

Abstract
Salidroside, the main active ingredient extracted from Rhodiola crenulata, has been shown to be neuroprotective in ischemic cerebral injury, but the underlying mechanism for this neuroprotection is poorly understood. In the current study, the neuroprotective effect of salidroside on cerebral ischemia-induced oxidative stress and the role of the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway was investigated in a rat model of middle cerebral artery occlusion. Salidroside (30 mg/kg) reduced infarct size, improved neurological function and histological changes, increased activity of superoxide dismutase and glutathione-S-transferase, and reduced malon-dialdehyde levels after cerebral ischemia and reperfusion. Furthermore, salidroside apparently increased Nrf2 and heme oxygenase-1 expression. These results suggest that salidroside exerts its neuroprotective effect against cerebral ischemia through anti-oxidant mechanisms and that activation of the Nrf2 pathway is involved. The Nrf2/antioxidant response element pathway may become a new therapeutic target for the treatment of ischemic stroke.
Keywords: nerve regeneration, traditional Chinese medicine, salidroside, cerebral ischemia and reperfusion, nuclear factor erythroid 2-related factor 2, heme oxygenase-1, middle cerebral artery occlusion model, superoxide dismutase, neuroprotection, neural regeneration
Introduction
Salidroside is the main active ingredient extracted from the root of Rhodiola crenulata, a traditional Tibetan medicine used as an adaptogen to enhance the body's resistance to a variety of chemical, biological, and physical stressors (Kelly, 2001). Salidroside has a wide range of pharmacological functions, relieves stress (Mattioli et al., 2009), promotes radiation-resistance (Zhou et al., 2012), enhances immunity (Lu et al., 2013), eliminates fatigue (Huang et al., 2009) and is anti-aging (Mao et al., 2010). Recently, a growing body of evidence suggests that salidroside may have a neuroprotective effect against ischemic cerebral injury. Salidroside has been shown to protect PC12 cells from glucose and serum depletion-induced apoptosis (Yu et al., 2008). In another study, salidroside significantly prevented ischemic cerebral injury induced by middle cerebral artery occlusion and reperfusion in rats (Shi et al., 2012). Pretreatment with salidroside was shown to reduce cellular damage resulting from global cerebral ischemia/reperfusion injury in rats (Zou et al., 2009). However, the mechanisms underlying the neuroprotective effects of salidroside remain unclear.
Strong experimental data support the notion that an imbalance between reactive oxygen species production and their detoxification takes place in the ischemic zone following a stroke (Manzanero et al., 2013). The brain has an endogenous system which combats oxidative stress, including dietary-free radical scavengers (ascorbate and α-tocopherol), endogenous tripeptide glutathione, and enzymatic antioxidants, such as superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) (Chen et al., 2011). Recently, there has been a lot of evidence indicating that the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) is essential for regulating endogenous enzymatic antioxidants in the brain (Zhang et al., 2013b). Nrf2 binds to the anti-oxidant response element (ARE) with high affinity. Activation of this transcription factor can induce the expression of Nrf2-dependent phase enzymes, such as heme oxygenase-1 (HO-1), NAD(P)H, quinone oxidoreductase 1, glutathione reductase, and GSH-Px (Shinkai et al., 2006). These enzymes have been proven to combat oxidative stress and are neuroprotective in ischemic cerebral injury (Son et al., 2010; Li et al., 2013). The Nrf2/ARE pathway is now considered to be a new therapeutic target for treating of ischemic stroke. Salidroside is known to be effective in the prevention of oxidative stress following many kinds of diseases (Xu and Li, 2012; Leung et al., 2013; Yuan et al., 2013; Zhang et al., 2013a). Whether the Nrf2 pathway is involved in the antioxidant effects of salidroside is still unclear.
To further characterize and understand the neuroprotective mechanisms of salidroside, we designed experiments to reveal the effects of salidroside on the endogenous antioxidant pathway and investigate a battery of endogenous enzymes such as SOD, GSH-Px, glutathione-S-transferase (GST) and heme oxygenases which are Nrf2-modulated proteins (McCord and Edeas, 2005), and determine the levels of Nrf2.
Materials and Methods
Animals
Male Sprague-Dawley rats aged 7–8 weeks and weighing 250–280 g were provided by the SLAC Laboratory Animal Co., Ltd., Shanghai, China (certificate No. SCXK (Hu) 2012-0002). The animals were housed in groups of five, in a 12-hour light/dark cycle with free access to a standard diet and water. The procedures followed were in accordance with the international laws on animal experimentation and approved by the Ethical Committee of Fujian Academy of Traditional Chinese Medicine, China.
All surgeries were performed under chloral hydrate (10% w/v) anesthesia, and all pain and distress of the experiment animals were minimized. All animal experiments were carried out in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animal (NIH Publication No. 85-23, revised 1986).
Experimental groups
For infarct volume measurement and neurological deficit evaluation, the rats were divided into four groups (seven rats in each group) as follows: sham operation group, vehicle group, and two salidroside groups (Sal15 and Sal30). Salidroside (C14H20O7, MW: 300.3, CAS:10338-51-9, purity > 99%; Tauto Biotech Co., Shanghai, China) was dissolved in 0.9% NaCl before use. Salidroside (15, 30 mg/kg, intraperitoneally) was administered twice to the Sal15 and Sal30 groups: once immediately prior to cerebral ischemia and once immediately after reperfusion. Rats in the sham operation group and the vehicle group received 0.9% saline. For immunohistochemical staining, western blot assay and real-time reverse transcription (RT)-PCR, the rats were divided into three groups: sham operation, vehicle and salidroside (Sal), with 3–7 rats per group. A dose of 30-mg/kg salidroside was administrated to the Sal group. All rats were subjected to middle cerebral artery occlusion (MCAO), except those in the sham operation group.
MCAO models
Focal cerebral ischemia and reperfusion was induced as described by Longa et al. (1989). The rats were anesthetized with chloral hydrate (10% w/v). A ventral median incision was made on the neck to expose the right common carotid artery, the external carotid artery and the internal carotid artery. The external carotid artery and its branches were then isolated and coagulated. A 3–0 nylon suture with a blunt tip was inserted into the internal carotid artery, through the external carotid artery stump, approximately 17–18-mm distal to the carotid bifurcation, until a mild resistance was felt. At this point, the suture had been advanced to the anterior cerebral artery and occluded the middle cerebral artery. Body temperature was monitored rectally and maintained between 36.5°C and 37.0°C, during the surgical procedure, using a heat lamp. After 120 minutes, the suture was removed from the internal carotid artery to allow middle cerebral artery reperfusion. The distal internal carotid artery was then immediately coagulated. The rats in the sham operation group underwent all surgical procedures, but no suture was made.
Neurological deficit scores
The animals were neurologically assessed at 24 hours after reperfusion by an investigator who was unaware of animal grouping. The deficits were scored using a modified scoring system based on that developed by Longa et al. (1989) as follows: 0, no deficits; 1, difficulty in fully extending the contralateral forelimb; 2, unable to extend the contralateral forelimb; 3, mild circling to the contralateral side; 4, severe circling; and 5, falling to the contralateral side.
Measurement of infarct volume
Infarct volume was measured using 2,3,5-triphenyltetrazolium chloride staining. After neurological deficit scoring, the rats were deeply anesthetized with 10% chloral hydrate solution. The brains were quickly removed and frozen for 5 minutes at −20°C. Each brain was then sliced into five coronal slices of the same thickness. The sliced sections were subsequently stained with 1.5% 2,3,5-triphenyltetrazolium chloride (w/v) at 37°C for 30 minutes, then fixed in 4% paraformaldehyde (w/v) at 4°C for 24 hours. Finally, images of brain slices were captured using a digital scanner and a per-son masked to the treatment groups manually delineated the areas of unstained tissue (representing the infarct area) using Image Pro Plus 6.0 software. Infarct areas in all sections were combined to obtain a whole infarct area. The whole infarct area was then multiplied by the thickness of brain sections to calculate the volume of infarction. Volume was carried out using the following equation: volume correction = infarct volume × contralateral volume/ipsilateral volume × 100%. The size of the infarct volume was represented by the percentage of the volume of infarction.
Nissl staining
At 24 hours after reperfusion, the rat was deeply anesthetized with chloral hydrate (10% w/v), rapidly perfused with 0.9% physiological saline and fixed with 4% paraformaldehyde solution from the heart until the rat limbs were stiff. Subsequently, the brain was removed and fixed in 4% paraformaldehyde, dehydrated in sucrose gradient until it submerged to the bottom of the vessel. For Nissl staining, coronal brain frozen sections at 30-µm thickness containing the cortex and striatum (from anteroposterior +1.7 to −0.3, relative to bregma) were sliced and mounted to the slides and treated with 70% ethylalcohol for at least 4 hours to degrease. After washing with distilled water for several minutes, the sections were incubated in cresyl violet solution (Beyotime Institute of Biotechnology, Haimen, China) for 5 minutes, and dehydrated in 100% ethylalcohol for 1 minute. The images were captured on an inverted microscope (IX70; Olympus America, Melville, NY, USA).
Enzyme assays
The rats were euthanized by decapitation at 24 hours after reperfusion. The brains were removed quickly and the cortex of the ischemic hemisphere was dissected on ice. The dissected part of the brain was homogenized (10%, w/v) in normal saline. The homogenate was centrifuged at 1,000 × g for 10 minutes at 4°C and the levels of SOD, GST, GSH-Px, and MDA in the supernatant were determined. The level of SOD was determined using the WST-1 method (Kit A001, Jiancheng Bioengineering Institute, Nanjing, China). The levels of GST and GSH-Px were assayed using the spectrophotometric method (Kit A004 and Kit A005, Jiancheng Bioengineering Institute). The MDA content was determined using the TBA method (Kit A003, Jiancheng Bioengineering Institute). The protein concentration was determined by bicinchoninic acid assay with bovine serum albumin as the standard. The levels of SOD, GST and GSH-Px were represented by its enzyme activity unit per mg protein, and MDA content was represented by its bicinchoninic acid per mg protein. All enzymes were measured according to the manufacturers’ protocols.
Immunohistochemical staining
Coronal brain frozen sections at 30-µm thickness were prepared as described before for Nissl staining. Immunohistochemical staining was performed on floating sections. The slices were treated with antigen retrieval reagent (Beyotime Institute of Biotechnology) for 20 minutes in a 90°C water bath, and slowly cooled to room temperature. The sections were then incubated in 3% hydrogen peroxidase for 10 minutes and in blocking reagent (Solution A, SP kit, ZSGB Biological Co., Beijing, China) for 15 minutes. Subsequently, sections were reacted with a rabbit Nrf2 polyclonal antibody (1:100; Abcam, New Territories, HK, China), or a rabbit anti-HO-1 polyclonal antibody (1:100; Abcam) diluted in 0.01-M phosphate-buffered saline at 4°C overnight. After incu-bation, the sections were washed with PBS and reacted with biotinylated conjugates secondary anti-rabbit antibodies (Solution B, SP kit, ZSGB Biological Co.) at 37°C for 15 minutes. Finally, the immunoreactive products were visualized using 0.01% 3,3′-diaminobenzidine tetrachloride as the chromogen reagent. Images of immunostained sections were captured using an optical microscope. Three discontinuous sections of each rat were selected, and five non-overlapping visual fields of each section were selected and photographed under an inverted microscope (IX70; Olympus America, Melville, NY, USA) for measurement. For each captured photo, the immunostaining intensity was represented with the integrated optical density of the whole photo, which was analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Bethesda, MD, USA). The immunostaining intensity of one animal was calculated from the average integrated optical density of all acquired photos of the animal.
Western blot assay
Cortical tissue of the ischemic hemisphere was used for western blot assay. The protein was extracted in ice-cold radioimmune precipitation assay lysis buffer (50-mM Tris-HCl, pH 7.4, 150-mM sodium chloride, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1-mM phenylmethylsulfonyl fluoride. Protein concentrations were determined using the bicinchoninic acid Protein Assay Kit (Beyotime Institute of Biotechnology). The following methods were used for protein electrophoresis and immunoblotting. Equal amounts of protein (80 µg) were loaded on a 10% sodium dodecyl sulfate-polyacrylamide gel for electrophoresis, then transferred onto polyvinylidene fluoride transfer membranes (Millipore, Billerica, MA, USA) at 110 V for 50 minutes. Membranes were blocked for 2 hours with 5% non-fat milk, dissolved in Tris-buffered saline with Tween 20 buffer (20-mM Tris, 0.14 M NaCl, and 0.1% Tween-20, pH 7.6), at room temperature, then incubated in polyclonal rabbit anti-Nrf2 antibody (ab31163, Abcam, New Territories, HK, China) (1:1,000 diluted in 5% non-fat milk-Tris-buffered saline with Tween 20) or anti-HO-1 antibody (ab13243, Abcam) (1:500 diluted in 5% non-fat milk-Tris-buffered saline with Tween 20) at 4°C overnight. Membranes were then incubated in goat anti-rabbit-horseradish peroxidase secondary antibody (1:1,000, Thermo-Pierce, Rockford, IL, USA) for 2 hours at room temperature and visualized using an enhanced chemiluminescence kit (Beyotime Institute of Biotechnology). Images were captured with a FluorChem M System (ProteinSimple, Santa Clara, CA, USA). Semiquantification of western blot bands was achieved by analyses of optical density using AlphaView SA software (Version 3.4.0; Protein-Simple).
Real-time RT-PCR
Total RNA was extracted from the cortex of the ischemic hemisphere using RNApure Tissue Kit (CWbio Co., Ltd., Beijing, China) according to the manufacturer's protocol. The quantity of total RNA was measured using Nano-Drop 2000 (Thermo, MA, USA). Reverse transcription was performed using SuperRT cDNA Kit (CWbio Co., Ltd.). Quantitative RT-PCR was carried out on a ABI 7500 real-time fluorescence quantitative PCR apparatus (Applied Biosystems, Foster City, CA, USA) using a UltraSYBR Mixture (With ROX) kit (CWbio Co., Ltd.). Amplification procedure was as follows: 95°C for 10 minutes, 45 cycles of 95°C for 15 seconds and 60°C for 60 seconds. The oligonucleotide primer sequences were designed as follows: HO-1 (92 bp), forward: TCA CTG GCA GGA AAT CAT CC, reverse: CTG AGA GGT CAC CCA GGT A; GADPH (138 bp), forward: TGG AGT CTA CTG GCG TCT T, reverse: TGT CAT ATT TCT CGT GGT TCA. All samples were assayed in triplicate, and relative gene expression was quantified using the 2-ΔΔCt method as previously described (Livak and Schmittgen, 2001).
Statistical analysis
Data are presented as the mean ± SD. All analyses were performed using the SPSS 13.0 software (SPSS, Chicago, IL, USA). Measurement data were first analyzed for normal distribution test. Comparisons among multiple groups were assessed by one-way analysis of variance, followed by the least significant difference post hoc test. P values less than 0.05 were considered statistically significant.
Results
Neuroprotective effects of salidroside on cerebral ischemia/reperfusion
Significantly reduced infarct volume and decreased neurological deficit scores were observed in the Sal30 group compared with the vehicle group (P < 0.05). No significant changes in infarct volume and decreased neurological deficit scores were detected in the Sal15 group (P > 0.05; Figure 1).
Figure 1.

Effects of salidroside on infarct volume and neurological deficit scores in rats with cerebral ischemia/reperfusion.
(A) 2,3,5-Triphenyltetrazolium chloride staining of brain slices. The white represents ischemic areas. Salidroside decreased infarct volume (B) and decreased neurological deficit scores (C) at 24 hours after ischemia and reperfusion (#P < 0.05). Data are expressed as the mean ± SD (n = 7; one-way analysis of variance, followed by the least significant difference post hoc test). *P < 0.05, vs. sham. Sham: Sham operation group; vehicle: 0.9% saline-treated group; Sal15: 15 mg/kg salidroside group; Sal30: 30 mg/kg salidroside group.
Salidroside improved the morphology of neurons in the cortex and striatum
Neural cells in the sham operation group were arranged tightly with regular shape and had an intact cell structure. The blue Nissl bodies in neural cells were visible and distinct. After cerebral ischemia and reperfusion, the morphology of the cells in the cortex and the striatum clearly changed; the cells in the cortex were disarranged and a large number of cells were swollen. The shape of the cells changed from triangular to rounded, and normal cell structures were invisible (Figure 2). Compared with the vehicle group, the morphology of the cells in the cortex and striatum was regular, and the number of normal cells increased in the Sal group (Figure 2). The results indicated that salidroside attenuated injury-induced histological change.
Figure 2.
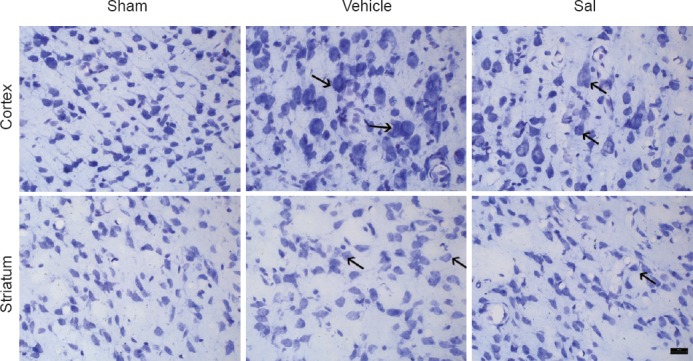
Salidroside improved the morphology of neurons in the cortex and striatum after ischemia /reperfusion (Nissl staining, × 400).
Histological changes in the cortex and the striatum were evaluated by Nissl staining. In the vehicle group (vehicle + MCAO), cells in the cortex and striatum were disarranged and a large number of cells were swollen. Cellular shapes changed from triangular to rounded, and normal cell structures were invisible. In the Sal group (salidroside + MCAO), the morphology of neurons was regular. Arrows show swelling and distension. Sham: Sham operation group; vehicle: 0.9% saline-treated group; Sal: 30 mg/kg salidroside group; MCAO: middle cerebral artery occlusion. Scale bar: 20 μm.
Effects of salidroside on SOD, GST, GSH-Px activities and MDA content in ischemic cortex
As shown in Figure 3A, the activity of SOD and GST were significantly higher 24 hours after cerebral ischemia and reperfusion (Figure 3A, P < 0.01) and there was a higher trend for GSH-Px activity. Treatment with 30-mg/kg salidroside significantly increased the activity of SOD and GST (P < 0.05). Furthermore, as shown in Figure 3A, salidroside seemed to increase the activity of GSH-Px, but this was not significant. MDA, the product of lipid peroxidation, is an indicator of the severity of oxidative stress. The cerebral content of MDA was significantly diminished in the vehicle group (P < 0.01; Figure 3B), while salidroside further decreased MDA content com-pared with the vehicle group (P < 0.05; Figure 3B).
Figure 3.

Effects of salidroside on SOD, GST, and GSH-Px activities and MDA content in rats with cerebral ischemia.
(A) Effects of salidroside on cerebral antioxidant enzyme activity after ischemia and reperfusion. (B) Effects of salidroside on MDA content after ischemia and reperfusion. Data are expressed as the mean ± SD (n = 7; one-way analysis of variance, followed by the least significant difference post hoc test). **P < 0.01, vs. sham; #P < 0.05, vehicle. Sham: Sham operation group; vehicle: 0.9% saline-treated group; Sal: 30 mg/kg salidroside group; SOD: superoxide dismutase; GST: glutathione-S-transferase; GSH-Px: glutathione peroxidase; MDA: malondialdehyde.
Effects of salidroside on Nrf2 and HO-1 immunoreactivity in the cortex and striatum
In the MCAO model, the striatum is the ischemic core and its surrounding cortex is regarded as the ischemic periphery. Nrf2 positive cells were observed in the striatum and cortex of rats in the sham operation, vehicle and Sal groups. The Nrf2-immunoreactive products were distributed in the cytoplasm of neural cells (Figure 4A). In the cortex, the intensity of the immunostaining was higher in the vehicle group than in the sham operation group (P < 0.01; Figure 4B). Treatment of salidroside significantly enhanced immunostaining intensity in the cortex (P < 0.05; Figure 4B). In the striatum, an obvious increase of immunostaining intensity was seen in the vehicle group compared with the sham operation group (P < 0.01; Figure 4B), but there was no change after treatment with salidroside.
Figure 4.

Effects of salidroside on Nrf2 and HO-1 immunoreactivity in the cortex and striatum of rats with cerebral ischemia (× 400).
Immunohistochemical staining was used to detect the distribution of Nrf2 and HO-1 in cortex and striatum at 24 hours after cerebral ischemia and reperfusion. (A) Representative images of immunohistochemistry for Nrf2. Brown Nrf2-stained cells were prominently observed in the vehicle group. In the cortex, the Nrf2 immunoreactivity was enhanced in the Sal group compared with the vehicle group. Arrows show Nrf2-immunoreactive cells. (B) Quantification of the IOD for Nrf2. (C) Representative images of immunohistochemistry for HO-1. Brown HO-1-stained cells were prominently observed in the vehicle group. In the striatum, the HO-1 immunostaining intensity was enhanced in the Sal group compared with the vehicle group. Arrows show HO-1-immunoreactive cells. (D) Quantification of IOD for HO-1. Data are expressed as the mean ± SD (n = 5; one-way analysis of variance, followed by the least significant difference post hoc test). **P < 0.01, vs. sham; #P < 0.05, vs. vehicle. Sham: Sham operation group; vehicle: 0.9% saline-treated group; Sal: 30-mg/kg salidroside group; IOD: integrated optical density; Nrf2: nuclear factor erythroid 2-related factor 2; HO-1: heme oxygenase-1. Scale bars: 20 μm. Experiments were conducted in triplicate.
The change in HO-1 was different. HO-1-positive cells were visible in the sham operation, vehicle and Sal groups in the region of interest. Immunoreactive products were mainly distributed in the cytoplasm (Figure 4C). In the sham operation group, the immunostaining reaction was weak in the cortex and striatum. In the striatum, the immunostaining intensity was significantly higher in the vehicle group than in the sham operation group (P < 0.01; Figure 4D). The size and shape of immunoreactive neural cells in the striatum were altered (Figure 4C). Treatment of salidroside significantly enhanced the immunostaining intensity in the striatum (P < 0.05; Figure 4D). In the cortex, the changes were not so evident, and the immunostaining intensity was greater in the vehicle group compared with the sham operation group (P < 0.01; Figure 4D). Treatment with salidroside enhanced im-munostaining intensity, but statistical analysis did not show a significance.
Effects of salidroside on Nrf2 and HO-1 protein levels
To verify the results of the immunohistochemical staining, western blot assay was used to determine the protein levels of the transcriptional factor Nrf2 and HO-1. The results showed that the expression of Nrf2 and HO-1 protein was significantly enhanced in the vehicle group (P < 0.01; Figure 5B, C), while salidroside treatment significantly increased protein levels of Nrf2 and HO-1 (P < 0.05; Figure 5B, C).
Figure 5.

Salidroside increased the protein level of Nrf2 and HO-1 and up-regulated HO-1 mRNA expression in the ischemic cortex.
(A) Representative images of western blot assay for Nrf2, HO-1. (B) Quantification of optical density for Nrf2, normalized to β-actin. (C) Quantification of optical density for HO-1, normalized to β-actin. (D) Effect of salidroside on relative HO-1 mRNA level in the ischemic brain. Salidroside (30 mg/kg) significantly reduced HO-1 mRNA expression compared with the vehicle group. Data are expressed as the mean ± SD (n = 3 for western blot assay; n = 5 for reverse transcription-PCR test; one-way analysis of variance, followed by the least significant difference post hoc test). **P < 0.01, vs. sham; #P < 0.05, ##P < 0.01, vs. vehicle. Sham: Sham opera-tion group; vehicle: 0.9% saline-treated group; Sal: 30-mg/kg salidroside group; Nrf2: nuclear factor erythroid 2-related factor 2; HO-1: heme oxygenase-1. Immunoblots were repeated at least twice and no less than three samples were used per test.
Effect of salidroside on mRNA expression of HO-1
HO-1 is one of the most important downstream enzymes of the Nrf2 pathway modulation. The mRNA expression of HO-1 is an indicator of Nrf2 pathway activation. Thus, mRNA expression of HO-1 was measured by real-time RT-PCR. There was an increase of HO-1 mRNA expression in the vehicle group at 24 hours after reperfusion (P < 0.01; Figure 5D) and treatment of salidroside significantly enhanced the mRNA expression of HO-1 (P < 0.01; Figure 5D).
Discussion
The neuroprotective effects of salidroside and the underlying mechanisms were examined in a rat model of transient focal cerebral ischemia and reperfusion. We demonstrated the effectiveness of salidroside for reducing infarct volume and improving neurological deficit scores after cerebral ischemic injury. In addition, the influence of salidroside on cerebral antioxidant enzymes after cerebral ischemia and reperfusion was explored. Three enzymes, SOD, GSH-Px and GST, which play a key role in ischemia/reperfusion injury, were tested. Our results showed that salidroside increased the activity of the antioxidant enzyme SOD and GST and reduced the content of MDA in the cerebral ischemic cortex. To further clarify the mechanism of the antioxidant activity, the effect of salidroside on the expression of Nrf2 and its downstream enzyme HO-1 was examined after cerebral ischemia and reperfusion. Results from immunohistochemical staining and western blot assays showed that cellular Nrf2 was higher in the cortex of the ischemic hemisphere and that salidroside further increased Nrf2 levels. HO-1, a ubiquitous and redox-sensitive inducible stress protein that degrades heme to CO, iron and biliverdin, is the phase II gene that is directly modulated by Nrf2. The level of HO-1 is an indicator of Nrf2 pathway activation. Previous studies have demonstrated that HO-1 protein is induced after focal cerebral ischemia and reperfusion, which may augment oxidative defense mechanisms that are compromised by this insult (Tanaka et al., 2011). In the current study, HO-1 protein and mRNA levels were elevated 24 hours after cerebral ischemia and reperfusion, and administration of salidroside further enhanced HO-1 protein level and mRNA expression. These results indicate that salidroside may be able to activate the Nrf2-ARE endogenous antioxidant system.
Recently, several research groups reported salidroside activates the Nrf2/ARE pathway. Shao et al. (2012) reported that salidroside activated Nrf2-ARE-dependent gene expression in an in vitro Nrf2-ARE screening model. Salidroside activates Nrf2-regulated antioxidant signaling in its protective effect against OGD/re-oxygenation-induced H9c2 cell injury (Zheng et al., 2014). In MPP+ -induced PC12 cells, salidroside attenuated cell apoptosis by the activation of Nrf2 (Wang et al., 2013). Salidroside protects against myocardial damage after exhaustive exercise and peroxisome proliferator-activated receptor-gamma coactivator 1 alpha and Nrf2 protein expression increase-induced mitochondrial biogenesis were involved in its mechanism (Cui et al., 2014). The results from the current study provided further proof that Nrf2 activation was involved in the protective effect of salidroside in ischemic cerebral injury. As the study was performed in vivo, it has some significance in the development of salidroside as a new neuroprotectant against ischemic stroke.
Zou et al. (2009) and Shi et al. (2012) analyzed the neuroprotective effects of salidroside on cerebral ische-mia/reperfusion injury in vivo. They concluded that salidroside prevented cerebral ischemia/reperfusion injury after pretreatment of salidroside (12 mg/kg per day) for 7 consecutive days. In our study, we did not pretreat and salidroside was administered twice, immediately before MCAO and immediately after reperfusion. Additionally, 2,3,5-triphenyltetrazolium chloride staining and neurological deficit scoring showed that 15 mg/kg salidroside was not as effective as 30 mg/kg, so we used the effective dosage 30 mg/kg to analyze the mechanisms.
Ischemic stroke promotes the upregulation of many cellular antioxidant defense systems including enzymes such as SOD and HO-1 (Takizawa et al., 1998; Fukui et al., 2002). Despite their neutralizing power of reactive oxygen species, they are not sufficient to prevent the deleterious stroke-induced generation of reactive oxygen species. The present results showed that at 24 hours after ischemia and reperfusion, the activity of the anti-oxidant enzymes SOD and GST increased in the ischemic cortex (considered to be the peripheral region of the ischemic core) (Tanaka et al., 2011). Correspondingly, MDA levels decreased, and the expression of Nrf2 and HO-1 increased. It is well-documented that the transcriptional control of the expression of these enzymes is mediated, at least in part, through the ARE found in the regulatory regions of the corresponding genes. The transcription factor, Nrf2, binds to ARE and appears to be essential for the induction of prototypical phase 2 enzymes such as HO-1 and GSTs (Ramos-Gomez et al., 2001). The increase in Nrf2 and its downstream enzymes indicate that the brain's endogenous antioxidant system was activated 24 hours after cerebral ischemia and reperfusion. This result is consis-tent with other reports (Tanaka et al., 2011; Chen et al., 2012). Results of this study also indicated that salidroside may increase the expression of Nrf2 and its downstream phase enzymes after cerebral ischemia and reperfusion.
Interestingly, our results showed that the Nrf2 and HO-1 immunoreactivity showed different patterns in the cortex and striatum. One possible explanation may be that the expression levels of Nrf2 and HO-1 were different in different cell types in the brain. Double-staining immunofluorescence can be used to show in which cell types Nrf2 and HO-1 expressed. Further research is needed to fully clarify the pathway(s) involved in Nrf2 elevation and its associated downstream enzymes.
In summary, salidroside has neuroprotective effects after cerebral ischemia/reperfusion injury, and may involve activation of the Nrf2 pathway and its endogenous antioxidant system. Salidroside may be a potential therapeutic agent for ischemic stroke. The Nrf2/ARE pathway could be regarded as a potent therapeutic target for oxidative stress-related neurological disorders.
Footnotes
Funding: This work was supported by the Independent Research Project of Fujian Academy of Traditional Chinese Medicine in China, No. 2012fjzyyk-4; the Natural Science Foundation of Fujian Province in China, No. 2014J01340; the Research Project of Fujian Provincial Health and Family Planning Commission, No. 2014-ZQN-JC-32; a grant from the Platform for Preclinical Studies of Traditional Chinese Medicine and Quality Control Engineering Technology Research Center of Fujian Province in China, No. 2009Y2003.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Copyedited by Paul P, Pack M, Wang J, Qiu Y, Li CH, Song LP, Zhao M
References
- 1.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, Chan PH. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14:1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen L, Wang L, Zhang X, Cui L, Xing Y, Dong L, Liu Z, Li Y, Zhang X, Wang C, Bai X, Zhang J, Zhang L, Zhao X. The protection by Octreotide against experimental ischemic stroke: up-regulated transcription factor Nrf2 HO-1 and down-regulated NF-kappaB expression. Brain Res. 2012;1475:80–87. doi: 10.1016/j.brainres.2012.07.052. [DOI] [PubMed] [Google Scholar]
- 3.Cui Y, Zhang L, Ping Z, Cao X. Effect of salidroside on key regulatory factors of the myocardial mitochondrial biogenesis in rats after acute exhaustive exercise. Jiefangjun Yiyao Zazhi. 2014;26:6–10. [Google Scholar]
- 4.Fukui S, Ookawara T, Nawashiro H, Suzuki K, Shima K. Post-ischemic transcriptional and translational responses of EC-SOD in mouse brain and serum. Free Radic Biol Med. 2002;32:289–298. doi: 10.1016/s0891-5849(01)00804-8. [DOI] [PubMed] [Google Scholar]
- 5.Huang SC, Lee FT, Kuo TY, Yang JH, Chien CT. Attenuation of long-term Rhodiola rosea supplementation on exhaustive swimming-evoked oxidative stress in the rat. Chin J Physiol. 2009;52:316–324. doi: 10.4077/cjp.2009.amh029. [DOI] [PubMed] [Google Scholar]
- 6.Kelly GS. Rhodiola rosea: a possible plant adaptogen. Altern Med Rev. 2001;6:293–302. [PubMed] [Google Scholar]
- 7.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 8.Leung SB, Zhang H, Lau CW, Huang Y, Lin Z. Salidroside improves homocysteine-induced endothelial dysfunction by reducing oxidative stress. Evid Based Complement Alternat Med. 2013;2013:679–635. doi: 10.1155/2013/679635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013;1497:32–39. doi: 10.1016/j.brainres.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 10.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ÄÄCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 11.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 12.Lu L, Yuan J, Zhang S. Rejuvenating activity of salidroside (SDS): dietary intake of SDS enhances the immune response of aged rats. Age. 2013;35:637–646. doi: 10.1007/s11357-012-9394-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manzanero S, Santro T, Arumugam TV. Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochem Int. 2013;62:712–718. doi: 10.1016/j.neuint.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Mao GX, Deng HB, Yuan LG, Li DD, Li YY, Wang Z. Protective role of salidroside against aging in a mouse model induced by D-galactose. Biomed Environ Sci. 2010;23:161–166. doi: 10.1016/s0895-3988(10)60047-5. [DOI] [PubMed] [Google Scholar]
- 15.Mattioli L, Funari C, Perfumi M. Effects of Rhodiola rosea L. extract on behavioural and physiological alterations induced by chronic mild stress in female rats. J Psychopharmacol. 2009;23:130–142. doi: 10.1177/0269881108089872. [DOI] [PubMed] [Google Scholar]
- 16.McCord JM, Edeas MA. SOD, oxidative stress and human pathologies: a brief history and a future vision. Biomed Pharmacother. 2005;59:139–142. doi: 10.1016/j.biopha.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shao S, Ma ZC, Hong Q, Wang YG, Wang XY, Gao Y. Screening antioxidants to protect radiation via Nrf2-ARE reporter gene. Chin Pharmacol Bull. 2012;28:29–33. [Google Scholar]
- 19.Shi TY, Feng SF, Xing JH, Wu YM, Li XQ, Zhang N, Tian Z, Liu SB, Zhao MG. Neuroprotective effects of Salidroside and its analogue tyrosol galactoside against focal cerebral ischemia in vivo and H2O2-induced neurotoxicity in vitro. Neurotox Res. 2012;21:358–367. doi: 10.1007/s12640-011-9290-7. [DOI] [PubMed] [Google Scholar]
- 20.Shinkai Y, Sumi D, Fukami I, Ishii T, Kumagai Y. Sulforaphane an activator of Nrf2 suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006;580:1771–1774. doi: 10.1016/j.febslet.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 21.Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, Moore TA, Luo W, Yu QS, Johnson DA, Johnson JA, Greig NH, Mattson MP. Plumbagin a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem. 2010;112:1316–1326. doi: 10.1111/j.1471-4159.2009.06552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takizawa S, Hirabayashi H, Matsushima K, Tokuoka K, Shinohara Y. Induction of heme oxygenase protein protects neurons in cortex and striatum but not in hippocampus, against transient forebrain ischemia. J Cereb Blood Flow Metab. 1998;18:559–569. doi: 10.1097/00004647-199805000-00011. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka N, Ikeda Y, Ohta Y, Deguchi K, Tian F, Shang J, Matsuura T, Abe K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011;1370:246–253. doi: 10.1016/j.brainres.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Wang S, He H, Li X, Wang L, He G, Chen J. Salidroside attenuated MPP+-induced appoptosis in PC12 cells by the activation of Nrf2. Chin J Neuroanat. 2013;29:651–655. [Google Scholar]
- 25.Xu J, Li Y. Effects of salidroside on exhaustive exerciseinduced oxidative stress in rats. Mol Med Report. 2012;6:1195–1198. doi: 10.3892/mmr.2012.1060. [DOI] [PubMed] [Google Scholar]
- 26.Yu S, Liu M, Gu X, Ding F. Neuroprotective effects of salidroside in the PC12 cell model exposed to hypoglycemia and serum limitation. Cell Mol Neurobiol. 2008;28:1067–1078. doi: 10.1007/s10571-008-9284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan Y, Wu SJ, Liu X, Zhang LL. Antioxidant effect of salidroside and its protective effect against furan-induced hepatocyte damage in mice. Food Funct. 2013;4:763–769. doi: 10.1039/c3fo00013c. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Zhen YF, Pu Bu Ci R, Song LG, Kong WN, Shao TM, Li X, Chai XQ. Salidroside attenuates beta amyloid-induced cognitive deficits via modulating oxidative stress and inflammatory mediators in rat hippocampus. Behav Brain Res. 2013a;244:70–81. doi: 10.1016/j.bbr.2013.01.037. [DOI] [PubMed] [Google Scholar]
- 29.Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. 2013b;100:30–47. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng K, Sheng Z, Li Y, Lu H. Salidroside inhibits oxygen glucose deprivation (OGD)/re-oxygenation-induced H9c2 cell necrosis through activating of Akt-Nrf2 signaling. Biochem Biophys Res Commun. 2014;451:79–85. doi: 10.1016/j.bbrc.2014.07.072. [DOI] [PubMed] [Google Scholar]
- 31.Zhou MJ, Zheng L, Guo L, Liu WL, Lv C, Jiang LH, Ou CS, Ding ZH. Differential responses to UVB irradiation in human keratinocytes and epidermoid carcinoma cells. Biomed Environ Sci. 2012;25:583–589. doi: 10.3967/0895-3988.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 32.Zou YQ, Cai ZY, Mao YF, Li JB, Deng XM. Effects of salidroside-pretreatment on neuroethology of rats after global cerebral ischemia-reperfusion. Zhong Xi Yi Jie He Xue Bao. 2009;7:130–134. doi: 10.3736/jcim20090207. [DOI] [PubMed] [Google Scholar]