

Inhibition of the transcription factor NF-κB or its target genes should be considered for the treatment of patients with glioblastoma multiforme.
Keywords: Gliomas, NF-κB inhibitors, TIMP1, Xenografts
Abstract
Glioblastoma multiforme (GBM) is the most common and lethal form of intracranial tumor. We have established a lentivirus-induced mouse model of malignant gliomas, which faithfully captures the pathophysiology and molecular signature of mesenchymal human GBM. RNA-Seq analysis of these tumors revealed high nuclear factor κB (NF-κB) activation showing enrichment of known NF-κB target genes. Inhibition of NF-κB by either depletion of IκB kinase 2 (IKK2), expression of a IκBαM super repressor, or using a NEMO (NF-κB essential modifier)–binding domain (NBD) peptide in tumor-derived cell lines attenuated tumor proliferation and prolonged mouse survival. Timp1, one of the NF-κB target genes significantly up-regulated in GBM, was identified to play a role in tumor proliferation and growth. Inhibition of NF-κB activity or silencing of Timp1 resulted in slower tumor growth in both mouse and human GBM models. Our results suggest that inhibition of NF-κB activity or targeting of inducible NF-κB genes is an attractive therapeutic approach for GBM.
INTRODUCTION
Glioblastoma multiforme (GBM) is one of the deadliest human cancers, with an average survival of around one year from the time of diagnosis (1). This poor prognosis is due to therapeutic resistance and tumor recurrence following surgical removal. GBM is remarkably heterogeneous and may represent several unique entities with distinct cell types of origin and various genetic lesions, resulting in different clinical behaviors (2, 3).
Recent advances in molecular technology, mainly high-density microarrays and next-generation sequencing, have allowed the classification of glioblastomas into subtypes based not only on histological level but also on gene expression signatures. Although several reports have attempted to define the different molecular subtypes of GBM (4–7), two subtypes, termed proneural and mesenchymal (MES), appear robust and generally consistent among the different classifications proposed. Tumors classified into the MES subtype exhibit worse prognosis and have been correlated with poor radiation responses, high expression of CD44, and nuclear factor κB (NF-κB) activation (8). Despite the progress in genetic analysis and classification of GBM, there remains an insufficient understanding of the underlying mechanisms of progression and recurrence of gliomagenesis.
NF-κB comprises a family of transcription factors that can form different heterodimers or homodimers and bind to consensus DNA sequences at promoter regions of target genes. NF-κB is a master regulator of cell survival, inflammation, and immunity, and is involved in a myriad of activities related to cellular functions. NF-κB activation pathways can be triggered by a variety of stimuli, including cytokines (tumor necrosis factor–α and interleukin-1β), pathogen-associated molecular patterns, ultraviolet and ionizing radiation, reactive oxygen species, growth factors, DNA damage, and oncogenic stress (9).
In a canonical pathway, NF-κB homodimers or heterodimers are retained in the cytoplasm through a noncovalent interaction with an inhibitory protein, IκB. In response to an external signal, the IκB protein is phosphorylated by the IκB kinase (IKK) complex, ubiquitinated, and degraded, leading NF-κB proteins to the nucleus. The IKK complex contains IKK1/IKKα, IKK2/IKKβ, and the NF-κB essential modifier (NEMO)/IKKγ. Upon activation, the IKK complex phosphorylates the IκB inhibitor, marking it for degradation by the proteasomal degradation machinery. Once translocated to the nucleus, NF-κB dimers can bind to DNA and regulate the transcription of various genes involved in several aspects of cellular activities.
Although NF-κB has been shown to play a major role in cancer development by inducing and maintaining a proinflammatory microenvironment, constitutive NF-κB activation appears to promote tumor initiation and progression through mechanisms such as cell proliferation, apoptosis, angiogenesis, tumor metastasis, and reprogramming of metabolism (9). In GBM, constitutive activation of NF-κB has been shown to promote growth and survival. Inhibition of NF-κB activity by sulfasalazine induces glioma cell apoptosis (10), and a strategy to inhibit NF-κB using a decoy oligonucleotide resulted in a significant reduction in cell numbers (11). NF-κB activity has been recently associated with the transition from proneural glioma stem cell (GSC) to MES differentiation, with an associated radioresistant phenotype. Here, MES differentiation parallels NF-κB activation, radioresistance, and poor prognosis (8). RelB has also been reported to be an oncogenic driver of MES glioma tumor growth and invasion (12).
Here, we confirmed the constitutive activity of the transcription factor NF-κB in mouse and human GBM models and found that specific inhibition of NF-κB in tumor cells by knocking down IKK2, overexpressing the super repressor IκBαM, or using a small peptide targeting NEMO [called NEMO-binding domain (NBD)] (13–15) decreased tumor cell proliferative capacity and extended mice survival. We further identified Timp1, one of the NF-κB target genes highly expressed in our mouse GBM tumors, to be involved in the mechanisms of NF-κB–dependent tumor cell proliferation. Furthermore, inhibition of NF-κB using NBD peptide or by knocking down Timp1 with short hairpin RNA (shRNA) in human GBM tumors reduced cell proliferation and resulted in slower tumor progression.
RESULTS
Activation of NF-κB in glioblastoma
MES GBM subtype is characterized by a high frequency of NF1 mutation/deletion, in combination with p53 mutations and loss of heterozygosity (7). In addition, The Cancer Genome Atlas (TCGA) analysis showed specific enrichment of genes in the NF-κB pathway in MES tumors (fig. S1) (7, 16). We modeled the MES subtype by stereotaxic injections of Cre-inducible oncogenic lentiviral vectors containing either shNF1-shp53 or HRasV12-shp53 in the cortex of glial fibrillary acidic protein (GFAP) Cre mice (17). Both oncogenic vectors gave rise to tumors with similar histopathological characteristics and molecular signatures. RNA-Seq analysis of HRasV12-shp53–induced glioma tumors revealed a high expression of NF-κB target genes in comparison to normal brain tissue (Fig. 1A). These results were further validated by quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis using six representative NF-κB target genes in either HRasV12-shp53– or shNF1-shp53–induced glioma tumors (Fig. 1B and fig. S2). Immunofluorescence analysis showed the presence of Nestin, the MES marker CD44, and nuclear p65+ cells, as well as interleukin-6 (IL-6) secretion in tumor sections (Fig. 1C). The expression of additional MES markers (18) was validated by qRT-PCR (fig. S3).
Fig. 1. Constitutive NF-κB activation in the lentivirus-induced GBM mouse model.
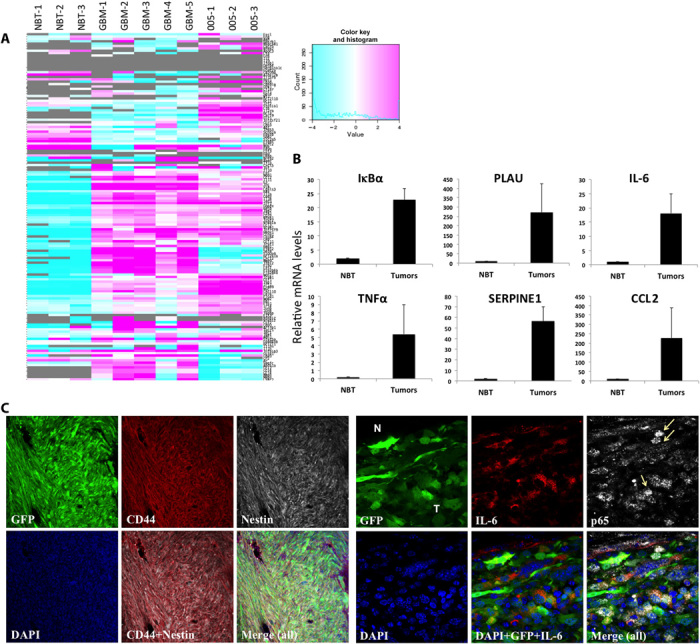
(A) Heat map of the RNA-Seq analysis of NF-κB target genes (list of 150 genes; http://bioinfo.lifl.fr/NF-KB/) comparing normal brain tissue (NBT; n = 3), HRas-shp53–induced GBM tumors (n = 5), and 005 tumors (n = 3). (B) qRT-PCR analysis of six representative NF-κB target genes in NBT and tumor samples. (C) Immunofluorescence staining of CD44 (red) and Nestin (white) in tumor sections shows MES subtype (left) and nuclear p65 (white) staining, and IL-6 (red) confirms NF-κB activation in these tumors (right). Arrows point to p65 staining in the nucleus of cells. N, normal tissue; T, tumor; PLAU, plasminogen activator, urokinase; SERPINE1, serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1; CCL2, chemokine (C-C motif) ligand 2; DAPI, 4′,6-diamidino-2-phenylindole.
To further characterize the extent of constitutive NF-κB activation in tumor cells, we examined NF-κB activity in 005 cells, a tumor-derived mouse cell line that we previously showed to have all the characteristics of a brain tumor–initiating stem cell (BTIC) (19). Nuclear extracts from 005 cells showed a high p65 translocation in comparison to control neural progenitor cells (NPCs) (Fig. 2A). We also transduced 005 cells and NPCs with a κB-mCherry reporter and found a higher expression of mCherry in 005 tumor cells (Fig. 2B and fig. S4). In addition, we performed a qRT-PCR analysis of representative NF-κB target genes and found a higher expression on 005 cells compared to NPCs (Fig. 2C). Finally, RNA-Seq analysis of 005 tumors revealed a high expression of NF-κB target genes in comparison to normal brain tissue (Fig. 1A). Together, these results indicate that our lentivirus-induced MES glioma tumors have constitutive NF-κB activation.
Fig. 2. NF-κB activation in BTICs.
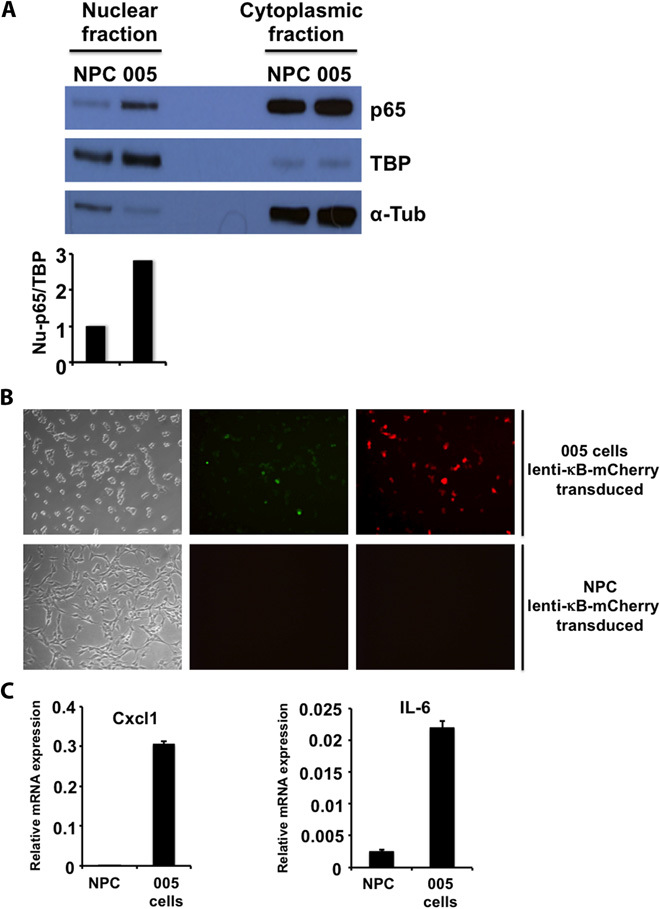
(A) Western blot analysis of nuclear and cytoplasmic fractions of 005 BTICs and control NPCs for p65 and for loading control TATA binding protein (TBP; nuclear protein) and α-tubulin (α-Tub; cytoplasmic protein). (B) Bright and immunofluorescence imaging of 005 BTICs and NPCs transduced with a lenti-κB-mCherry reporter virus. Transduced cells were selected for puromycin resistance. Green, green fluorescent protein (GFP); red, mCherry. (C) qRT-PCR analysis of two representative NF-κB target genes in 005 BTICs and control NPCs. Cxcl1, chemokine (C-X-C motif) ligand 1.
Silencing IKK2 in glioma cells impairs tumor proliferation
The NF-κB pathway has been reported to play an important role in tumor cell development, survival, invasion, and chemoresistance (8), and IKK2 is a critical kinase in the NF-κB activation cascade. We sought to explore the effects of IKK2 inhibition on glioma cells as a therapeutic approach to blocking the NF-κB pathway. We transduced 005 tumor cells with a lenti-shIKK2-mCherry vector and confirmed the knockdown of IKK2 in sorted cells by Western blot analysis and the decreased expression of NF-κB target genes by qRT-PCR analysis (Fig. 3, A and B). We next analyzed the proliferation capacity of IKK2 knockdown cells in comparison to parental 005 cells or 005 cells transduced with a scrambled lenti-shRNA (shIRR), and we found that silencing IKK2 in 005 cells impaired their proliferative capacity (Fig. 3C). To examine the effects of IKK2 inhibition on tumor growth, we performed in vivo experiments by transplanting 005-shIKK2 cells and 005-shIRR control cells into the cortex of new mice. Compared with 005-shIRR transplanted mice, 005-shIKK2 mice developed glioma tumors with a longer latency (Fig. 3D). Similar results were obtained when the same line of experiments was conducted in a BTIC derived from an shNF1-shp53 tumor (fig. S5). We next decided to work with an inducible system where primary cortical astrocytes isolated from IKK2fl/fl transgenic mice were transduced with a lenti-HRasV12-iresCre-ErT2-shp53 construct (see the diagram of a lentivector in Fig. 3E). Transduced astrocytes (AR53Cre-ErT2) lose IKK2 expression in the presence of 4-hydroxytamoxifen (4-ohtmx; the active metabolite of tamoxifen), as shown by Western blot analysis and qRT-PCR analysis of NF-κB target genes (Fig. 3, F and G). In addition, we confirmed the knockdown of IKK2 by immunofluorescence staining of p65 and electrophoretic mobility shift assay for NF-κB binding (fig. S6, A and B). As shown before for 005 cells, inhibition of IKK2 by a Cre-inducible deletion of IKK2 in AR53Cre-ErT2 cells reduced their proliferative capacity (Fig. 3H). Finally, orthotopic transplantation of AR53Cre-ErT2 cells and cells transduced with the IκBαM super repressor (nondegradable mutant form of IκBα; fig. S6) showed that blocking NF-κB activity, either by Cre-inducible deletion of IKK2 or by use of the IκBαM super repressor, prolongs mice survival (Fig. 3I).
Fig. 3. IKK2 depletion affects the proliferative capacity of tumor cells and prolongs mice survival.
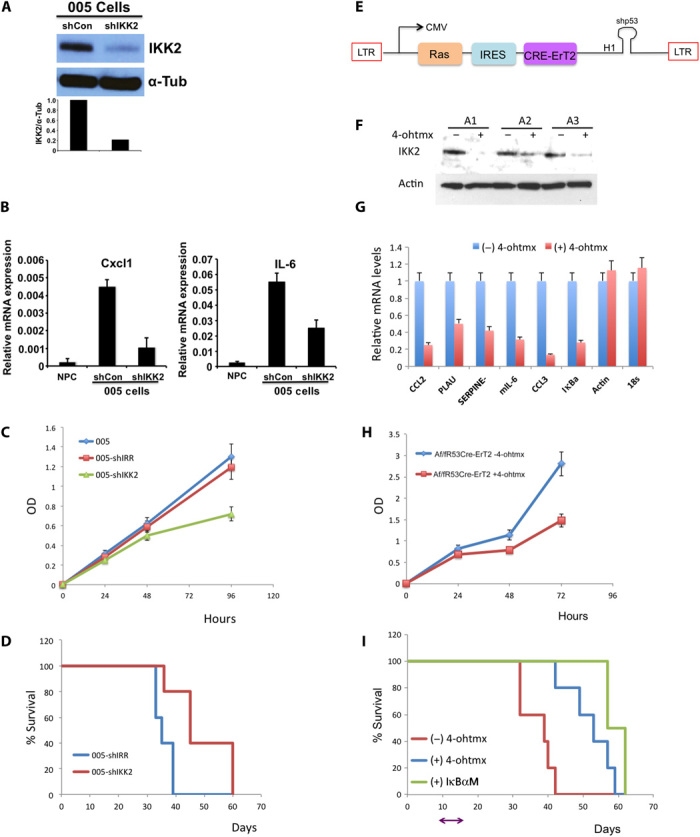
(A) Western blot analysis of the silencing of IKK2 in 005 BTICs. 005 cells were transduced with either a scrambled shRNA control (shCon) or an shRNA targeting IKK2 (shIKK2). α-Tubulin (α-Tub) was used as loading control. (B) Silencing of IKK2 was further validated by decreased relative mRNA levels of representative NF-κB target genes (qRT-PCR analysis). Actin and 18S were used as control genes not affected by silencing of IKK2. (C) 005 control, 005-shIRR (transduced with scrambled shRNA), and 005-shIKK2 were seeded on culture plates (in triplicate), and WST-1 cell proliferation assay reagent was used to monitor cell proliferation at the indicated time points. (D) Kaplan-Meier curves show the survival of mice implanted with 005-shIRR or 005-shIKK2 at 3 × 105 cells per mouse. (E) Diagram of lentivector design: the GFP cassette in the HRas-shp53 vector was replaced by Cre-ErT2. (F) Induced silencing of IKK2 in primary cortical astrocytes from IKK2fl/fl transgenic mice. Astrocytes from three IKK2fl/fl mice were isolated (A1, A2, and A3) and transduced with HRas-shp53-Cre-ErT2. Western blot analysis confirmed IKK2 deletion by addition of 4-ohtmx to the culture media. (G) Deletion of IKK2 by addition of 4-ohtmx was further validated by qRT-PCR of representative NF-κB target genes. (H) Af/fR53Cre-ErT2 cells were seeded on culture plates (in triplicate) in the absence or presence of 4-ohtmx, and WST-1 cell proliferation assay reagent was used to monitor cell proliferation at the indicated time points. (I) Kaplan-Meier curve shows the survival of mice implanted with Af/fR53Cre-ErT2 or Af/fR53Cre-ErT2 transduced with lenti-IκBαM (Af/fR53Cre-ErT2-IκBαM) at 3 × 105 cells per mouse. On day 10 after transplantation, one group of Af/fR53Cre-ErT2–implanted mice received 4-ohtmx for 5 days to delete IKK2. OD, optical density; LTR, long terminal repeat; CMV, cytomegalovirus; IRES, internal ribosomal entry site; CCL3, chemokine (C-C motif) ligand 3.
NBD peptide inhibits NF-κB activation in gliomas
We have so far used a genetic approach to block NF-κB activity in glioma cells. Next, we sought to explore the selective inhibition of NF-κB activation by a peptide that blocks the interaction of the regulatory protein NEMO with the IKK complex (13–15, 20–22). Activation of NF-κB requires the activity of the IKK complex that includes IKK1 and IKK2 and the regulatory protein NEMO (23). Ghosh et al. (14) showed that the NBD peptide (Fig. 4A) specifically blocked the cytokine-induced NF-κB activity and the NF-κB target gene expression without affecting the basal NF-κB activity. Moreover and relevant to our study, they showed that the NBD peptide can enter the central nervous system (CNS) and inhibit NF-κB activity in vivo (fig. S7) (14).
Fig. 4. Effects of the NBD peptide on mouse glioma cell lines.
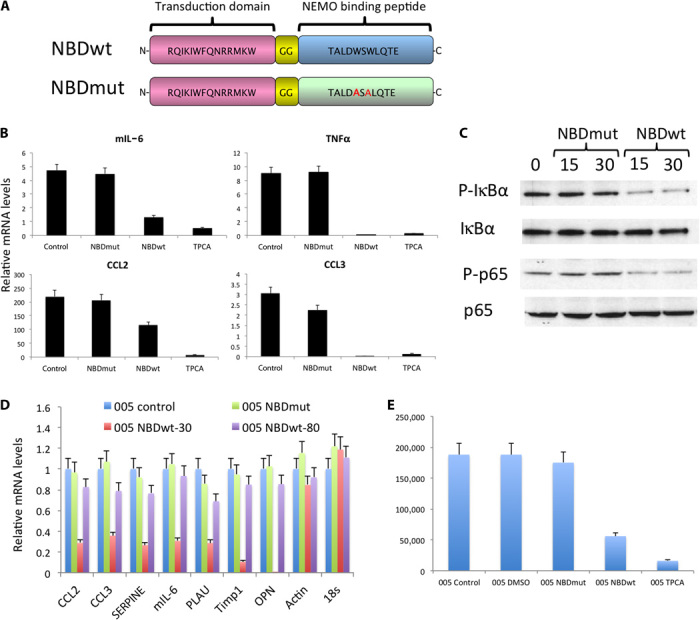
(A) Diagram of the NBDwt and NBDmut peptides. The W amino acids in the NBDwt peptide were substituted by A in the NBDmut (highlighted in red). (B) qRT-PCR analysis of NF-κB representative genes. Af/fR53Cre-ErT2 cells were incubated with NBDmut (50 μM), NBDwt (50 μM), or TPCA inhibitor (5 μM) for 24 hours. (C) Af/fR53Cre-ErT2 cells were incubated with either NBDmut or NBDwt at a concentration of 50 μM for the indicated times. Cell lysates were analyzed by Western blot analysis. (D) qRT-PCR analysis of NF-κB representative genes. 005 BTICs were incubated with either NBDmut or NBDwt at a concentration of 50 μM for 24 hours. (E) 005 cells were incubated with either NBDmut or NBDwt at a concentration of 50 μM or with TPCA inhibitor (5 μM) for 48 hours. Cell numbers (viable cells) were counted.
To test the inhibitory effects of NBD on glioma cell lines, we incubated AfR53Cre-ErT2 cells with either the NBD peptide or TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), a small-molecule inhibitor of IKK2. Analysis by qRT-PCR showed a decreased expression of NF-κB target genes (Fig. 4B). NBDwt, but not a mutant form of the peptide (NBDmut), reduced NF-κB activity, as shown by decreased P-IκBα and P-p65 (Fig. 4C). Similar results were obtained when 005 BTICs were incubated with NBDwt peptide, as validated by qRT-PCR analysis and proliferation assay (Fig. 4, D and E).
We next examined the effects of NBD treatment using an orthotopic model for 005 BTICs. We started treating the mice 10 days after 005 cells were stereotaxically transplanted into the brains of the mice. Each mouse received a daily dose (10 mg/kg) of either NBDwt or NBDmut peptide intraperitoneally for 20 days. Compared to the nontreated or NBDmut-treated mice, the NBDwt-treated mice developed tumors with a much longer latency (Fig. 5A). At the end of the 20-day treatment, we sacrificed two mice in the NBDwt-treated group and extracted RNA from the small GFP+ lesion in their brain. Next, we compared the expression of NF-κB target genes by qRT-PCR analysis with two representative tumors from mice in the nontreated group, two tumors from mice in the NBDmut-treated group, and two representative tumors from mice in the NBDwt-treated group that succumbed to the disease (end point). The results show that NBDwt was capable of inhibiting the activity of NF-κB in the tumor during the duration of the treatment, which was evident in the lower expression of representative NF-κB target genes (Fig. 5B). Although NBDwt-treated mice had a small tumor lesion at an early time point (fig. S8), they eventually developed comparable tumors at the end points and recovered the expression of NF-κB target genes (Fig. 5B). Taking into account the antiproliferative effect of NBDwt on glioma cell lines in vitro, we examined tumor sections at the end point for the proliferation marker Ki67 and found that NBDwt-treated mice had a low expression of Ki67 compared to the control group (Fig. 5, C and D). These results support the idea that the major contribution of the NF-κB pathway to our glioma model is most likely pro-proliferation.
Fig. 5. Treatment of 005 tumors with the NBD peptide attenuated tumor growth.
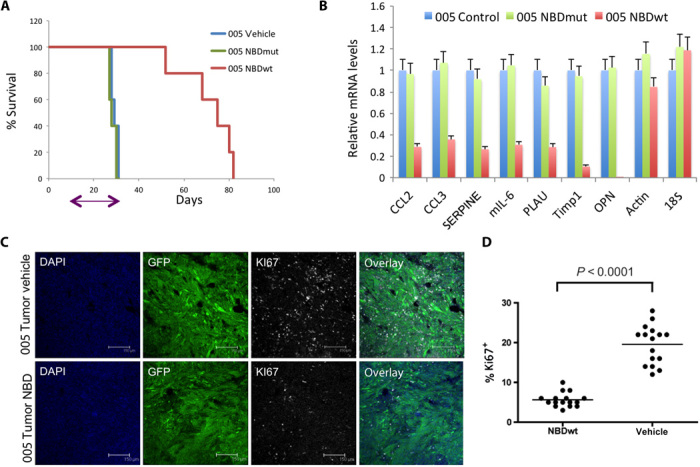
(A) Kaplan-Meier curve shows the survival of mice implanted with 005 cells at 3 ×105 cells per mouse. On day 10 after transplantation, mice received a daily dose of either NBDmut or NBDwt (10 mg/kg) intraperitoneally for 20 days (n = 10); arrow indicates length of treatment (P = 0.01). (B) RNA was isolated from GFP+ tumors of two representative mice from the NBDwt-treated group sacrificed on day 30 (NBDwt-30; small lesion), together with two mice from the NBDwt (NBDwt-80), NBDmut, and control groups all at end point. qRT-PCR analysis shows decreased mRNA levels of representative NF-κB genes in NBDwt-treated mice. (C) Tumors from NBDwt and vehicle-treated mice (end point) were collected, and sections were immunostained with Ki67 and analyzed by confocal microscopy. (D) Quantification of Ki67+ cells at multiple fields.
To further validate our results in a human setting, we examined the constitutive NF-κB activity in patient-derived glioma cell lines. qRT-PCR of representative NF-κB target genes confirmed NF-κB activity (Fig. 6A) and RNA-Seq analysis of these cell lines. Next, we evaluated the effects of the NBD peptide on U87 cells and found that the expression of NF-κB target genes is decreased in the presence of NBDwt but not when the cells were incubated with NBDmut peptide (Fig. 6B). To examine the effects of NBDwt in vivo, we transduced U87 cells with a lentivirus expressing luciferase and injected into the brain of NOD-SCID (nonobese diabetic–severe combined immunodeficient) mice (the experimental design is outlined in fig. S9A). We initiated treatment with NBDwt peptide 10 days after U87-luc injection, and bioluminescent imaging results showed that NBDwt treatment resulted in slower tumor growth and reduced tumor size (Fig. 6C). Mice in the control group succumbed to the disease after 30 days of injection, and this time, we decided to continue the treatment with NBDwt peptide. Almost 40 days after treatment, mice started to show symptoms of sickness (they lost 20 to 25% of their original weight), and we were not able to continue with the experiment. Toxicology analysis revealed liver toxicity, as shown by an elevated serum level of the liver enzyme l-alanine-2-oxoglutarate aminotransferase (ALT) (fig. S9B). Although we were not able to continue treatment beyond 40 days owing to the toxic side effects, NBDwt treatment almost doubled the median survival time from less than 30 days to more than 50 days (Fig. 6D). Together, these results suggest that NBDwt peptide slows tumor growth in both mouse and human GBM models and extends survival.
Fig. 6. Treatment of human GBM with the NBD peptide prolongs mice survival.
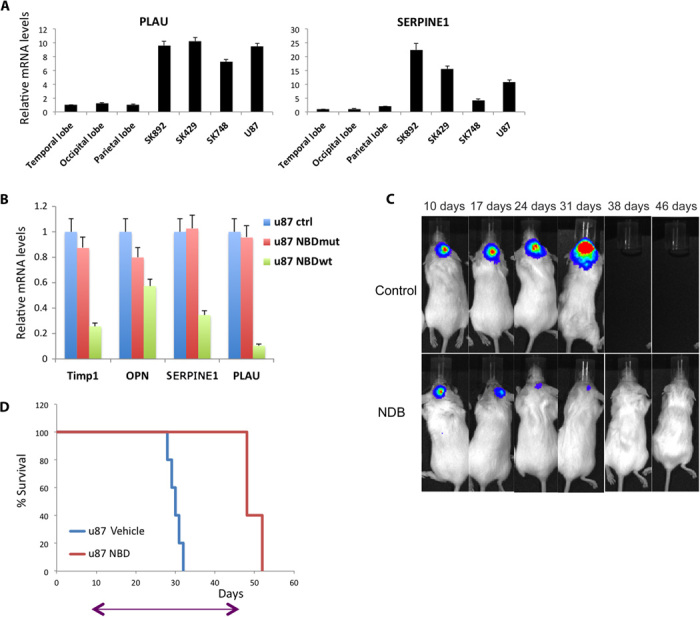
(A) qRT-PCR analysis of PLAU and SERPINE1 in human-derived glioma cell lines and normal brain tissue from the indicated regions. Error bar indicates ±SD. (B) qRT-PCR analysis of NF-κB target genes after treatment with either NBDmut or NBDwt for 24 hours. Error bar indicates ±SD. (C) Mice were monitored by IVIS imaging at the indicated time points; one representative mouse from each of the indicated groups is shown. (D) Kaplan-Meier curve shows the survival of mice implanted with U87 cells at 3 × 105 cells per mouse. On day 10 after transplantation, mice received a daily dose of NBDwt (10 mg/kg) intraperitoneally for 36 days (n = 10); arrow indicates length of treatment.
Timp1 is one of the NF-κB target genes involved in tumor proliferation
The pleiotropic toxicity of NF-κB inhibitors is known (24), so we next sought to find downstream NF-κB target genes involved in NF-κB–induced tumor proliferation as therapeutic targets. We examined the expression levels of NF-κB target genes, and Timp1 was among the genes significantly up-regulated in the lentivirus-induced GBM tumors (table S1). Xia et al. (25) recently identified Timp1 as one of the mediators for NF-κB–induced lung tumor growth. Here, we validated the expression of Timp1 in mouse glioma tumors and mouse GBM cell lines by qRT-PCR (Fig. 7A). In addition, when 005 and NF5310 BTICs were treated in vitro with the NF-κB inhibitor TPCA-1, Timp1 expression levels decreased (Fig. 7B). The same results were observed when NF-κB activity was inhibited by either deletion of IKK2 (fig. S10A) or expression of the IκBαM super repressor in AR53Cre-ErT2 cells (fig. S10B). As Xia et al. (25) showed previously in lung cancer, a lack of NF-κB activation leads to a low expression of Timp1 and impaired phosphorylation of Erk, supporting the idea that, as in lung cancer, NF-κB and its target Timp1 play an important role in glioma cell proliferation (fig. S10B). Reduced levels of Timp1 were also found in 005 tumors treated with NBDwt peptide compared to the control group (fig. S10C).
Fig. 7. Timp1 expression in mouse and human glioma cells.
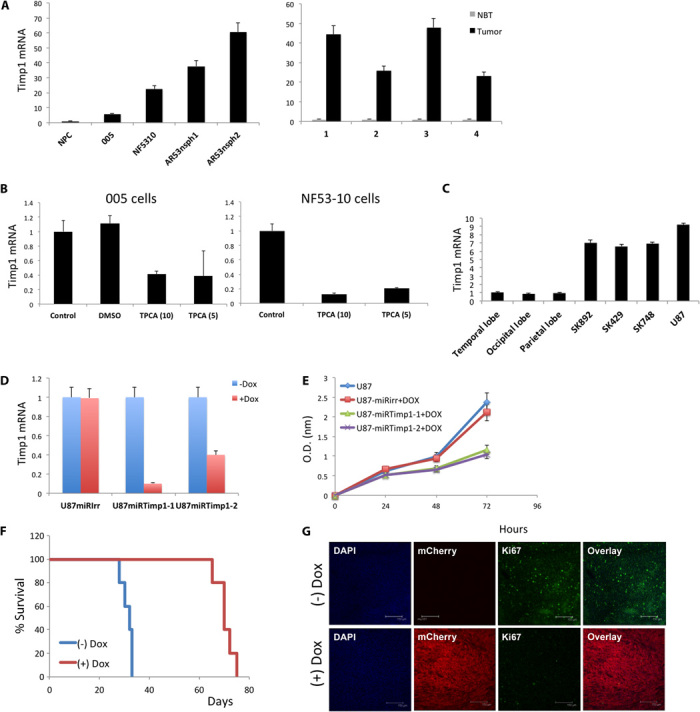
(A) qRT-PCR analysis of Timp1 in mouse glioma cell lines and NPCs as control (left graph) and four different tumors induced by injection of HRas-shp53 and the corresponding normal brain tissue (control; right graph). Error bar indicates ±SD. (B) qRT-PCR analysis of Timp1 after treatment with the TPCA inhibitor in 005 and NF5310 cells for 24 hours. Error bar indicates ±SD. (C) qRT-PCR analysis of Timp1 in human-derived glioma cell lines and normal brain tissue from the indicated regions. Error bar indicates ±SD. (D) qRT-PCR analysis of Timp1 after knockdown of Timp1 with an inducible shTimp1 vector (miRTimp1-1 and miRTimp1-2; a scrambled miRIrr was used as control) in U87 cells. Timp1 knockdown was induced by adding Dox to the culture media. Error bar indicates ±SD. (E) U87 cells transduced with either miRIrr, miRTimp1-1, or miRTimp1-2 were seeded on culture plates (in triplicate), and WST-1 cell proliferation assay reagent was used to monitor cell proliferation at the indicated time points. Timp1 knockdown was induced by Dox. Error bar indicates ±SD. (F) Kaplan-Meier plot of survival for mice fed normal chow [miRTimp1(−)Dox; n = 10] and Dox-containing chow [miRTimp1(+)Dox; n = 10]. The (+)Dox group was fed the special chow 10 days after the transplantation of U87-miRTimp1 cells [3 × 105 cells per mouse; arrow indicates the beginning of (+)Dox chow diet]. (G) Tumors from each group were collected at the end of the experiment, and brain sections (40 nm) were immunostained with Ki67 and analyzed using confocal microscopy. mCherry is a fluorescent reporter expressed in the lentiviral microRNA construct. DAPI nuclei. Scale bar, 150 μm. DMSO, dimethyl sulfoxide.
We next investigated the role of Timp1 in human GBM by assessing the expression of the gene in TCGA data sets. Timp1 is highly expressed in glioma tumors and is significantly up-regulated (approximately 60% of the tumors) in comparison to pooled normal controls (fig. S11, A and B). In addition, we confirmed by qRT-PCR that our patient-derived cell lines also express high levels of Timp1 (Fig. 7C). To test the functional consequence of silencing Timp1, we designed an inducible lentivirus to knock down Timp1 (miRTimp1; fig. S12, A and B, and Fig. 7D). Silencing Timp1 in U87 cells significantly decreased their proliferative capacity (Fig. 7E). The addition of the matrix metalloproteinase inhibitor GM6001 had no effect on the proliferative capacity of U87 cells, suggesting that the original role of Timp1 is not responsible for its pro-proliferative activity, in agreement with what was previously reported for the lung cancer model (25). Finally, U87-miRTimp1 cells were transplanted into the brain of NOD-SCID mice. Ten days after the transplantation, one group of mice [miRTimp1(+)Dox] received doxycycline (Dox) in their chow. The control group continued to feed on normal chow. As shown in Fig. 7F, specific inhibition of Timp1 prolonged mice survival compared with the control groups. Depletion of Timp1 in tumor cells resulted in slower tumor growth (fig. S13), and confocal analysis of tumor sections showed that U87-miRTimp1(+)Dox expressed lower levels of Ki67 (Fig. 7G). qRT-PCR analysis confirmed that inducible shTimp1 tumors [miRTimp1(+)Dox] had less Timp1 expression than control mice [miRTimp1(−)Dox] (fig. S14).
To further investigate the correlation between Timp1 expression and patient survival, we looked through the REMBRANDT (REpository for Molecular BRAin Neoplasia DaTa) data set. Glioma patients with down-regulated expression of Timp1 have better prognosis than patients with up-regulation of Timp1 expression (fig. S15). Our data strongly support the possibility of using NF-κB inhibitors or targeting NF-κB downstream target genes (for example, Timp1) as therapeutic approaches for the treatment of glioblastoma.
DISCUSSION
Here, we have confirmed the constitutive NF-κB activity in our MES GBM mouse model and in patient-derived cell lines. Inhibition of NF-κB activity has been shown to impair tumor progression (9). Several groups have proposed the idea of targeting the NF-κB pathway as a therapeutic approach to treating GBM. Preclinical studies using sulfasalazine, an anti-inflammatory drug, showed promising results (10); however, a phase 1/2 clinical trial using this drug in advanced-grade gliomas was terminated as a result of lack of response (26). Other drugs that target NF-κB have been used in preclinical studies either alone or in combination with temozolomide, with the latter being the standard-of-care chemotherapeutic treatment for GBM (27). Radiation therapy has also been extensively used for GBM treatment in the past decades, although the mechanisms responsible for GBM radioresistance remain unknown. Cancer stem cells, a population of cells that sustain the growth of GBM and other types of cancer, have been previously reported to be resistant to radiation therapy (28–30). A recent study by Bhat et al. (8) showed the correlations among MES differentiation and plasticity, NF-κB activation, and poor radiation response accompanied by shorter survival. They suggested that tumor microenvironmental factors induce a MES phenotype in GSCs, rendering these cells radioresistant in an NF-κB–dependent fashion, and they proposed that NF-κB inhibition could directly affect radioresistance and therefore constitute an attractive therapeutic approach for GBM.
Here, we primarily focused on the inhibition of NF-κB activity in tumor cells. We observed a significant decrease in proliferative capacity among cells lacking IKK2 or expressing the IκBαM super repressor. This impaired proliferation was translated in vivo into slower tumor growth. To achieve a better effect in vivo, we used the NBD peptide, a peptide corresponding to the NEMO binding domain that specifically inhibits the induction of NF-κB activity (15). The NBD peptide has been used previously to inhibit inflammation in animal models such as acute inflammation in carrageenan-induced mouse paw edema and collagen-induced arthritis (31) or Duchenne muscular dystrophy (13). One of our main reasons for choosing the NBD peptide to treat GBM is the reported ability of NBD to enter into the CNS and exert an effect on NF-κB activation in a mouse model of Parkinson’s disease (14). Indeed, our results show that NBD treatment of mice with GBM significantly extended their survival, probably as a result of the specific inhibition of NF-κB not only in the tumor cells but also in the tumor microenvironment. The NBD peptide has a potential advantage over other currently available anti–NF-κB drugs that target the IKK kinases. Most of the available small inhibitors of IKK kinases compete for the adenosine triphosphate binding pocket, which is known to be shared by many other kinases. The pleiotropic effect of anti–NF-κB drugs cannot be ruled out, and our attempt to continuously treat glioma-bearing mice intraperitoneally for almost 40 days resulted in toxic effects. In the future, this toxicity effect can be reduced if the NBD peptide is specifically targeted to the tumor. We have previously successfully administered an otherwise highly toxic antiapoptotic peptide (KLAKLAK) coupled to tumor-homing nanoparticles (32).
In our search for an NF-κB target gene, we identified Timp1 as being highly up-regulated in our GBM mouse model and in patient-derived cells. Selective inhibition of Timp1 in tumor cells affected their proliferative capacity, and silencing Timp1 in tumor cells in vivo prolonged mice survival. These results further support our previous findings in lung cancer, where NF-κB and Timp1 were shown to play an important role in tumor cell proliferation and lung cancer progression (9). As discussed previously, many pharmaceutical companies have focused their efforts on developing small inhibitors targeting NF-κB. However, safety concerns arise from the involvement of NF-κB in so many biological functions (8). Identifying target genes of NF-κB that play a role in gliomagenesis may offer an alternative approach to targeting this pathway. We have recently identified Spp1/OPN to be an important hub in the molecular network regulating dedifferentiation of tumor cells. Silencing OPN specifically in tumor cells resulted in longer survival (33). OPN has been reported to have putative NF-κB binding sites in its promoter (34), and our results show decreased levels of OPN when GBM-bearing mice are treated with the NBD peptide (Fig. 5). Further experiments are required to show the link between NF-κB activation, OPN secretion, and dedifferentiation in glioma cells.
In summary, we validated the constitutive NF-κB activity in MES gliomas both in our GBM mouse model and in patient-derived cells. Our results suggest that NF-κB activation has a pro-proliferative function in GBM and that inhibition of this pathway, by inhibiting either NF-κB activity or preferentially NF-κB–inducible genes, presents an attractive therapeutic approach to treating GBM. Suppression of NF-κB activity or target genes by drugs administered locally following surgery will block tumor cell growth and recurrence and will reduce toxicity.
MATERIALS AND METHODS
Mice, lentiviral injections, and orthotopic transplants
hGFAP-Cre and NOD-SCID mice were purchased from the Jackson Laboratory. The IKK2fl/fl mouse was obtained from Pasparakis et al. (University of Cologne) (35). All mice were maintained under pathogen-free conditions at the Salk Institute, and all procedures performed in this study were approved by the Institutional Animal Care and Use Committee.
Lentivirus [HRas-shp53 or shNF1-shp53; (17)], prepared as described previously (36), or cells (3 × 105 cells per mouse) were stereotaxically injected into the brain of mice (transgenic or NOD-SCID, respectively). The shRNA sequences used were 5′-CCACCTTATACCAGCGTTATA-3′ (IKK2), 5′-ATTCCAGTCCCGTCACCTTGCTA-3′ (hTimp1-1), and 5′-AAAGTCAACCAGACCACCTTAT-3′ (hTimp1-2). For in vivo bioluminescent imaging, U87 cells were engineered to express luciferase by transduction with a lenti-CMV-luciferase vector. Tumor growth was monitored using the Xenogen IVIS 100 Imaging System (Caliper Life Sciences). NBD-treated mice were administered 10 mg/kg intraperitoneally for the indicated number of days.
RNA-Seq and bioinformatics analysis
Once mice had reached clinical end points, they were anesthetized and perfused with phosphate-buffered saline. The brain was removed from the skull, and normal brain tissue was trimmed away from GFP+ tumor tissue using a dissecting microscope. Tumor tissue was cut into small pieces and stored in RNAlater (Qiagen). RNA was isolated using the RNeasy kit (Qiagen) according to the manufacturer’s protocol, with the addition of deoxyribonuclease. cDNA libraries were prepared using the TruSeq RNA Sample Prep kit (Illumina). RNA sequencing was performed using a HiSeq 2500 Sequencing System (Illumina). Sequence reads were mapped to the mm9 mouse genome build. Raw reads of each individual gene were counted and normalized to fpkm using Cuffdiff v2.1.1. The NF-κB target gene list was obtained from http://bioinfo.lifl.fr/NF-KB/. A heat map was drawn using heatmap.2 from the R package.
Cell culture
Primary astrocytes were obtained from GFAP-Cre mice pups 10 days after birth and maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Primary astrocytes were transduced at early passage. 005 and NF5310 cells were maintained in N2-supplemented (Invitrogen) DMEM/Ham’s F12 (Omega Scientific) containing fibroblast growth factor-2 (20 ng/ml), epidermal growth factor (20 ng/ml; Promega), and heparin (50 μg/ml; Sigma). SK892, SK429, and SK748 patient-derived cell lines were provided by S. Kesari (University California, San Diego) and maintained in NeuroCult NS-A Basal Medium (StemCell Technologies) supplemented with NeuroCult NS-A Proliferation Supplement, recombinant human epidermal growth factor (20 ng/ml), recombinant human basic fibroblast growth factor (10 ng/ml), and heparin (2 μg/ml). U87 glioma cells were maintained in DMEM containing 10% FBS.
Quantitative reverse transcription polymerase chain reaction
Total RNA isolated from homogenized tumor samples or cell lines was reverse-transcribed using qScript cDNA Synthesis kit (Quanta BioSciences). qRT-PCRs were performed with the 7900HT Fast Real-Time PCR System using the PerfeCTa SYBR Green SuperMix (Quanta BioSciences). Data are presented after normalization with glyceraldehyde-3-phosphate dehydrogenase. The primers are listed in table S2.
Flow cytometry and fluorescent staining
Cells were analyzed on a Becton Dickinson LSR II flow cytometer using FlowJo software. For fluorescent staining, coronal sections (40 μm) were cut on a microtome, and images were obtained using a confocal laser-scanning microscope (Leica TCS SP2 ABS). All of the antibodies are listed in table S3.
WST-1 assay
Cell proliferation was measured using the WST-1 reagent (Roche). Cells were seeded (10,000 to 25,000 cells per well) on a 96-well plate in triplicate, and 10 μl of WST-1 reagent was added per well. Absorbance was measured at 450 nm after 30 min.
Western blot analysis
Protein lysates were obtained by cell lysis using a radioimmunoprecipitation assay buffer supplemented with protease inhibitors (Roche). The antibodies used are listed in table S3.
Toxicity study
Serum was collected from mice before treatment and after treatment (almost 40 days). ALT levels in serum were determined using Infinity ALT (GPT) reagent (Thermo Scientific) according to the manufacturer’s instructions.
Supplementary Material
Acknowledgments
We thank G. Estepa for technical assistance, R. Alvarez Rodriguez for the design of the inducible microRNA lentivector, D. Guttridge for providing NBD peptide and P. Robbins for initial discussions on the use of NBD peptide, E. Ke and C. Benner for bioinformatics analysis, and V. Bhargava for the analysis of Timp1 expression in TCGA data sets. Funding: I.M.V. is an American Cancer Society professor of molecular biology and holds the Irwin and Joan Jacobs Chair in Exemplary Life Science. This work was supported in part by the NIH (grant HL053670 to I.M.V.), the Cancer Center Core Grant (P30 CA014195-38), the H.N. and Frances C. Berger Foundation, and the Leona M. and Harry B. Helmsley Charitable Trust (grant 2012-PG-MED002 to I.M.V.). Author contributions: D.F.-M. and I.M.V. designed the research. D.F.-M., R.N., Y.X., C.M., and Y.S. performed the experiments. D.F.-M. and Y.X. analyzed the results. D.F.-M. and I.M.V. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. HRas-shp53 or shNF1-shp53 lentiviral vectors require materials transfer agreement. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/1/e1501292/DC1
Fig. S1. Heat map of the RNA-Seq analysis of NF-κB target genes (list of 150 genes; http://bioinfo.lifl.fr/NF-KB/) comparing the four molecular subtypes of human GBM (TCGA data, downloaded as level 3 normalization counts; classification from www.cbioportal.org).
Fig. S2. qRT-PCR analysis of six representative NF-κB target genes in normal brain tissue, HRas-shp53 tumor (n = 5), and shNF1-shp53 tumor (n = 5) samples.
Fig. S3. qRT-PCR analysis of six representative MES markers in normal brain tissue and four tumor samples (HRasV12-shp53).
Fig. S4. Flow cytometry analysis of 005 BTICs either left untransduced (control) or transduced with a lenti-κB-mCherry reporter.
Fig. S5. Silencing IKK2 in NF5310 BTICs.
Fig. S6. Silencing IKK2 in Af/fIKK2R53-Cre-ErT2 cells.
Fig. S7. Entry of the NBD peptide into the CNS.
Fig. S8. Hematoxylin and eosin staining of a tumor section from (A) a representative mouse from the control group at clinical end point and (B) an NBDwt-treated mouse that was sacrificed when the control mice had reached clinical end point.
Fig. S9. Treatment of U87 xenograft mice with the NBD peptide.
Fig. S10. Timp1 expression.
Fig. S11. Timp1 expression in human GBM.
Fig. S12. Inducible knockdown of Timp1.
Fig. S13. Bioluminescent imaging of a representative mouse from the U87-miRTimp1(−)Dox and U87-miRTimp1(+)Dox groups at the indicated time points.
Fig. S14. qRT-PCR analysis of Timp1 in U87luc-miRTimp1 tumors in the absence or presence of Dox in the chow.
Fig. S15. Kaplan-Meier survival plot for human glioma samples with differential Timp1 gene expression (National Cancer Institute, REMBRANDT Web site).
Table S1. Expression levels of NF-κB target genes in lentivirus-induced GBM.
Table S2. List of primers.
Table S3. List of antibodies.
REFERENCES AND NOTES
- 1.Furnari F. B., Fenton T., Bachoo R. M., Mukasa A., Stommel J. M., Stegh A., Hahn W. C., Ligon K. L., Louis D. N., Brennan C., Chin L., DePinho R. A., Cavenee W. K., Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 21, 2683–2710 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Huse J. T., Holland E. C., Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 10, 319–331 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Stiles C. D., Rowitch D. H., Glioma stem cells: A midterm exam. Neuron 58, 832–846 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Cooper L. A. D., Kong J., Gutman D. A., Wang F., Cholleti S. R., Pan T. C., Widener P. M., Sharma A., Mikkelsen T., Flanders A. E., Rubin D. L., Van Meir E. G., Kurc T. M., Moreno C. S., Brat D. J., Saltz J. H., An integrative approach for in silico glioma research. IEEE Trans. Biomed. Eng. 57, 2617–2621 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huse J. T., Phillips H. S., Brennan C. W., Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 59, 1190–1199 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Phillips H. S., Kharbanda S., Chen R., Forrest W. F., Soriano R. H., Wu T. D., Misra A., Nigro J. M., Colman H., Soroceanu L., Williams P. M., Modrusan Z., Feuerstein B. G., Aldape K., Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Verhaak R. G. W., Hoadley K. A., Purdom E., Wang V., Qi Y., Wilkerson M. D., Miller C. R., Ding L., Golub T., Mesirov J. P., Alexe G., Lawrence M., O’Kelly M., Tamayo P., Weir B. A., Gabriel S., Winckler W., Gupta S., Jakkula L., Feiler H. S., Hodgson J. G., James C. D., Sarkaria J. N., Brennan C., Kahn A., Spellman P. T., Wilson R. K., Speed T. P., Gray J. W., Meyerson M., Getz G., Perou C. M., Hayes D. N., Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhat K. P. L., Balasubramaniyan V., Vaillant B., Ezhilarasan R., Hummelink K., Hollingsworth F., Wani K., Heathcock L., James J. D., Goodman L. D., Conroy S., Long L., Lelic N., Wang S., Gumin J., Raj D., Kodama Y., Raghunathan A., Olar A., Joshi K., Pelloski C. E., Heimberger A., Kim S. H., Cahill D. P., Rao G., Den Dunnen W. F. A., Boddeke H. W. G. M., Phillips H. S., Nakano I., Lang F. F., Colman H., Sulman E. P., Aldape K., Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia Y., Shen S., Verma I. M., NF-κB, an active player in human cancers. Cancer Immunol. Res. 2, 823–830 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robe P. A., Bentires-Alj M., Bonif M., Rogister B., Deprez M., Haddada H., Khac M.-T. N., Jolois O., Erkmen K., Merville M.-P., Black P. M., Bours V., In vitro and in vivo activity of the nuclear factor-κB inhibitor sulfasalazine in human glioblastomas. Clin. Cancer Res. 10, 5595–5603 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Gill J. S., Zhu X., Moore M. J., Lu L., Yaszemski M. J., Windebank A. J., Effects of NFκB decoy oligonucleotides released from biodegradable polymer microparticles on a glioblastoma cell line. Biomaterials 23, 2773–2781 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Lee D. W., Ramakrishnan D., Valenta J., Parney I. F., Bayless K. J., Sitcheran R., The NF-κB RelB protein is an oncogenic driver of mesenchymal glioma. PLOS One 8, e57489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acharyya S., Villalta S. A., Bakkar N., Bupha-Intr T., Janssen P. M. L., Carathers M., Li Z.-W., Beg A. A., Ghosh S., Sahenk Z., Weinstein M., Gardner K. L., Rafael-Fortney J. A., Karin M., Tidball J. G., Baldwin A. S., Guttridge D. C., Interplay of IKK/NF-κB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Invest. 117, 889–901 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh A., Roy A., Liu X., Kordower J. H., Mufson E. J., Hartley D. M., Ghosh S., Mosley R. L., Gendelman H. E., Pahan K., Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 104, 18754–18759 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May M. J., D’Acquisto F., Madge L. A., Glöckner J., Pober J. S., Ghosh S., Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science 289, 1550–1554 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Riddick G., Fine H. A., Integration and analysis of genome-scale data from gliomas. Nat. Rev. Neurol. 7, 439–450 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedmann-Morvinski D., Bushong E. A., Ke E., Soda Y., Marumoto T., Singer O., Ellisman M. H., Verma I. M., Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carro M. S., Lim W. K., Alvarez M. J., Bollo R. J., Zhao X., Snyder E. Y., Sulman E. P., Anne S. L., Doetsch F., Colman H., Lasorella A., Aldape K., Califano A., Iavarone A., The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marumoto T., Tashiro A., Friedmann-Morvinski D., Scadeng M., Soda Y., Gage F. H., Verma I. M., Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 15, 110–116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habineza Ndikuyeze G., Gaurnier-Hausser A., Patel R., Baldwin A. S., May M. J., Flood P., Krick E., Propert K. J., Mason N. J., A phase I clinical trial of systemically delivered NEMO binding domain peptide in dogs with spontaneous activated B-cell like diffuse large B-cell lymphoma. PLOS One 9, e95404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tilstra J. S., Gaddy D. F., Zhao J., Davé S. H., Niedernhofer L. J., Plevy S. E., Robbins P. D., Pharmacologic IKK/NF-κB inhibition causes antigen presenting cells to undergo TNFα dependent ROS-mediated programmed cell death. Sci. Rep. 4, 3631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibata W., Maeda S., Hikiba Y., Yanai A., Ohmae T., Sakamoto K., Nakagawa H., Ogura K., Omata M., Cutting edge: The IκB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks inflammatory injury in murine colitis. J. Immunol. 179, 2681–2685 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Hayden M. S., Ghosh S., Signaling to NF-κB. Genes. Dev. 18, 2195–2224 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Bottero V., Withoff S., Verma I. M., NF-κB and the regulation of hematopoiesis. Cell Death Differ. 13, 785–797 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Xia Y., Yeddula N., Leblanc M., Ke E., Zhang Y., Oldfield E., Shaw R. J., Verma I. M., Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 14, 257–265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robe P. A., Martin D. H., Nguyen-Khac M. T., Artesi M., Deprez M., Albert A., Vanbelle S., Califice S., Bredel M., Bours V., Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 9, 372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puliyappadamba V. T., Hatanpaa K. J., Chakraborty S., Habib A. A., The role of NF-κB in the pathogenesis of glioma. Mol. Cell. Oncol. 1, e963478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao S., Wu Q., McLendon R. E., Hao Y., Shi Q., Hjelmeland A. B., Dewhirst M. W., Bigner D. D., Rich J. N., Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Phillips T. M., McBride W. H., Pajonk F., The response of CD24−/low/CD44+ breast cancer–initiating cells to radiation. J. Natl. Cancer Inst. 98, 1777–1785 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Woodward W. A., Chen M. S., Behbod F., Alfaro M. P., Buchholz T. A., Rosen J. M., WNT/β-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 104, 618–623 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.di Meglio P., Ianaro A., Ghosh S., Amelioration of acute inflammation by systemic administration of a cell-permeable peptide inhibitor of NF-κB activation. Arthritis Rheum. 52, 951–958 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Agemy L., Friedmann-Morvinski D., Kotamraju V. R., Roth L., Sugahara K. N., Girard O. M., Mattrey R. F., Verma I. M., Ruoslahti E., Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 108, 17450–17455 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedmann-Morvinski D., Bhargava V., Gupta S., Verma I. M., Subramaniam S., Identification of therapeutic targets for glioblastoma by network analysis. Oncogene 10.1038/onc.2015.119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Samant R. S., Clark D. W., Fillmore R. A., Cicek M., Metge B. J., Chandramouli K. H., Chambers A. F., Casey G., Welch D. R., Shevde L. A., Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-κB activation. Mol. Cancer 6, 6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pasparakis M., Courtois G., Hafner M., Schmidt-Supprian M., Nenci A., Toksoy A., Krampert M., Goebeler M., Gillitzer R., Israel A., Krieg T., Rajewsky K., Haase I., TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 417, 861–866 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Ikawa M., Tanaka N., Kao W. W.-Y., Verma I. M., Generation of transgenic mice using lentiviral vectors: A novel preclinical assessment of lentiviral vectors for gene therapy. Mol. Ther. 8, 666–673 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/1/e1501292/DC1
Fig. S1. Heat map of the RNA-Seq analysis of NF-κB target genes (list of 150 genes; http://bioinfo.lifl.fr/NF-KB/) comparing the four molecular subtypes of human GBM (TCGA data, downloaded as level 3 normalization counts; classification from www.cbioportal.org).
Fig. S2. qRT-PCR analysis of six representative NF-κB target genes in normal brain tissue, HRas-shp53 tumor (n = 5), and shNF1-shp53 tumor (n = 5) samples.
Fig. S3. qRT-PCR analysis of six representative MES markers in normal brain tissue and four tumor samples (HRasV12-shp53).
Fig. S4. Flow cytometry analysis of 005 BTICs either left untransduced (control) or transduced with a lenti-κB-mCherry reporter.
Fig. S5. Silencing IKK2 in NF5310 BTICs.
Fig. S6. Silencing IKK2 in Af/fIKK2R53-Cre-ErT2 cells.
Fig. S7. Entry of the NBD peptide into the CNS.
Fig. S8. Hematoxylin and eosin staining of a tumor section from (A) a representative mouse from the control group at clinical end point and (B) an NBDwt-treated mouse that was sacrificed when the control mice had reached clinical end point.
Fig. S9. Treatment of U87 xenograft mice with the NBD peptide.
Fig. S10. Timp1 expression.
Fig. S11. Timp1 expression in human GBM.
Fig. S12. Inducible knockdown of Timp1.
Fig. S13. Bioluminescent imaging of a representative mouse from the U87-miRTimp1(−)Dox and U87-miRTimp1(+)Dox groups at the indicated time points.
Fig. S14. qRT-PCR analysis of Timp1 in U87luc-miRTimp1 tumors in the absence or presence of Dox in the chow.
Fig. S15. Kaplan-Meier survival plot for human glioma samples with differential Timp1 gene expression (National Cancer Institute, REMBRANDT Web site).
Table S1. Expression levels of NF-κB target genes in lentivirus-induced GBM.
Table S2. List of primers.
Table S3. List of antibodies.