

Abstract
Objective
Obesity-induced diabetes has increased over the years and has become one of the risk factors for stroke. We investigated the influence of diet-induced obesity and hyperglycemia on permanent distal middle cerebral artery occlusion (pMCAO) induced ischemic stroke in mice.
Methods
Male C57/Bl6 mice were treated with a high fat/ high carbohydrate diet [HFCD/obese and hyperglycemia (O/H)] or a normal diet (control) for 3.5 months, subjected to pMCAO and sacrificed after 7 days.
Results
Infarct volume analysis showed no differences between the O/H and control group, whereas neurological deficits were significantly higher in the O/H group compared to the control group. Sirtuin (Sirt1) was overexpressed and NADPH oxidase was reduced in the O/H group. O/H mice had significantly lower expression of Wnt and glycogen synthase kinase 3 α and β, a key component in the Wnt signaling pathway. Translocation of apoptosis inducing factor (AIF) to the nucleus was observed in both the O/H and control groups, but O/H mice showed a higher expression of AIF in the nucleus.
Conclusions
Our data suggest that impaired Wnt signaling and active apoptosis results in reduced post-stroke recovery in obese and hyperglycemic mice.
Keywords: Hyperglycemia, obesity, Apoptotic inducing factor, Wnt, ischemic stroke
Introduction
In the United States and similar industrialized nations, obesity caused by long term consumption of a high fat diet is an independent risk factor for acute ischemic stroke (1, 2). Recent studies have suggested that obesity is linked to type II diabetes, hypertension, neurodegenerative disease, lipid disorders and cognitive impairments (3, 4). Obesity is an autonomous predictor of unfavorable functional outcome and mortality in ischemic patients treated with tissue plasminogen activator (tPA), which is the only therapy available for ischemic stroke (5). Concurrent pathological conditions in obesity compromise the metabolic syndrome and increase the risk of vascular disease and type II diabetes mellitus, which are the risk factors for stroke. Neovascularization is considered protective in stroke, but in high fat diet-induced type II diabetes mice excessive angiogenesis resulted in impaired neovascularization and subsequent coronary and peripheral complications (6)..
Wnt proteins are extracellular factors that are secreted as glycoproteins and play an important role in developing an adult central nervous system. They regulate proliferation and differentiation of neural progenitor cells to neuronal cells. Activation of Wnt signaling initiates a neuroprotective mechanism in Alzheimer’s disease (7). Activation of Wnt signaling provides an appropriate environment for neuronal differentiation and migration of newborn neurons towards the ischemic lesion, thus helping in functional recovery. Moreover, Wnt signaling is critically involved in neurogenesis, and inhibition of Wnt signaling reduces newly formed neurons and impairs object recognition in rodents (8, 9).
In this study, we explore whether high fat diet induced-obesity exacerbates ischemic injury and post ischemic recovery. Age matched animals were fed with either a high fat, high carbohydrate diet (HFCD) or a normal diet (ND) for 3.5 months prior to ischemia, and subsequent functional and histological evaluations were conducted 7 days following stroke. We questioned whether mice exposed to HFCD for several months would have increased functional deficits and larger infarct volume following permanent distal middle cerebral artery occlusion (pMCAO) compared to animals maintained on a ND. In addition, we investigated the cortical protein expression levels of various proteins that play a critical role in the injury and repair phase of stroke, such as histone deacetylase sir2 (silent information regulator 2), orthologue sirtuin-1 (Sirt1), Wnt signaling, glycogen synthase kinase 3 α and β (GSK-3β), apoptosis inducing factor (AIF), non-amyloidogenic amyloid β precursor protein (APP), and NADPH oxidase 4 (NOX4).
Material and methods
Animals and Diet
All animal protocols were approved by The University of Toledo Institutional Animal Care and Use Committee, and the guidelines of the National Institutes of Health were followed throughout the study. C57BL/6 male mice, 8–10 weeks old and weighing about 20–25 grams, were procured from Charles River Laboratories, USA. Animals were housed at 22 ± 1°C with 12 hours light and 12 hours dark cycle; water and food were available ad libitum. Mice were randomized into different treatment groups, and personnel working on the study were blinded from the experimental design. Animals were fed a high fat/high carbohydrate diet (HFCD) (Research Diet Inc., NJ, USA #12079B) or a normal diet (ND) for 3.5 months, and fasting blood glucose was monitored prior to permanent ischemia. In agreement with our previous study, (10) animals on HFCD diet gained significantly more weight and had significantly higher fasting blood glucose levels than ND fed mice (P value < 0.0001). Therefore, after the 3.5 months of feeding, the HFCD fed mice were obese and hyperglycemic (O/H). Animals were divided into 4 groups: 1) sham (no obesity/no hyperglycemia/no pMCAO) 2) sham obesity/hyperglycemic (O/H) (no obesity/hyperglycemia/no pMCAO) 3) control (no obesity/no hyperglycemia/pMCAO) 4) O/H/pMCAO (obesity/hyperglycemia/pMCAO). Animals that were fed a HFCD prior to the pMCAO, continued on the HFCD for the seven days post ischemia. Likewise, ND fed mice continued a ND for the seven days following pMCAO. Therefore, fasting blood glucose was only tested prior to the pMCAO.
Permanent Middle Cerebral Artery Occlusion (pMCAO)
Control (n=12) and O/H/pMCAO (n=15) groups were subjected to pMCAO as per our previously optimized method to occlude the distal part of the MCA (11). Mice initially anesthetized with 5% isoflurane, and then maintained at 1.5%, were operated on under a surgical microscope. A thin 1.0-cm vertical skin incision given between the right eye and ear exposed the temporal bone after moving the temporal muscles aside. A 2.0-mm hole drilled with a dental drill precisely over the region of the distal MCA was followed by removing dura mater over the distal part of MCA. With the help of a bipolar coagulator, the distal part of the MCA was severed to interrupt the blood flow to the area supplied by the artery. A rectal probe was inserted to monitor the core body temperature, which was maintained at 37.0 ± 0.5°C during the procedure with a heating blanket. After the end of the surgical procedure, mice were transferred to a temperature-regulated incubator and kept for 2 hours to recover from surgery and finally shifted to home cages. All behavioral parameters were evaluated by an expert person blinded to the treatment groups.
Motor activity
Motor functions were evaluated by using rotarod equipment (Columbus Instrument, OH). Each animal was given three trials 24 hours before performing the actual task. Locomotor activity was monitored 4 hours before surgery (baseline), 24 hours post-surgery and 7 days after pMCAO surgery.
Neurological deficit scoring (NDS)
Each group was evaluated on a 28-point scoring pattern after 7 days following pMCAO. Both sensory and motor deficits, such as body symmetry, gait, climbing, circling behavior, front limb symmetry, compulsory circling, and whisker response, were evaluated in the task. Each of the seven tests was graded from 0–4, with higher scores indicating more severe deficits.
Grip Strength
Front limb grip strength in mice was determined at 4hours prior to ischemia, 24hours and 7 days after pMCAO using grip strength equipment (Columbus Instrument, OH). Each animal was tested three times per trial at 4hours before, 2 days after and 7 days after pMCAO, and the scores were averaged.
Infarct volume analyses
Animals from control and O/H/pMCAO groups were euthanized by a carbon dioxide overdose 7 days after pMCAO for evaluation of infarct volume. Brains were carefully removed and placed on dry ice for sectioning into five 2-mm-thick coronal sections before incubating in 2% triphenyltetrazolium chloride (TTC) (Sigma Co., MI). After sectioning the digital images of the caudal and rostral sides of each stained coronal section, the sections were obtained and analyzed. The infarct area was estimated from five slices of each brain, measuring rostral and caudal sides of each individual slice in conjunction with the thickness, and expressed as a percentage of the volume of the contralateral hemisphere. Infarct volume analysis was performed using ImageJ software provided by NIH.
Western Blot Analysis
A separate cohort of 4 groups of mice (n=6/group) subjected to same experimental protocol were used for protein analysis. The ipsilateral brain cortices (including infarcted tissues) from sham (control and O/H) and pMCAO (control and O/H) groups were dissected out, weighed, and homogenized. Fractionation was carried out to separate nuclear and cytosolic fractions of brains tissues. Protein concentrations were determined by Bio-Rad Bradford reagent (Bio-Rad Laboratories, Hercules, CA), and samples were analyzed by loading equivalent amounts of total cytoplasmic or nuclear proteins (25μg) onto 10% or 12% SDS-polyacrylamide gels. Proteins were transferred from the gel to the PVDF membrane followed by overnight incubation at 4°C with the following antibodies: mouse anti-actin (Millipore), rat ant-histone (Thermo scientific, IL), rabbit anti-Wnt (Abcam, MA), rabbit anti-p-GSK-3 α/β (Thermo Scientific), mouse anti-GSK-3 α/β (Thermo Scientific), mouse anti-Sirt1 (Santa Cruz Biotech, TX) rabbit anti-AIF (Millipore), rabbit anti-APP (Genscript, NJ) and rabbit anti-NOX4 (Thermo scientific) using the following dilutions:1:2,000, 1;2,000, 1:2,000, 1:400, 1:1,000, 1:1,000, 1:1,000, 1;1,000 and 1:1,000 respectively. After washing, membranes were incubated with the secondary antibody, goat anti-rabbit, and/or goat anti-mouse, at a dilution of 1:4,000 (Jackson Immuno-Research Laboratories, PA). Images were analyzed using Bio-Rad chemi Doc™ XRS+ image analyzer. The densitometric values were normalized with respect to the values of actin immunoreactivity to correct for any loading and transfer differences between samples.
Statistical Analysis
All behavioral parameters and infarct volumes between different groups were analyzed by one-way ANOVA with Newman–Keuls post hoc test. For the protein analysis, the differences between groups were determined by Student’s t test. In all statistical analyses performed, a value of p<0.05 was considered to be significant.
Results
Diet-induced obesity and hyperglycemia
Mice fed a HFCD for 3.5 months have a significant weight gain (P<.001), and significantly increased fasting blood glucose level (P <.001) compared to mice fed a ND (see Table 1). Therefore, after 3.5 months of feeding, the HFCD fed mice are obese and hyperglycemic.
Table 1.
| Groups | Weight ± SD | Blood glucose ± SD |
|---|---|---|
| Normal Sham | 29.41 ± 0.64 | 83.75 ± 2.98 |
| O/H sham | 35.22 ± 0.70 | 118 ± 3.68 |
Mice with obesity and hyperglycemia (O/H) suffer from higher neurological deficits and impaired locomotor activity after 7 days of pMCAO
Here, we wanted to determine the effect of HFCD on mouse brains after permanent ischemia. Mice were sacrificed after 7 days of pMCAO. The O/H group suffered from significantly higher neurologic deficits (Fig. 1A) and impaired locomotor activity (Fig. 1B), but grip strength was not significantly affected (Fig. 1C) on day 7, and the infarct size was similar to that of the control group (Fig 1D). Our data are consistent with previous findings, (6) suggesting that the permanent model of ischemia in early diabetic condition causes a smaller infarct volume and no hemorrhage, which was observed possibly due to no reperfusion in the permanent ischemia or possibly due to neovascularization in the brain (12).
Figure 1.

HFCD-induced O/H affects recovery after 7 days of permanent ischemia. A) O/H group showed significantly higher neurological deficits as compared to control group. B) O/H animals showed significantly lower locomotor activity after 7 days of pMCAO as compared to control group. C) Grip strength was lower, but not significantly in O/H group as compared to control group. D) Infarct volume analysis in control and O/H groups showed no significant differences after 7 days of pMCAO. Data are expressed as mean ± SEM; * p<0.05, vs. control; Control n=12 and O/H n=13
Sirt 1 expression in O/H group following pMCAO
The sirtuin proteins are class III histone deacetylases (HDAC), and Sirt1 has been shown to play a central role in metabolic homeostasis. Here we observed that Sirt1 expression goes down in control pMCAO mice, but was significantly higher in the O/H group subjected to pMCAO (Fig. 2A–B) and compared to the control pMCAO group. However, in sham O/H and pMCAO control groups, NOX4 expression was significantly lower as compared to sham control which was observed to be further downregulated in the O/H pMCAO group (Fig. 2C–D).
Figure 2.

HFCD treatment induced Sirt1 expression and after 7 days of pMCAO. A) Sirt1 expression was significantly reduced in control pMCAO mice. Sirt1 expression was significantly elevated in O/H pMCAO group as compared to control pMCAO group. C) NAD(P)H oxidase (NOX4) expression in sham O/H and control pMCAO was reduced significantly as compared to control group and O/H pMCAO group showed further significant decrease in the expression levels. (B and D) corresponding graph shows the densitometric analysis normalized to actin. Data are expressed as mean ± SEM; @ p<0.05, vs. sham-control; # p<0.05, vs. sham-O/H; * p<0.05, vs. pMCAO-control; Control n=6 and O/H n=6 (@@/##/** represents p<0.001)
Impaired Wnt signaling and GSK-3αβ after pMCAO in control mice is further decreased in O/H mice
Wnt proteins are the secreted cysteine rich glycosylated proteins that play an important role in embryonic development and in the matured central nervous system involved in cell proliferation and cell survival. Our Western blot data suggest that the control pMCAO group had significantly lower Wnt1 expression which was further down regulated in the O/H pMCAO group (Fig. 3A–B) as compared to sham control animals. Similarly, p-GSK 3 α/β (Fig. 3C–D) expression levels were observed to be lower in sham O/H and control pMCAO as compared to sham control group and the expression was observed to be further down regulated in O/H pMCAO group. These results indicate that obesity and hyperglycemia worsens the post ischemic recovery in O/H mice possibly due to lower expression of Wnt and p-GSK 3 α/β expression levels.
Figure 3.

Effect of HFCD-induced O/H on Wnt and GSK-3 α/β expression following pMCAO. A) Wnt expression was reduced non-significantly in sham O/H and significantly on control pMCAO as compared to sham control. On subjecting O/H mice to pMCAO, Wnt expression was significantly altered after 7 days as compared to control pMCAO group. C) Similarly, phosphorylation of GSK-3 α/β was significantly reduced in sham O/H and control pMCAO which were further reduced in O/H pMCAO. (B and D) corresponding graph shows the densitometric analysis normalized to actin and t-GSK. Data are expressed as mean ± SEM; @ p<0.05, vs. sham-control; # p<0.05, vs. sham-O/H; * p<0.05, vs. pMCAO-control; Control n=6 and O/H n=6 (@@/##/** represents p<0.001)
AIF translocation to nucleus after pMCAO in O/H results in apoptosis
Oxidative stress causes apoptosis by either a caspase-dependent or caspase-independent mechanism. AIF is a caspase-independent protein which, on activation, is released from mitochondria and translocates to the nucleus, leading to cell death (13). Expression of AIF and translocation to the nucleus was significantly higher in O/H pMCAO and as compared to sham control or sham O/H (Fig. 4A–B). These results suggest that the caspase-independent apoptosis mechanism is prevalent in O/H mice even after 7 days of ischemia.
Figure 4.
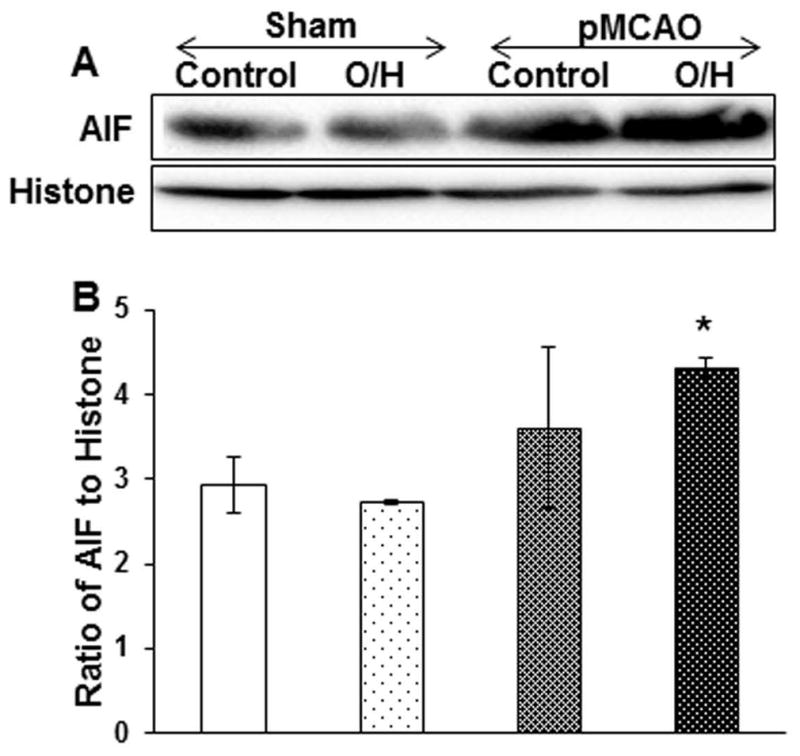
O/H induced by HFCD upregulates caspase independent mechanism via AIF translocation to nucleus after permanent ischemia. A) O/H group when subjected to pMCAO activated caspase independent mechanism by significantly upregulating AIF translocation to nucleus as compared to control pMCAO group. B) Corresponding graph shows the densitometric analysis normalized to histones. Data are expressed as mean ± SEM; * p<0.05, vs. control; Control n=6 and O/H n=6
HFCD induced O/H reduces non-amyloidogenic APP expression following pMCAO
Amyloid precursor protein (APP) is a type I transmembrane protein expressed in a soluble form as sAPP. In the central nervous system, an increase in the expression of APP coincides with the peak of neuronal differentiation and neurite outgrowth (14). Here we investigated the role of a short N terminal sequence of non-amyloidogenic APP (44–63) in control and O/H mice subjected to pMCAO; we observed no significant changes in APP expression in the sham control and sham O/H mice. However, the non-amyloidogenic APP levels were significantly reduced in both the groups when compared to their basal levels in sham (Fig. 5A–B).
Figure 5.
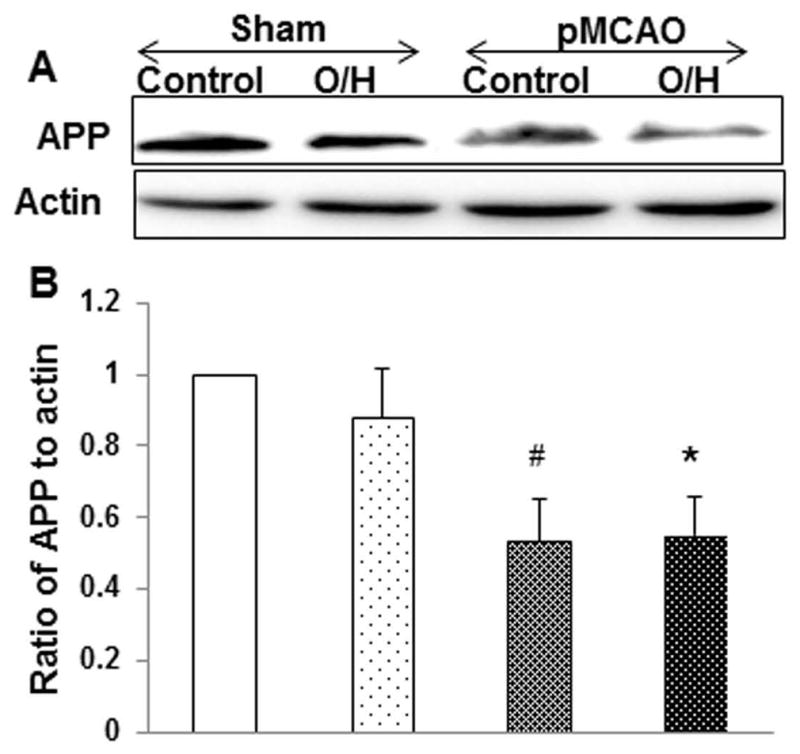
HFCD alters non-amyloidogenic APP expression after 7 days of pMCAO. A) O/H group of mice showed no significant difference compared to control group subjected to pMCAO, however APP expression was significantly reduced as compared to respective sham groups. B) Corresponding graph shows the densitometric analysis normalized to actin. Data are expressed as mean ± SEM; # p<0.05, vs. sham-O/H; * p<0.05, vs. control; Control n=6 and O/H n=6
Discussion
In the present study, we demonstrated that mice fed with HFCD showed significantly lower post stroke recovery in terms of reduced locomotor activity as well as higher NDS as compared to the control group after 7 days of permanent ischemia. Infarct volume analysis showed no differences between the control group and O/H mice. We also demonstrated the possible association between brain Sirt1 and NOX4 expression in HFCD fed mice. In addition, Wnt1 and pGSK-3α/β expression were completely abrogated in HFCD fed mice subjected to permanent ischemia. Lower expression of non-amyloidogenic APP was also observed in both the control and O/H groups after pMCAO. Finally, translocation of AIF to the nucleus was significantly elevated in the O/H group, which is a major pathway for apoptosis. Together, these results suggest that no differences in infarct volume in the O/H group compared to that of the control group might be possibly due to the protective role of sirt1 but impaired signaling of AIF and Wnt partly contributed to diminished recovery after 7 days of permanent ischemia.
A diet with a high amount of saturated fat has been linked to obesity, type II diabetes and metabolic syndrome, but the effect of these co-morbidities has not been widely studied in human or animal models (15). Clinical as well as pathological studies have shown that elevated blood glucose due to obesity-induced type II diabetes at the time of stroke is associated with higher cerebral infarction and hemorrhagic transformation (16). Previous studies on rats have shown that the activity of matrix metalloprotease-9 (MMP-9) is increased in diabetes. MMP inhibitors such as metformin and minocycline inhibit MMP-9 activity and attenuate cerebral macro-vessel formation, suggesting a direct role of MMP-9 and vascular remodeling in diabetes (17). Recent studies also suggest that diabetes modulates neovascularization, which can result from angiogenesis, vasculogenesis, collateral growth and/or arteriogenesis (6). It is also well established in colon cancer models that chronic consumption of a high fat diet increases tumor angiogenesis due to overexpression of vascular-endothelial growth factor (VEGF), leptin, monocyte chemoattractant protein-1, angiogenin, angiopoietin-1 and angiopoietin-3 (18). Sirt1 is an important modulator of angiogenesis, vasodilation, and blood supply. FOXO1 deacetylation causes Sirt1 to block endothelial cell senescence and promotes endothelial cell growth, vascular sprouting, branching morphogenesis, and blood vessel formation in mice. Deacetylation of endothelial nitric oxide activates Sirt1 thus resulting in overexpression of nitric oxide, leading to vasodilation, and increased blood supply (19). Numerous studies have reported the protective effect of Sirt1 in liver and muscle from detrimental metabolic consequences upon exposure to high fat diet (20). In a recent study, (21) inhibiting Sirt1 resulted in increased acetylation of eNOS activity and further reduced eNOS expression, initiating endothelial dysfunction. It was also reported that NO is inactivated by NAD(P)H oxidase derived O2o− (22). However, ours is the first study showing that Sirt1 overexpression in the brain is possibly associated with angiogenesis and cerebral remodeling in HFCD fed mice which might be the cause of not observing any differences in the infarct volume. There are conflicting reports on the differences between O/H and normal mice when subjected to different models of stroke. Chen et al (23) showed differences between db/db versus db+ (nondiabetic) mice at 24h after 1h of ischemia reperfusion whereas Kim et al (24) showed the differences only after 30 min of ischemia and 3 day of reperfusion in diet induced diabetic mice. However, the differences were not observed at 24h of permanent model of stroke (6) pertinently due to augmented neovascularization owing to angiogenesis and arteriogenesis. Our study is consistent with the permanent model of ischemia and supports our hypothesis (6).
The canonical Wnt signaling pathway is an extracellular factor crucial for mammalian central nervous system development and regulates diverse processes including cell proliferation, differentiation, and migration (11, 25). Binding of Wnt to the frizzled receptors and LRP co-receptors initiates the signaling cascade and inhibits GSK-3β activity. Reduced GSK-3β stabilizes and prevents β-catenin from proteasomal degradation, allowing unphosphorylated β-catenin to translocate to the nucleus for activation of developmentally important genes such as VEGF, bcl2, survivin, tight junctions (TJ) (26). Wnt proteins are significantly involved in embryonic development, cellular differentiation and cell survival (27). More importantly, β-catenin expression by Wnt signaling promotes renal cell survival during metabolic stress and may be involved in muscle-to-fat cell conversion (28). Recent studies showed that loss of Wnt1 during OGD (oxygen glucose deprivation) contributes to cell injury, apoptosis and DNA degradation. Studies also suggest that type 2 diabetes risk gene, transcription factor 7-like 2 (T-cell specific, HMG-box) (TCF7L2) is an effector of Wnt signaling pathway, and Wnt signaling controls the gut and brain glucose metabolism (29, 30). Wnt is the important component against neuronal degeneration, and blockade of Wnt inhibits neuroprotection and affects the functional recovery, thus highlighting the role of Wnt in neurodegeneration (31, 32). The results of our study conform to previous studies suggesting that ischemia abrogates Wnt1-mediated neuroprotection in the brain against oxidative stress, and we are the first to show the detrimental effect of a high fat diet on Wnt expression, which might have been the reason for slow post ischemic functional recovery. In a non-pathological condition, APP is cleaved within the β amyloid sequence by non- amyloidogenic α secretase to release a soluble form of sAPP which represents a major secretory pathway and is non-amyloidogenic (33). In the central nervous system, the basal level of APP peaks during neuronal differentiation and neural outgrowth. The soluble form of APP generated by cleaving within the β amyloid sequence has beneficial neuronal functions such as synaptogenesis, neurite outgrowth, cell survival and adhesion (33). The existence of adult neural stem cells and progenitors increases the possibility of sAPP’s role in adult neurogenesis (34). A study demonstrating Wnt inhibitor induced inhibition of non-amyloidogenic processing of APP, also suggests possible crosstalk between Wnt and non-amyloidogenic APP expression (35). Our results of reduced APP expression in both the control and O/H groups after ischemia are in agreement with the previous study.
In brain development as well as in neuronal tissue injury, the apoptotic cell death pathway plays an important role and is required for eliminating truncated neurons and maintaining healthy cells (36). Various cell death pathways have been elucidated during neuronal injury induced by ischemia, including p53, PARP, c-Jun and the plasma membrane death receptor ligand system (36, 37, 38). Although caspases are recognized as important mediators of apoptosis, accumulating evidence indicates the existence of caspase-independent mechanisms of neuronal cell death (13). AIF is a caspase-independent effector of cell death which, on activation in response to death signals, is released from mitochondria and translocated to the nucleus (39). Recent evidence on PARP-1 knockout mice and PARP-1 null neuronal culture suggests that PARP-1 activation signals mitochondrial AIF release. Apoptosis from cerebral ischemia involving nuclear translocation of cell death protein, AIF, occurs in neurons (40), but to our knowledge, this is the first report to examine the role of this cell death pathway in neurons of HFCD-fed animals after stroke. In this study, we showed that neuronal apoptosis in HFHC-fed animals after ischemia occurs due to the translocation of AIF to the nucleus as one of the possible mechanism of cell death.
Conclusion
In conclusion, we have demonstrated the importance of increased Sirt1 expression and its possible association in the angiogenesis of HFCD-induced O/H, which resulted in no changes in infarct volume. The reduced expression of Wnt possibly resulted in lower neurobehavioral and functional outcomes after 7 days of ischemia in the HFCD group. The HFCD fed group also showed translocation of AIF to the nucleus, which can be a possible mechanism of ongoing apoptosis. We recognize that there are some limitations to this study; for example, neurobehavioral parameters were assessed only after 7 days. Further studies are warranted to explore the underlying mechanism of neovascularization, blood flow measurements in HFCD and the use of Sirt1 and Wnt inhibitors on functional recovery after ischemia.
Known about the subject
Obesity is linked to type II diabetes, hypertension, neurodegenerative disease and cognitive impairments.
Obesity and hyperglycemia cause excessive angiogenesis, leading to neovascularization.
Elevated blood glucose at the time of stroke, is associated with higher brain infarction and hemorrhagic transformation.
New addition to the subject
Obesity and hyperglycemic conditions significantly lower post stroke recovery after cortical stroke.
Brain Wnt1 and pGSK-3α/β expression were completely abrogated in mice with obesity and hyperglycemia that were subjected to ischemia.
Translocation of AIF to the nucleus was significantly elevated in the brains of mice with obesity and hyperglycemia suggesting ongoing apoptosis.
Acknowledgments
Funding: The study was partly funded by grants from NIH (R00AT004197) to ZAS, startup funds from The University of Toledo to ZAS, USDA/NIFA grant (2010-38903-20740) to MFM, Wolfe Innovation Fund (University of Toledo) to MFM and NIH (R15DK10396) to MFM.
Footnotes
Conflict of Interest: Authors declare “no conflict of interest”;
References
- 1.Li W, Prakash R, Chawla D, Du W, Didion SP, Filosa JA, et al. Early effects of high-fat diet on neurovascular function and focal ischemic brain injury. American journal of physiology Regulatory, integrative and comparative physiology. 2013;304:R1001–1008. doi: 10.1152/ajpregu.00523.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hubert HB, Feinleib M, McNamara PM, Castelli WP. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation. 1983;67:968–977. doi: 10.1161/01.cir.67.5.968. [DOI] [PubMed] [Google Scholar]
- 3.George MG, Tong X, Kuklina EV, Labarthe DR. Trends in stroke hospitalizations and associated risk factors among children and young adults, 1995–2008. Annals of neurology. 2011;70:713–721. doi: 10.1002/ana.22539. [DOI] [PubMed] [Google Scholar]
- 4.Park HR, Park M, Choi J, Park KY, Chung HY, Lee J. A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neuroscience letters. 2010;482:235–239. doi: 10.1016/j.neulet.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 5.Sarikaya H, Arnold M, Engelter ST, Lyrer PA, Mattle HP, Michel P, et al. Outcome of intravenous thrombolysis in stroke patients weighing over 100 kg. Cerebrovascular diseases. 2011;32:201–206. doi: 10.1159/000328813. [DOI] [PubMed] [Google Scholar]
- 6.Li W, Prakash R, Kelly-Cobbs AI, Ogbi S, Kozak A, El-Remessy AB, et al. Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes. 2010;59:228–235. doi: 10.2337/db09-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toledo EM, Colombres M, Inestrosa NC. Wnt signaling in neuroprotection and stem cell differentiation. Progress in neurobiology. 2008;86:281–296. doi: 10.1016/j.pneurobio.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Shruster A, Melamed E, Offen D. Neurogenesis in the aged and neurodegenerative brain. Apoptosis : an international journal on programmed cell death. 2010;15:1415–1421. doi: 10.1007/s10495-010-0491-y. [DOI] [PubMed] [Google Scholar]
- 9.Varela-Nallar L, Inestrosa NC. Wnt signaling in the regulation of adult hippocampal neurogenesis. Frontiers in cellular neuroscience. 2013;7:100. doi: 10.3389/fncel.2013.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebke LA, Nestor-Kalinoski AL, Slotterbeck BD, Al-Dieri AG, Ghosh-Lester S, Russo L, et al. Tight association between macrophages and adipocytes in obesity: Implications for adipocyte preparation. Obesity. 2014;22:1246–1255. doi: 10.1002/oby.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nada SE, Tulsulkar J, Shah ZA. Heme oxygenase 1-mediated neurogenesis is enhanced by Ginkgo biloba (EGb 761(R)) after permanent ischemic stroke in mice. Molecular neurobiology. 2014;49:945–956. doi: 10.1007/s12035-013-8572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ergul A, Elgebaly MM, Middlemore ML, Li W, Elewa H, Switzer JA, et al. Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC neurology. 2007;7:33. doi: 10.1186/1471-2377-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rideout HJ, Stefanis L. Caspase inhibition: a potential therapeutic strategy in neurological diseases. Histol Histopathol. 2001;16:895–908. doi: 10.14670/HH-16.895. [DOI] [PubMed] [Google Scholar]
- 14.Hung AY, Koo EH, Haass C, Selkoe DJ. Increased expression of beta-amyloid precursor protein during neuronal differentiation is not accompanied by secretory cleavage. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:9439–9443. doi: 10.1073/pnas.89.20.9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langdon KD, Clarke J, Corbett D. Long-term exposure to high fat diet is bad for your brain: exacerbation of focal ischemic brain injury. Neuroscience. 2011;182:82–87. doi: 10.1016/j.neuroscience.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 16.Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: a vascular perspective. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27:435–451. doi: 10.1038/sj.jcbfm.9600355. [DOI] [PubMed] [Google Scholar]
- 17.Deng J, Zhang J, Feng C, Xiong L, Zuo Z. Critical role of matrix metalloprotease-9 in chronic high fat diet-induced cerebral vascular remodeling and increase of ischemic brain injury in mice. Cardiovascular research. 2014 doi: 10.1093/cvr/cvu154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park H, Kim M, Kwon GT, Lim do Y, Yu R, Sung MK, et al. A high-fat diet increases angiogenesis, solid tumor growth, and lung metastasis of CT26 colon cancer cells in obesity-resistant BALB/c mice. Molecular carcinogenesis. 2012;51:869–880. doi: 10.1002/mc.20856. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69:1702–1705. doi: 10.1158/0008-5472.CAN-08-3365. [DOI] [PubMed] [Google Scholar]
- 20.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarzuelo MJ, Lopez-Sepulveda R, Sanchez M, Romero M, Gomez-Guzman M, Ungvary Z, et al. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: implications for vascular aging. Biochem Pharmacol. 2013;85:1288–1296. doi: 10.1016/j.bcp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation research. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Cui X, Zacharek A, Cui Y, Roberts C, Chopp M. White matter damage and the effect of matrix metalloproteinases in type 2 diabetic mice after stroke. Stroke. 2011;42:445–452. doi: 10.1161/STROKEAHA.110.596486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim E, Tolhurst AT, Cho S. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J Neuroinflammation. 2014;11:83. doi: 10.1186/1742-2094-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valvezan AJ, Klein PS. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Frontiers in molecular neuroscience. 2012;5:1. doi: 10.3389/fnmol.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wada H, Ghysen A, Asakawa K, Abe G, Ishitani T, Kawakami K. Wnt/Dkk negative feedback regulates sensory organ size in zebrafish. Current biology : CB. 2013;23:1559–1565. doi: 10.1016/j.cub.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 27.Jozwiak J, Kotulska K, Grajkowska W, Jozwiak S, Zalewski W, Oldak M, et al. Upregulation of the WNT pathway in tuberous sclerosis-associated subependymal giant cell astrocytomas. Brain & development. 2007;29:273–280. doi: 10.1016/j.braindev.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Havasi A, Gall JM, Mao H, Schwartz JH, Borkan SC. Beta-catenin promotes survival of renal epithelial cells by inhibiting Bax. Journal of the American Society of Nephrology : JASN. 2009;20:1919–1928. doi: 10.1681/ASN.2009030253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanei-Ishii C, Nomura T, Tanikawa J, Ichikawa-Iwata E, Ishii S. Differential sensitivity of v-Myb and c-Myb to Wnt-1-induced protein degradation. The Journal of biological chemistry. 2004;279:44582–44589. doi: 10.1074/jbc.M407831200. [DOI] [PubMed] [Google Scholar]
- 30.Ip W, Shao W, Chiang YT, Jin T. The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. American journal of physiology Endocrinology and metabolism. 2012;303:E1166–1176. doi: 10.1152/ajpendo.00249.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chong ZZ, Shang YC, Hou J, Maiese K. Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways. Oxidative medicine and cellular longevity. 2010;3:153–165. doi: 10.4161/oxim.3.2.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chong ZZ, Hou J, Shang YC, Wang S, Maiese K. EPO relies upon novel signaling of Wnt1 that requires Akt1, FoxO3a, GSK-3beta, and beta-catenin to foster vascular integrity during experimental diabetes. Current neurovascular research. 2011;8:103–120. doi: 10.2174/156720211795495402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiological reviews. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 34.Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, et al. Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development. 2004;131:2173–2181. doi: 10.1242/dev.01103. [DOI] [PubMed] [Google Scholar]
- 35.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 36.Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. The Journal of cell biology. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell death and differentiation. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- 39.Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2000;14:729–739. [PubMed] [Google Scholar]
- 40.Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends in pharmacological sciences. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]