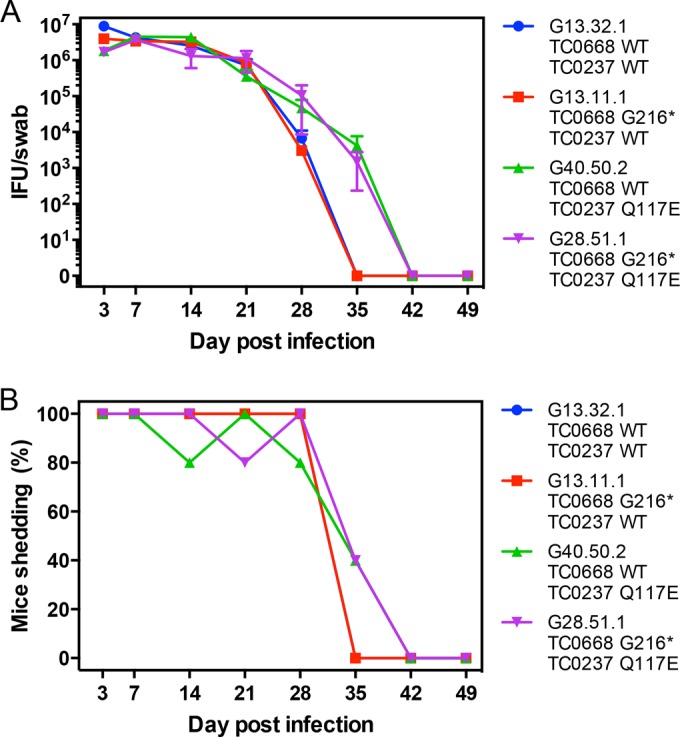
In vivo lower genital tract dynamics of C. muridarum Nigg3 mutant clones. (A) Lower genital tract shedding course of clones following intravaginal inoculation with 2 × 105 IFU. Shedding course was titrated from cervicovaginal swabs collected on the indicated days postinfection (dpi). Points represent means of 5 mice per group (n = 5) with standard errors of the means. (B) Percentage of mice in each infection group with positive cervicovaginal shedding on the day postinfection indicated.
By 56 dpi, infection with the control organism resulted in a high incidence and severity of hydrosalpinx (Fig. 6A to C). The TC0668 null mutant, on the other hand, induced almost no hydrosalpinx. Addition of the TC0237 Q117E mutation to the TC0668 G216* mutation rendered C. muridarum unable to elicit hydrosalpinx, while the TC0237 Q117E mutant (G40.50.2) displayed a dichotomous split between pathogenic and apathogenic outcomes within its infection group. Severity of hydrosalpinx was confirmed by microscopic examination of oviduct dilation of sectioned tissue (Fig. 7A and B). Since it is hypothesized that chronic oviduct inflammation is a contributing factor to hydrosalpinx formation, the level of inflammatory cell infiltrates into the tissue surrounding the oviduct was also scored (Fig. 8A and B). In support of the aforementioned hypothesis, the dilation of each oviduct studied was significantly correlated with the level of chronic oviduct inflammation (Fig. 8C). Thus, TC0668 is consistently associated with the ability of C. muridarum to stimulate chronic disease of the murine oviduct.
FIG 6.

Gross pathology of murine genital tracts infected with C. muridarum Nigg3 mutant clones. (A) Representative gross pathology images of whole genital tracts harvested at 56 dpi. Left and right oviducts and ovaries are amplified for detail. White arrows indicate hydrosalpinges. (B) Incidence of hydrosalpinx at 56 dpi (*, P < 0.05; ***, P < 0.001, by Fisher's exact test against the results for the control organism; n = 5 per group). (C) Severity of hydrosalpinx at 56 dpi (P = 0.021, by a Kruskal-Wallis test; *, P < 0.05, by Dunn's multiple-comparison test against the results for the control organism). Both individual bilateral scores (black dots) and medians (gray bars) are shown.
FIG 7.

Oviduct dilation of mice infected with C. muridarum Nigg3 mutant clones. (A) Representative micrographs of H&E-stained oviducts from infected mice at 56 dpi. Oviduct dilation is illustrated by white double-headed arrows. (B) Individual (black dots) and median (gray bars) bilateral oviduct dilation scores determined from H&E-stained tissue sections (P = 0.0363, by a Kruskal-Wallis test; *, P < 0.05, by Fisher's exact test against results for the control organism).
FIG 8.

Chronic oviduct inflammation of mice infected with C. muridarum Nigg3 mutant clones. (A) Representative micrographs of H&E-stained oviducts from infected mice at 56 dpi. Top panels (a to d) present a broad view of tissue sections. Chronic inflammatory cell infiltrate foci are illustrated by white arrows in bottom panels (a1 to d1). (B) Individual (black dots) and median (gray bars) bilateral chronic inflammatory cell infiltrate scores determined from H&E-stained tissue sections (P = 0.0533, by a Kruskal-Wallis test; *, P < 0.05, by Dunn's multiple-comparison test against results for the control organism). (C) Unilateral oviduct dilation scores plotted against corresponding unilateral oviduct chronic inflammation scores. Deming linear regression (black line) illustrates an unbiased trend of the overlapping ordinal score pairs. A majority of the points (23 of 40) lie within the (0, 1) coordinate (#). Spearman rank correlation is highly significant at a P value of <0.0001 with a correlation coefficient of ρ = 0.7637.
DISCUSSION
In this study, the individual contribution of the chromosome-encoded factor TC0668 to in vitro and in vivo fitness and pathogenicity was explored. Foremost, a G322R missense mutation of TC0668 was previously associated with attenuated pathogenicity following intravaginal inoculation of mice, but the in vitro-passaged organism containing this mutation also had a Q117E missense mutation of TC0237 that enhanced its in vitro infectivity. After NGS analysis of passaged populations and genotyping of plaque clones derived from the passages, it was discovered that a middle gene G216* nonsense mutation developed independently of TC0237 Q117E. This mutant was isolated alongside a control with the same TC0412 mutation (B2 proteoform), as well as a TC0237 Q117E single mutant and TC0668 G216* and TC0237 Q117E double mutants. These myriad techniques and strategies therefore resulted in a testable set of mutants to dissect the contribution of TC0668 and TC0237 to fitness and urogenital pathogenesis of C. muridarum.
The enhanced in vitro infectivity phenotype of TC0237 Q117E mutants was duplicated here by a separate research team with independent mutants, strengthening the previous observation. TC0236, a paralog of TC0237 with the same domain of unknown function 720 (DUF720) motif, with a Q119K missense mutation was discovered by another group and displays a similarly enhanced infectivity phenotype (34). However, whereas the TC0236 Q119K mutant developed larger cell monolayer plaques than a wild-type control, the TC0237 Q117E mutant in this study (G40.50.2) displayed consistently a smaller average plaque size than its control. The selective advantage of the TC0237 Q117E mutation during C. muridarum passage suggests that it significantly increases the ability of the organism to invade host cells before TC0237 wild-type organisms are able to. It can be argued from this assumption that TC0237 Q117E allows more immediate cell invasion, resulting in decreased spreading during plaque formation. However, further experimentation is necessary to determine the mechanisms behind infectivity enhancement by both TC0236 Q119K and TC0237 Q117E.
Previously, the role of the TC0668 G322R mutation in urogenital attenuation of C. muridarum was explored, albeit only in the presence of TC0237 Q117E. Here, the characterization of the G216* mutant permitted a more definitive measure of the contribution of TC0668 to C. muridarum phenotypes. Premature termination of the TC0668 ORF by the G216* missense mutation resulted in no obvious effect on in vitro fitness. TC0668 protein cannot be detected in this mutant, suggesting that it is dispensable for infection, intracellular growth, and life cycle progression in vitro. Following intravaginal inoculation, the TC0668 null mutant was equally capable of surviving in the lower genital tract as its genetic background control. Unlike the TC0237 Q117E mutant, the TC0668 null mutant elicited severely reduced levels of chronic inflammation and hydrosalpinx of the oviduct, supporting the hypothesis that TC0668, not TC0237, is a significant pathogenicity factor of C. muridarum.
Consistent with discoveries reported here, a C. muridarum strain with a TC0668 F386V missense mutation was mentioned in the literature due to its reduced urogenital pathogenicity in C57BL/6 mice (21). CmVar004, the TC0668 F386V mutant, and four relatives were isolated from the lower genital tract of mice inoculated with highly passaged C. muridarum. The only apparent causal variants for attenuation were an intergenic nucleotide transversion (T381636A) and intragenic TC0668 F386V missense mutation (T798169G). These lesions accompanied a 34 to 67% drop in disease incidence, but given the evidence provided here, the TC0668 F386V variant may be the more likely determinant. With this additional event, the role of TC0668 in pathogenesis is becoming more apparent.
Aside from being associated with the outer membrane complex of Chlamydia (40), not much else is known about TC0668. The N-terminal 70 amino acids (aa) consist of a series of transmembrane domains, and the remainder of the 408-aa protein contains the domain of unknown function 1207 (DUF1207) motif. Phylogenetic analysis of TC0668 protein indicates that C. muridarum inherited the DUF1207 motif from a distant chlamydial progenitor (Fig. 9). The evolution of the motif closely follows the diversification of chlamydial organisms themselves; however, it is also shared by an equally large and even more diverse group of bacterial chemoorganotrophs. Precisely how and why DUF1207 proteins are shared between such dissimilar organisms or what this may indicate about the role of TC0668 in chlamydial pathogenesis are unclear.
FIG 9.

Neighbor-joining tree of TC0668 homologs. Homologous protein sequences were discovered by a protein-protein BLAST search (38) against the NCBI nonredundant protein database. The neighbor-joining phylogenetic tree is rooted by the longest distance as calculated using the Grishin general method (39).
Tertiary protein structure prediction and a homology search of TC0668 (Fig. 10) with the I-TASSER suite (35–37) revealed that it shares structural homology with eukaryotic integrins alpha V beta 1 (α5β1, fibronectin receptor), alpha IIb beta 3 (αIIbβ3, fibrinogen receptor), and alpha V beta 3 (α5β3, vitronectin receptor). In the host, these receptors and their ligands engage in diverse cellular activities, including cell adherence, cell migration, cell signaling, cell differentiation, prevention of complement deposition, platelet activation, wound healing, and fibrosis. It is therefore difficult to predict which of these processes TC0668 could be involved in, but they offer tantalizing possibilities, such as stimulation of host pathways leading to a chronic fibrotic response within the oviduct. Interestingly, the G322 and F386 residues of TC0668 are predicted to be less than 8 Å from one another, suggesting that they may be components of a common molecular process. Further structural analysis of TC0668 is necessary in order to verify the computation prediction and determine its molecular function.
FIG 10.

Predicted model of TC0668 protein. TC0668 tertiary protein structure was predicted using the I-TASSER suite (35–37). Residues are labeled at the upper right by position and include the N-terminal M1 and C-terminal F408, G216, G322, and F386 residues. The distance in angstroms between the G322 and F386 residues is indicated by a dashed line (7.96 Å). Colors distinguish secondary structure features. Model depictions were acquired using the Jmol plug-in within the MATLAB Bioinformatics Toolbox.
In summary, the discovery of TC0668 as a major chromosome-encoded urogenital pathogenicity factor of C. muridarum is the first of its kind. However, this discovery raises many subsequent questions pertaining to the perplexing evolutionary origins, pathogenic modus operandi, and basis for spontaneous in vitro disruption of TC0668. Given that TC0668 shares 97% protein identity with its C. trachomatis serovar D counterpart, CT389, further evaluation of DUF1207-containing homologs may lead to a better understanding or means to combat highly prevalent urogenital C. trachomatis disease in humans.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health and the U.S. Army Research Office.
We thank the UTHSCSA Next Generation Sequencing and Nucleic Acids Core Facilities for sequencing counsel and support.
Funding Statement
The work was supported in part by US NIH grants R01AI047997 and R01AI064537. The funder had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01171-15.
REFERENCES
- 1.Nigg C. 1942. An unidentified virus which produces pneumonia and systemic infection in mice. Science 95:49–50. doi: 10.1126/science.95.2454.49. [DOI] [PubMed] [Google Scholar]
- 2.Nigg C, Eaton MD. 1944. Isolation from normal mice of a pneumotropic virus which forms elementary bodies. J Exp Med 79:497–510. doi: 10.1084/jem.79.5.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Utterback T, Berry K, Bass S, Linher K, Weidman J, Khouri H, Craven B, Bowman C, Dodson R, Gwinn M, Nelson W, DeBoy R, Kolonay J, McClarty G, Salzberg SL, Eisen J, Fraser CM. 2000. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res 28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B, Brandt P, Nyakatura GJ, Droege M, Frishman D, Rattei T, Mewes H-W, Wagner M. 2004. Illuminating the evolutionary history of Chlamydiae. Science 304:728–730. doi: 10.1126/science.1096330. [DOI] [PubMed] [Google Scholar]
- 5.Torrone E, Papp J, Weinstock H, Centers for Disease Control and Prevention . 2014. Prevalence of Chlamydia trachomatis genital infection among persons aged 14–39 years—United States, 2007–2012. MMWR Morb Mortal Wkly Rep 63:834–838. [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization. 2001. Global prevalence and incidence of selected curable sexually transmitted diseases: overview and estimates. World Health Organization, Geneva, Switzerland: http://www.who.int/hiv/pub/sti/who_hiv_aids_2001.02.pdf. [Google Scholar]
- 7.Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex Transm Dis 32:49–56. doi: 10.1097/01.olq.0000148299.14513.11. [DOI] [PubMed] [Google Scholar]
- 8.Lan J, Melgers I, Meijer CJ, Walboomers JM, Roosendaal R, Burger C, Bleker OP, van den Brule AJ. 1995. Prevalence and serovar distribution of asymptomatic cervical Chlamydia trachomatis infections as determined by highly sensitive PCR. J Clin Microbiol 33:3194–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baud D, Goy G, Jaton K, Osterheld M-C, Blumer S, Borel N, Vial Y, Hohlfeld P, Pospischil A, Greub G. 2011. Role of Chlamydia trachomatis in miscarriage. Emerg Infect Dis 17:1630–1635. doi: 10.3201/eid1709.100865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shao R, Wang X, Wang W, Stener-Victorin E, Mallard C, Brännström M, Billig H. 2012. From mice to women and back again: causalities and clues for Chlamydia-induced tubal ectopic pregnancy. Fertil Steril 98:1175–1185. doi: 10.1016/j.fertnstert.2012.07.1113. [DOI] [PubMed] [Google Scholar]
- 11.Maxion HK, Liu W, Chang M-H, Kelly KA. 2004. The infecting dose of Chlamydia muridarum modulates the innate immune response and ascending infection. Infect Immun 72:6330–6340. doi: 10.1128/IAI.72.11.6330-6340.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Zhang H, Zhou Z, Yang Z, Ding Y, Zhou Z, Zhong E, Arulanandam B, Baseman J, Zhong G. 2014. Chlamydial induction of hydrosalpinx in 11 strains of mice reveals multiple host mechanisms for preventing upper genital tract pathology. PLoS One 9:e95076. doi: 10.1371/journal.pone.0095076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell J, Huang Y, Liu Y, Schenken R, Arulanandam B, Zhong G. 2014. Bioluminescence imaging of Chlamydia muridarum ascending infection in mice. PLoS One 9:e101634. doi: 10.1371/journal.pone.0101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Zhou Z, Chen J, Wu G, Yang Z, Zhou Z, Baseman J, Zhang J, Reddick RL, Zhong G. 2014. Lack of long-lasting hydrosalpinx in A/J mice correlates with rapid but transient chlamydial ascension and neutrophil recruitment in the oviduct following intravaginal inoculation with Chlamydia muridarum. Infect Immun 82:2688–2696. doi: 10.1128/IAI.00055-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barron AL, White HJ, Rank RG, Soloff BL, Moses EB. 1981. A new animal model for the study of Chlamydia trachomatis genital infections: infection of mice with the agent of mouse pneumonitis. J Infect Dis 143:63–66. doi: 10.1093/infdis/143.1.63. [DOI] [PubMed] [Google Scholar]
- 16.de la Maza LM, Pal S, Khamesipour A, Peterson EM. 1994. Intravaginal inoculation of mice with the Chlamydia trachomatis mouse pneumonitis biovar results in infertility. Infect Immun 62:2094–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Huang Y, Yang Z, Sun Y, Gong S, Hou S, Chen C, Li Z, Liu Q, Wu Y, Baseman J, Zhong G. 2014. Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect Immun 82:5327–5335. doi: 10.1128/IAI.02576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y, Zhang Q, Yang Z, Conrad T, Liu Y, Zhong G. 2015. Plasmid-encoded Pgp5 is a significant contributor to Chlamydia muridarum induction of hydrosalpinx. PLoS One 10:e0124840. doi: 10.1371/journal.pone.0124840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramsey KH, Sigar IM, Schripsema JH, Denman CJ, Bowlin AK, Myers GAS, Rank RG. 2009. Strain and virulence diversity in the mouse pathogen Chlamydia muridarum. Infect Immun 77:3284–3293. doi: 10.1128/IAI.00147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jasper DK, Sigar IM, Schripsema JH, Sainvil CK, Smith CL, Yeruva L, Rank RG, Murthy AK, Widder JR, Ramsey KH. 2015. Genomic variant representation in a Chlamydia population is dynamic and adaptive with dependence on in vitro and in vivo passage. Pathog Dis 73:1–12. doi: 10.1093/femspd/ftv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeruva L, Myers GSA, Spencer N, Creasy HH, Adams NE, Maurelli AT, McChesney GR, Cleves MA, Ravel J, Bowlin A, Rank RG. 2014. Early microRNA expression profile as a prognostic biomarker for the development of pelvic inflammatory disease in a mouse model of chlamydial genital infection. mBio 5:e01241-14. doi: 10.1128/mBio.01241-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Zhou Z, Conrad T, Yang Z, Dai J, Li Z, Wu Y, Zhong G. 2015. In vitro passage selects for Chlamydia muridarum with enhanced infectivity in cultured cells but attenuated pathogenicity in mouse upper genital tract. Infect Immun 83:1881–1892. doi: 10.1128/IAI.03158-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in Chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv arXiv:13033997v2 [q-bio.GN] http://arxiv.org/abs/1303.3997. [Google Scholar]
- 25.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. 2002. From FASTQ data to high-confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 11:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Ciufo S, Li W. 2013. Prokaryotic genome annotation pipeline. In Beck J, Benson D, Coleman J, Hoeppner M, Johnson M, Maglott D, Mizrachi I, Morris R, Ostell J, Pruitt K, Rubinstein W, Sayers E, Sirotkin K, Tatusova (ed), The NCBI handbook, 2nd ed. National Center for Biotechnology Information, Bethesda, MD. [Google Scholar]
- 29.Smith LM, Kelleher NL, Consortium for Top Down Proteomics . 2013. Proteoform: a single term describing protein complexity. Nat Methods 10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borges V, Ferreira R, Nunes A, Sousa-Uva M, Abreu M, Borrego MJ, Gomes JP. 2013. Effect of long-term laboratory propagation on Chlamydia trachomatis genome dynamics. Infect Genet Evol 17:23–32. doi: 10.1016/j.meegid.2013.03.035. [DOI] [PubMed] [Google Scholar]
- 31.O'Connell CM, AbdelRahman YM, Green E, Darville HK, Saira K, Smith B, Darville T, Scurlock AM, Meyer CR, Belland RJ. 2011. Toll-like receptor 2 activation by Chlamydia trachomatis is plasmid dependent, and plasmid-responsive chromosomal loci are coordinately regulated in response to glucose limitation by C. trachomatis but not by C. muridarum. Infect Immun 79:1044–1056. doi: 10.1128/IAI.01118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sturdevant GL, Kari L, Gardner DJ, Olivares-Zavaleta N, Randall LB, Whitmire WM, Carlson JH, Goheen MM, Selleck EM, Martens C, Caldwell HD. 2010. Frameshift mutations in a single novel virulence factor alter the in vivo pathogenicity of Chlamydia trachomatis for the female murine genital tract. Infect Immun 78:3660–3668. doi: 10.1128/IAI.00386-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonner C, Caldwell HD, Carlson JH, Graham MR, Kari L, Sturdevant GL, Tyler S, Zetner A, McClarty G. 2015. Chlamydia trachomatis virulence factor CT135 is stable in vivo but highly polymorphic in vitro. Pathog Dis 73:ftv043. doi: 10.1093/femspd/ftv043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell M, Darville T, Chandra-Kuntal K, Smith B, Andrews CW, O'Connell CM. 2011. Infectivity acts as in vivo selection for maintenance of the chlamydial cryptic plasmid. Infect Immun 79:98–107. doi: 10.1128/IAI.01105-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER suite: protein structure and function prediction. Nat Methods 12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 39.Grishin NV. 1995. Estimation of the number of amino acid substitutions per site when the substitution rate varies among sites. J Mol Evol 41:675–679. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Afrane M, Clemmer DE, Zhong G, Nelson DE. 2010. Identification of Chlamydia trachomatis outer membrane complex proteins by differential proteomics. J Bacteriol 192:2852–2860. doi: 10.1128/JB.01628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.