

Abstract
Objective:
To investigate in vivo the relationship of regional brain β-amyloid (Aβ) to gait speed in a group of elderly individuals at high risk for dementia.
Methods:
Cross-sectional associations between brain Aβ as measured with [18F]florbetapir PET and gait speed were examined in 128 elderly participants. Subjects ranged from healthy to mildly cognitively impaired enrolled in the control arm of the multidomain intervention in the Multidomain Alzheimer Preventive Trial (MAPT). Nearly all participants presented spontaneous memory complaints. Regional [18F]florbetapir (AV45) standardized uptake volume ratios were obtained via semiautomated quantitative analysis using the cerebellum as reference region. Gait speed was measured by timing participants while they walked 4 meters. Associations were explored with linear regression, correcting for age, sex, education, body mass index (BMI), and APOE genotype.
Results:
We found a significant association between Aβ in the posterior and anterior putamen, occipital cortex, precuneus, and anterior cingulate and slow gait speed (all corrected p < 0.05). A multivariate model emphasized the locations of the posterior putamen and the precuneus. Aβ burden explained up to 9% of the variance in gait speed, and significantly improved regression models already containing demographic variables, BMI, and APOE status.
Conclusions:
The present PET study confirms, in vivo, previous postmortem evidence showing an association between Alzheimer disease (AD) pathology and gait speed, and provides additional evidence on potential regional effects of brain Aβ on motor function. More research is needed to elucidate the neural mechanisms underlying these regional associations, which may involve motor and sensorimotor circuits hitherto largely neglected in the pathophysiology of AD.
According to recent research, postmortem indices of Alzheimer disease (AD) pathology in older adults are associated with decline in gait speed prior to death, suggesting that AD pathology may account for a substantial proportion of not only cognitive but also motor decline.1 These findings are in line with prior reports of declining gait speed in patients with mild cognitive impairment (MCI)2 and in healthy adults converting to MCI years later.3 The mechanisms by which AD pathology may lead to motor dysfunction remain unknown.
The striatum plays a pivotal role in neurodegenerative disorders characterized by motor symptoms. Although this brain region is not conventionally associated with AD, striatal β-amyloid (Aβ) plaques have been observed in patients with AD4 and in people without dementia.5 The striatum is also one of the earliest brain sites showing Aβ deposition in presymptomatic familial AD,6 which is more commonly associated with extrapyramidal symptoms than sporadic AD. Composed anatomically of caudate nucleus and putamen, the striatum can be partialled further into subregions modulating functionally distinct neural circuits according to their cortical input.7,8 The dorsal posterior putamen receives its primary input from the motor and sensorimotor cortices and is hence particularly important in the modulation of motor circuits.
Our objective was to examine the association between regional Aβ measured in vivo and gait speed in a group of elderly individuals with high risk for dementia. We hypothesized that the disruption of motor circuits through focal Aβ toxicity, preferentially in the postcommissural putamen, constitutes a potential mechanism by which AD pathology leads to motor dysfunction.
METHODS
The Multidomain Alzheimer Preventive Trial: Standard protocol approvals, registrations, and patient consents.
Data were obtained from an ancillary [18F]florbetapir PET study carried out in the context of the larger phase III, multicenter, randomized, placebo-controlled Multidomain Alzheimer Preventive Trial. The objective of the trial was to assess the efficacy of omega-3 fatty acid supplementation in combination with a multidomain intervention (consisting of nutritional counseling, physical exercise, and cognitive stimulation) in slowing cognitive decline in older adults at risk of cognitive decline.9 The protocol is registered on a public-access clinical trial database (www.clinicaltrials.gov) (NCT00672685). Both MAPT and the PET substudy were approved by the ethical committee in Toulouse (CPP SOOM II). Written consent was obtained from all participants.
Participants.
A total of 271 participants participated in the MAPT-florbetapir PET study. At inclusion, participants were aged 70 years or older and presented any one of spontaneous memory complaint, limitation in one instrumental activity of daily living, or gait speed ≤0.8 m/s. Participants with dementia, severe depression, or limitations in basic activities of daily living were excluded. To rule out potential effects of the multidomain intervention on walking speed or amyloid levels, analyses were carried out only on individuals from the multidomain control group (n = 130). Two further participants were excluded due to having developed dementia at the time of the visit closest to the PET scan, leaving a total of 128 participants in the current analyses (see flowchart in figure 1).
Figure 1. Flowchart describing the participants/excluded in the current study.
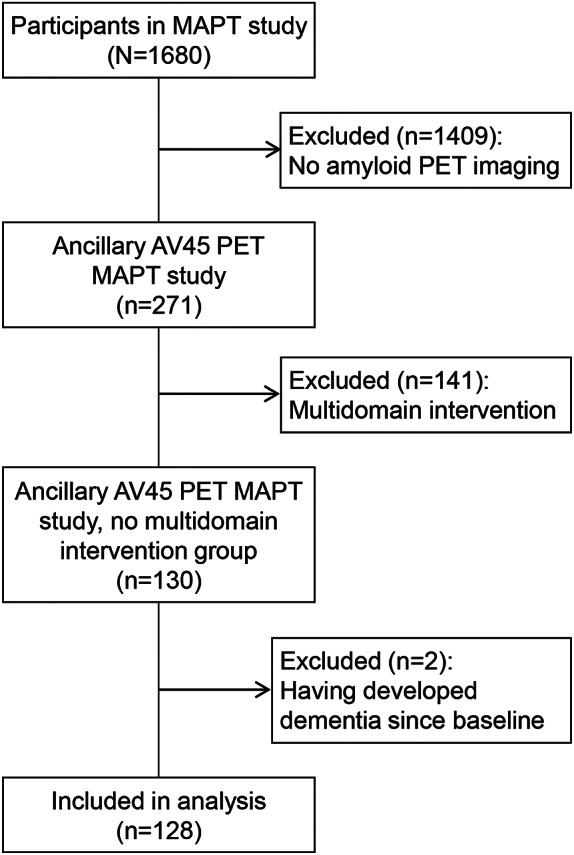
MAPT = Multidomain Alzheimer Preventive Trial.
Cognitive, functional, and physical assessments.
MAPT visits were carried out at baseline, at 6 months, and at 1, 2, and 3 years by independent research staff blinded to the participants' treatment as described in detail elsewhere.9 Cognitive and functional assessments included the Mini-Mental State Examination, the Clinical Dementia Rating scale (CDR), the Alzheimer's Disease Cooperative Study–Activities of Daily Living Inventory, and the Geriatric Depression Scale. Anthropometric measurements were performed by a trained technician using standardized techniques. Weight and height were measured by a beam balance scale and a wall-mounted stadiometer, respectively. Body mass index (BMI) was calculated as weight/height2 (kg/m2). Gait speed was measured by timing participants while they walked 4 meters at their usual walking pace.10 The procedure was carried out twice, and the faster of the 2 walking trials was used to calculate gait speed in meters/second. APOE status (ε4 carrier/noncarrier) was determined from blood samples and was available for 113 participants.
[18F]Florbetapir PET scanning.
[18F]Florbetapir scans were acquired on 5 different hybrid PET-CT scanners, including one PET CT 690 (GE Healthcare; Cleveland, OH), one Discovery RX VCT (General Electric; Fairfield, CT), 2 True Point HiRez (Siemens Medical Solutions; Malvern, PA), and one Biograph 4 Emission Duo LSO (Siemens Medical Solutions). PET sinograms were reconstructed with an iterative algorithm, with corrections for randomness, scatter, photon attenuation, and decay, which produced images with an isotropic voxel of 2 × 2 × 2 mm3 and a spatial resolution of approximately 5-mm full width at half maximum at the field of view center. The acquisition data were processed using the standard package delivered with each acquisition system. All cerebral emission scans began 50 minutes after a mean injection of 4 MBq/kg weight of [18F]florbetapir. For each participant, 10- or 15-minute frames were acquired to facilitate movement-free image acquisition.
[18F]Florbetapir PET analysis.
Regional standard uptake value ratios (SUVRs) were obtained via semiautomated quantitative analysis using the cerebellum as reference region. [18F]Florbetapir images were coregistered with statistical parametric mapping to a [18F]florbetapir template provided by Avid Radiopharmaceuticals (Philadelphia, PA).11 The following regions of interest were defined using the Avid and Montreal Neurological Institute templates: temporal cortex, parietal cortex, medial orbitofrontal cortex, occipital cortex, precuneus, anterior and posterior cingulate, anterior and posterior putamen, caudate, semioval center, and pons. The pons region was included as a negative control. A quality control procedure was carried out using a semi-quantification-based method also facilitated by Avid. The positivity threshold for amyloid PET was set at mean cortical SUVR ≥1.17 as described elsewhere.12
Statistical analyses.
The association between Aβ levels and gait speed was examined with linear regression using [18F]florbetapir SUVR as predictor variable (appendix e-1 on the Neurology® Web site at Neurology.org). First, each regional [18F]florbetapir SUVR was examined separately. Then, regional effects were explored by entering all regional [18F]florbetapir SUVRs as predictor variables in a multivariate regression model. Models were corrected for age, sex, education, BMI, APOE genotype (presence of at least one ε4 allele), days since baseline at PET visit, and days between PET scan and closest gait speed assessment (appendix e-2). Interaction terms were explored in all models to determine whether associations between amyloid burden and gait speed varied as a function of any of the above covariates or disease severity as measured with the CDR score.
Next, we quantified the proportion of variance in gait speed explained specifically by regional amyloid levels. For this purpose, we ran the following regression models for each regional SUVR and examined the change in R2 from one model to the other: Model 1 included age, sex, education, BMI, APOE, days since baseline at PET visit, and days between PET scan and closest gait speed assessment as predictor variables. Model 2 included, in addition to the aforementioned covariates, regional florbetapir SUVR. As we examined 13 regions of interest, there were 13 versions of model 2. A priori levels of significance were set at 0.05. For the examination of regional effects, p values were corrected using the Bonferroni method (by applying a p value of 0.05/13 to account for repeated comparisons across regions). Complete case analyses were carried out. Since APOE genotype was available for 113 participants (15 cases with missing data), analyses corrected for APOE genotype were carried out in 113 participants. Statistical analyses were performed using IBM (Armonk, NY) SPSS Statistics 22.
RESULTS
From the 128 participants included in the current analyses (60.2% female), 99% presented spontaneous memory complaints. A total of 53.9% and 46.1% of volunteers had a CDR of 0 and 0.5, respectively. Their demographic and clinical characteristics are shown in table 1. Mean gait speed was 1.06 (0.25) m/s. Overall, 48.4% were considered amyloid-positive. Time elapsed between baseline and the PET scan date ranged between 58 and 978 days, with most participants being scanned after the first year of being enrolled in the study (median 445 days). The clinical data reported are those acquired at the study visit closest to each individual's PET visit, between 3 and 77.8 days apart (median 71 days).
Table 1.
Participant characteristics
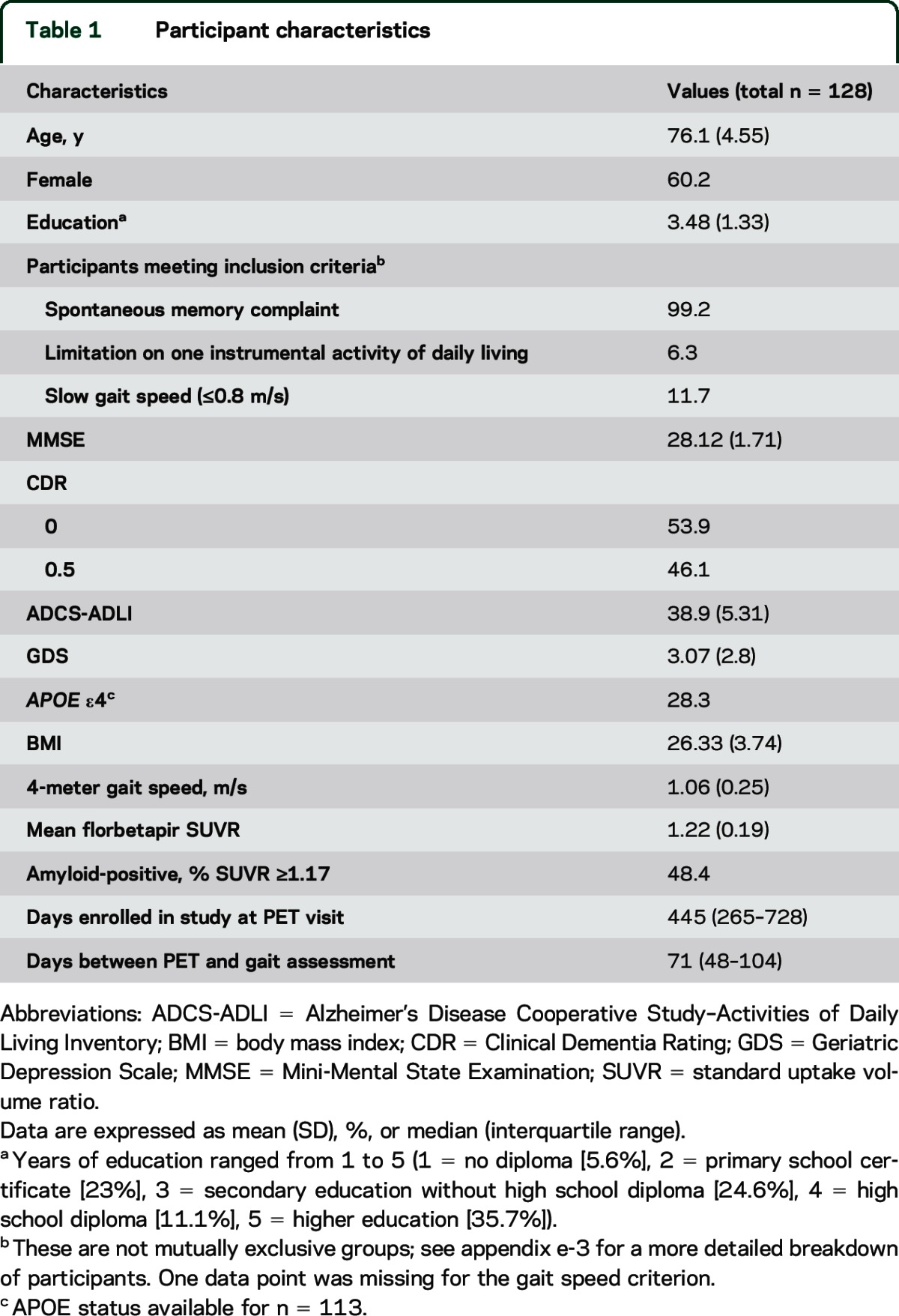
Figure 2 shows the average [18F]florbetapir SUVR in each region of interest (right column) and the corresponding regression coefficients (B) on gait speed (left). Results are corrected for age, sex, education, BMI, days between PET and baseline, and days between PET and closest clinical assessment. The 3 regions at the top of the graph correspond to the 3 subdivisions of the striatum: posterior putamen, anterior putamen, and caudate, with the posterior putamen showing the strongest association with gait speed. Regression coefficients were consistently negative across regions and showed overlapping 95% confidence intervals (CI). After correction for multiple comparisons, the association was significant for the posterior and anterior putamen, the occipital cortex, precuneus, and anterior cingulate (all corrected p < 0.05). The multivariate analysis including all regional SUVRs and covariates in the same model was consistent with a preferential association between gait and Aβ in the posterior putamen and the precuneus (B = −1.55, p = 0.006, 95% CI −2.64, −0.47; and B = −0.69, p = 0.02, 95% CI −1.28, −0.11, respectively).
Figure 2. Regression coefficients (B) of regional amyloid levels on gait speed.

Error bars represent 95% confidence intervals (CI). Results are corrected for age, sex, education, body mass index, APOE status, time of enrollment in study at PET visit, and time between PET visit and closest gait speed assessment. Striatal regions are indicated with darker markers. The right column provides means and SDs of regional florbetapir standard uptake value ratios (SUVRs). Associations that remained significant after multiple comparisons are indicated with an asterisk (all corrected p < 0.05).
The association between amyloid burden and gait speed did not vary as a function of age, sex, education, APOE genotype, or disease severity (CDR) (all interaction terms were p > 0.05). Potential confounding effects of interval events were ruled out (appendix e-4).
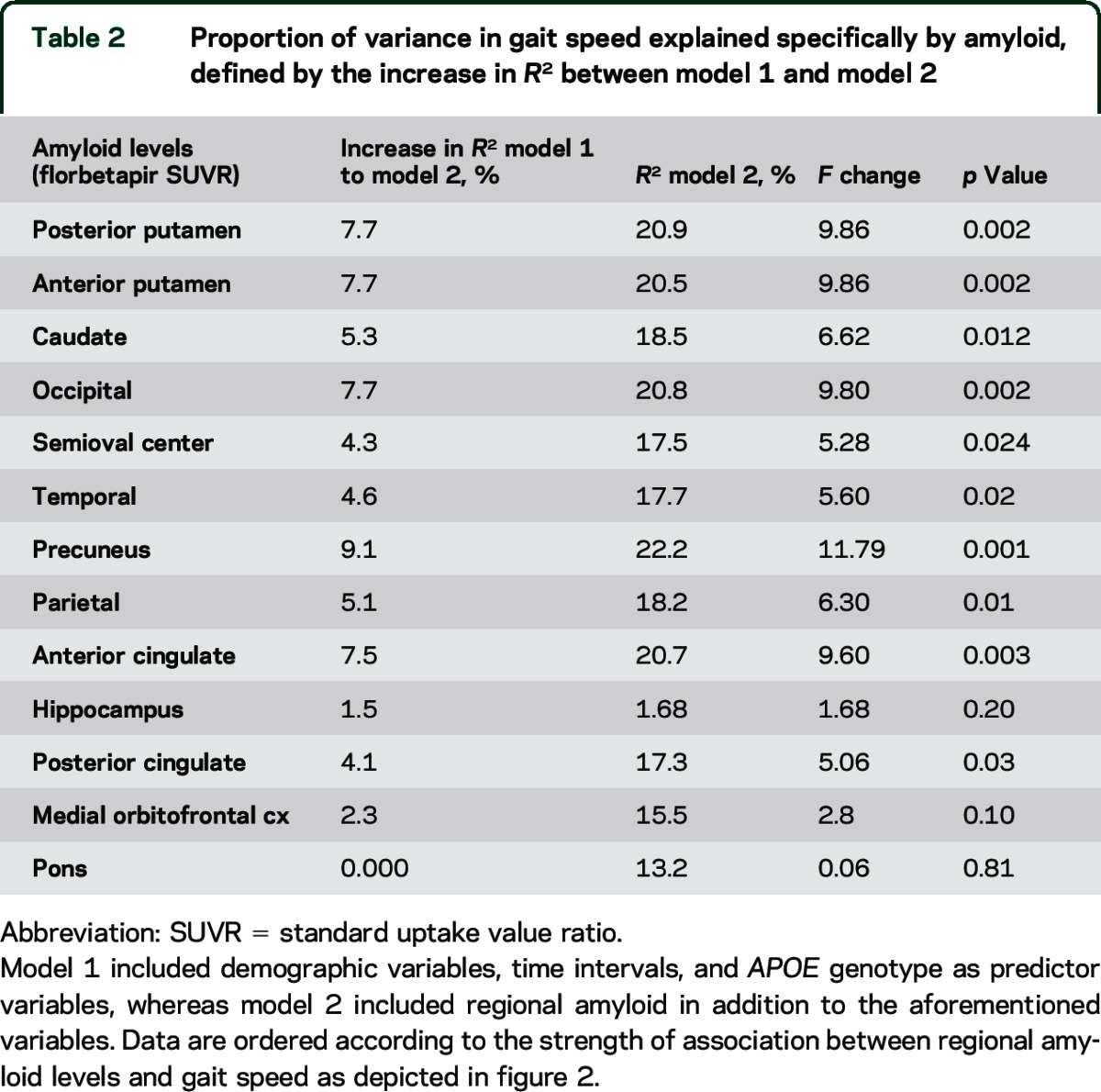
Table 2 shows the proportion of variance in gait speed explained by regional amyloid and APOE genotype. Amyloid pathology accounted for between 1% and 9% of the variance observed in gait speed, depending on the brain region examined. The contribution of explained variance was significant for most of the regions.
Table 2.
Proportion of variance in gait speed explained specifically by amyloid, defined by the increase in R2 between model 1 and model 2
DISCUSSION
The major findings of this study were a significant association between gait speed and brain Aβ (measured with amyloid PET) in the posterior and anterior putamen, the occipital cortex, precuneus, and anterior cingulate, independent of age, APOE genotype, and disease stage. Aβ burden explained up to 9% of the variance in gait speed, and significantly improved regression models already containing demographic variables, BMI, and APOE. Our findings are consistent with previous neuropathologic evidence showing a relationship between postmortem AD pathology and rate of decline in gait speed prior to death.1 The brain region showing the strongest association (i.e., greatest regression coefficient) with gait speed was the dorsal posterior putamen. This region was also emphasized by the multivariate analysis with all regions of interest included. These findings partially support our a priori hypothesis concerning the disruption of motor corticostriatal circuits through focal Aβ toxicity. However, more research is needed, ideally using whole brain analyses, to confirm and help interpret regional effects of brain Aβ in relation to gait speed.
The observed relationship between Aβ and gait speed has several potential interpretations. First, it is possible that Aβ accumulation and slow gait speed co-occur as the result of a common lifestyle factor such as diet through childhood or adulthood, physical activity, or smoking, or a common underlying metabolic or cardiovascular factor, for example, diabetes or hypertension.13 Exposure to the latter cluster of risk factors, particularly during midlife, has been shown to be predictive of dementia risk14 and poor motor performance15 later in life, although causality has not been established. Prevention trials addressing these factors with methodologic rigor are needed to dissect the relative contribution of these risk factors to the independent vs joint development of AD dementia and late-life motor dysfunction.
Second, slow gait speed may constitute a risk factor for AD. Gait speed is a marker of the frailty phenotype, which is thought to result from an age-related reduction in physiologic reserve.16 Studies have consistently shown that gait speed predicts major health-related events, including future disability, hospitalization, and death,17 but also dementia.18 Slow gait speed possibly reflects a state characterized by multisystemic changes that may turn the brain more vulnerable to the accumulation of AD pathology and subsequent damage. While this hypothesis has not been tested, there is strong evidence that, conversely, high levels of physical activity and cardiovascular fitness, 2 parameters underlying gait speed, have protective effects against brain aging.19 In the current study, all participants except for 2 scored within normal ranges of gait speed according to Fried et al.16 This suggests that the aforementioned changes may already be detectable during the pre-frail stages.
A third potential explanation is that Aβ pathology in the brain causes slowing of gait speed, by a direct neurotoxic effect, by accelerating tau deposition, or by other mechanisms. According to the prevailing amyloid cascade hypothesis, Aβ leads to the formation of tau tangles, which are the primary cause responsible for local synaptic dysfunction, neurodegeneration, and neuronal loss.20,21 Consistent with this view, Aβ-induced tau tangles but not amyloid per se would be expected to have local neurotoxic effects with implications for the regulation of motor and sensorimotor circuits. However, there have been reports of in vitro and animal studies that Aβ, independent of tau tangles, disrupts synaptic function in the immediate vicinity of Aβ plaques altering the organization of related neural networks,22–24 supporting the notion that Aβ toxicity can also cause neuronal dysfunction. This latter hypothesis is further corroborated by studies of combined resting-state functional MRI and amyloid PET showing an association between Aβ burden and functional connectivity within large-scale intrinsic functional connectivity networks in elderly individuals,25 even within neural circuits devoid of early tau accumulation.26
The striatum is pivotal in the modulation of basal ganglia–thalamocortical networks implicated in a range of behaviors. These networks are organized into functionally segregated parallel circuits involving different subregions of the striatum.7 Out of all striatal subregions, the dorsal posterior putamen is uniquely interlinked with primary motor, premotor, supplementary motor, and primary somatosensory cortical areas and therefore plays a pivotal role in the modulation of motor circuits.8,27,28 Supported by evidence that the distribution of Aβ in the striatum mirrors the topographic pattern of the functionally distinct corticostriatal circuits,29 we hypothesized that focal Aβ, preferentially in the dorsal posterior putamen, leads to slow gait speed by disrupting motor circuits through its neurotoxic effects. The importance of the dorsal posterior putamen in motor function is further underscored by the clinical observations in a range of neurologic conditions known to affect this brain region selectively (appendix e-5). Although we observed a significant association between posterior putamen Aβ and gait speed, significant associations were also observed for other brain regions, including the precuneus and occipital cortex. The precuneus is functionally connected with the motor, sensorimotor, and visual cortices and plays a multimodal, integrative functional role.30 The occipital cortex participates in visual–motion perception.31
In addition to the identification of anatomical regions that may play a role in the decline of mobility in AD, a further key finding is that the Aβ–gait speed relationship was independent of the participants' CDR status. A potential clinical implication of this observation is that slow gait, in the presence of subjective memory complaints, may represent an early marker of AD pathology even in fully asymptomatic individuals. This notion is consistent with previous research showing that the co-presence of slow gait and cognitive complaints is a better predictor of cognitive decline than its individual components.32,33 Based on this evidence, a novel clinical entity has recently emerged entitled motoric cognitive risk syndrome.33 The syndrome has been defined as the presence of cognitive complaints and slow gait in older individuals without dementia or mobility disability, and has been shown to be a strong and early risk factor for dementia. However, the pathophysiologic mechanisms underlying the prognostic value of this syndrome remain unknown. Our findings are an important contribution to this line of research in that they suggest that the deposition of brain Aβ in selected brain regions may be one potential mechanism by which slow gait speed (in the co-presence of subjective cognitive impairment) is a powerful predictor of future cognitive status. They also imply that even gait speed scores considered as normal according to current conventions (e.g., gait speed threshold <0.08 m/s) may already index incipient neuropathologic processes.
This PET study measured brain Aβ, but not other common co-pathologies that are also likely to contribute to the slowing of gait speed. High-resolution MRI data allowing quantification of atrophy and white matter disease were not available in the current analyses. Our findings might be explained by the neurotoxic effects of tau tangles causing neurodegeneration and neuronal death along neural circuits involved in motor function. According to neuropathologic evidence in older persons, gait speed is associated with neurofibrillary tangles in the substantia nigra, a region known to be key in the modulation of corticostriatal circuits.34 Relationships of gait speed with tau in other brain areas have not been explored. However, Aβ plaques but not neurofibrillary tangles have been reported in the striatum of individuals predementia.5,35 Moreover, the topographic cortical distribution pattern of neurofibrillary degeneration in AD is thought to spare the primary sensory and motor cortices in the early stages of the disease.36,37 It is thus possible that both Aβ and tau exert neurotoxic effects on motor networks via different pathways and at different time points of the disease.
This was a cross-sectional study, which does not allow drawing conclusions about the direction, causality, or prognostic value of the observed association between amyloid and gait. Also, our data were drawn from an intervention study aiming to investigate the effects of a multidomain intervention and a dietary supplement of omega-3 in participants at risk of developing AD dementia. We included only participants in the PET ancillary study who were enrolled in the control arm of the multidomain intervention. However, we could not control for between-participant differences due to the omega-3 supplementation, as the randomization remains currently blinded.
Accumulating evidence suggests that gait speed represents a key indicator of frailty with predictive value of adverse health-related events in older persons, even in well-functioning older people.17 Negative outcomes include, in addition to dementia, falls, morbidity, and mortality.17 In this context, gait dysfunction is typically explained as resulting from a range of pathophysiologic processes that are inherently complex and multifactorial.38 In the present work, we focused on the specific contribution of amyloid pathology to gait performance, without taking into account other pathways known to also independently contribute to frailty such as macroinfarcts or nigral neuronal loss.1
This study showed an association between brain Aβ amyloid and slow gait speed in a population of elderly participants with high risk for dementia ranging from healthy to MCI. We provide several possible interpretations of this association, including a neural network perspective of how the neurotoxic effects of regional Aβ may be involved in the pathogenesis of motor dysfunction. More research is needed to elucidate the neural mechanisms underlying this association, ideally involving longitudinal study designs that might enable cause-effect conclusions.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants and all study contributors of the MAPT/DSA Study Group.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- BMI
body mass index
- CDR
Clinical Dementia Rating scale
- CI
confidence interval
- MAPT
Multidomain Alzheimer Preventive Trial
- MCI
mild cognitive impairment
- SUVR
standard uptake value ratio
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: MAPT/DSA Study Group, Bruno Vellas, Sophie Guyonnet, Isabelle Carrié, Sandrine Andrieu, Jean-François Dartigues, Marie-Noëlle-Cuffi, Lawrence Bories, Françoise Desclaux, Thierry Dantoine, Kristelle Sudres, Jacques Touchon, Philippe Robert, Yves Gasnier, Serge Bordes, Pierre Payoux, and Julien Delrieu
AUTHOR CONTRIBUTIONS
N. del Campo performed the statistical analyses and wrote the manuscript. P. Payoux supported the PET analysis and revised the manuscript. A. Djilali performed the PET analysis and revised the manuscript. J. Delrieu helped with data interpretation and revised the manuscript. E. Hoogendijk supported the statistical analysis and revised the manuscript. Y. Rolland revised the manuscript. M. Cesari supported the statistical analysis and revised the manuscript. M. Weiner helped with data interpretation and contributed to the drafting of the manuscript. S. Andrieu contributed to the design of the study and revised the manuscript. B. Vellas was Principal Investigator of MAPT, contributed to the conceptualization of the study, and revised the manuscript.
STUDY FUNDING
MAPT was funded by the French Ministry of Health (PHRC 2008) and the Institut de Recherche Pierre Fabre. The [18F]florbetapir PET scans were supported by Avid Radiopharmaceuticals/Eli Lilly and Company. Biological sample collection was supported by Exhonit Therapeutics. The promotion of this study was supported by the University Hospital Center of Toulouse.
DISCLOSURE
N. del Campo, P. Payoux, A. Dijilali, J. Delrieu, E. Hoogendijk, and Y. Rolland report no disclosures relevant to the manuscript. M. Cesari reports grants from Pfizer and Innovative Medicines Initiative and personal fees from Nestlé and Novartis. M. Weiner has served on the scientific advisory boards for Pfizer, BOLT International, Neurotrope Bioscience, and Eli Lilly; and provided consulting to Synarc, Pfizer, Janssen, KLJ Associates, Easton Associates, Harvard University, University of California, Los Angeles (UCLA), Alzheimer's Drug Discovery Foundation (ADDF), Avid Radiopharmaceuticals, Clearview Healthcare Partners, Perceptive Informatics, Smartfish AS, Decision Resources, Inc., Araclon, Merck, Defined Health, and Genentech. The following entities have provided funding for travel: Pfizer, Paul Sabatier University, MCI Group France, Travel eDreams, Inc., Neuroscience School of Advanced Studies (NSAS), Danone Trading, BV, CTAD Ant Congres, Kenes, Intl., ADRC, UCLA, UCSD, Sanofi-Aventis Groupe, University Center Hospital, Toulouse, Araclon, AC Immune, Eli Lilly, New York Academy of Sciences (NYAS), and National Brain Research Center, India, for Johns Hopkins Medicine. He served on the editorial boards for Alzheimer's & Dementia and Magnetic Resonance Imaging. He received honoraria from Pfizer, Tohoku University, and Danone Trading, BV; and research support from Merck, Avid, the Veterans Administration (VA), and Department of Defense (DOD). S. Andrieu reports personal fees from Beaufour Ipsen Pharma SAS, Esai Inc., Pierre Fabre Laboratories, Pfizer, Eli Lilly and Company, Lundbeck Inc., Nestle S.A., Novartis, Roche, Servier, Janssen, Exhonit, Sanofi, and Beaufour Ipsen Pharma SAS; and grants from Beaufour Ipsen Pharma SAS, Eli Lilly and Company, Lundbeck Inc., and Nestle S.A. B. Vellas reports personal fees from Lilly, MSD, Nestlé, Roche, Sanofi; and grants from Abbvie, Affiris, Avid, Eisai, Envivo, Exhonit, Genentech, GSK, Lilly, MSD, Nutricia, Otsuka, Pharnext, Pfizer, Pierre-Fabre, Régénéron, Roche, Sanofi, Servier, TauRx Therapeutics, and Wyeth. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Buchman AS, Yu L, Wilson RS, Schneider JA, Bennett DA. Association of brain pathology with the progression of frailty in older adults. Neurology 2013;80:2055–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montero-Odasso M, Oteng-Amoako A, Speechley M, et al. The motor signature of mild cognitive impairment: results from the gait and brain study. J Gerontol A Biol Sci Med Sci 2014;69:1415–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buracchio T, Dodge HH, Howieson D, Wasserman D, Kaye J. The trajectory of gait speed preceding mild cognitive impairment. Arch Neurol 2010;67:980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Alzheimer's disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol 1990;49:215–224. [PubMed] [Google Scholar]
- 5.Wolf DS, Gearing M, Snowdon DA, Mori H, Markesbery WR, Mirra SS. Progression of regional neuropathology in Alzheimer disease and normal elderly: findings from the Nun study. Alzheimer Dis Assoc Disord 1999;13:226–231. [DOI] [PubMed] [Google Scholar]
- 6.Knight WD, Okello AA, Ryan NS, et al. Carbon-11-Pittsburgh compound B positron emission tomography imaging of amyloid deposition in presenilin 1 mutation carriers. Brain 2011;134:293–300. [DOI] [PubMed] [Google Scholar]
- 7.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986;9:357–381. [DOI] [PubMed] [Google Scholar]
- 8.Draganski B, Kherif F, Klöppel S, et al. Evidence for segregated and integrative connectivity patterns in the human basal ganglia. J Neurosci 2008;28:7143–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vellas B, Carrie I, Gillette-Guyonnet S, et al. MAPT study: a multidomain approach for preventing Alzheimer's disease: design and baseline data. J Prev Alzheimer's Dis 2014;1:13–22. [PMC free article] [PubMed] [Google Scholar]
- 10.Gill TM. Assessment of function and disability in longitudinal studies. J Am Geriatr Soc 2010;58(suppl 2):S308–S312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer's disease and cognitively normal subjects. J Nucl Med 2012;53:378–384. [DOI] [PubMed] [Google Scholar]
- 12.Fleisher AS, Chen K, Liu X, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol 2011;68:1404–1411. [DOI] [PubMed] [Google Scholar]
- 13.Barnett JH, Hachinski V, Blackwell AD. Cognitive health begins at conception: addressing dementia as a lifelong and preventable condition. BMC Med 2013;11:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu WL, Atti AR, Gatz M, Pedersen NL, Johansson B, Fratiglioni L. Midlife overweight and obesity increase late-life dementia risk: a population-based twin study. Neurology 2011;76:1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elbaz A, Shipley MJ, Nabi H, Brunner EJ, Kivimaki M, Singh-Manoux A. Trajectories of the Framingham general cardiovascular risk profile in midlife and poor motor function later in life: the Whitehall II study. Int J Cardiol 2014;172:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001;56:M146–M156. [DOI] [PubMed] [Google Scholar]
- 17.Cesari M, Kritchevsky SB, Penninx BWHJ, et al. Prognostic value of usual gait speed in well-functioning older people: results from the Health, Aging and Body Composition Study. J Am Geriatr Soc 2005;53:1675–1680. [DOI] [PubMed] [Google Scholar]
- 18.Aggarwal NT, Wilson RS, Beck TL, Bienias JL, Bennett DA. Motor dysfunction in mild cognitive impairment and the risk of incident Alzheimer disease. Arch Neurol 2006;63:1763–1769. [DOI] [PubMed] [Google Scholar]
- 19.Burzynska AZ, Chaddock-Heyman L, Voss MW, et al. Physical activity and cardiorespiratory fitness are beneficial for white matter in low-fit older adults. PLoS One 2014;9:e107413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gómez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 1997;41:17–24. [DOI] [PubMed] [Google Scholar]
- 21.Josephs KA, Whitwell JL, Ahmed Z, et al. Beta-amyloid burden is not associated with rates of brain atrophy. Ann Neurol 2008;63:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2012;2:a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009;457:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008;14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedden T, Van Dijk KRA, Becker JA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci 2009;29:12686–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim HK, Nebes R, Snitz B, et al. Regional amyloid burden and intrinsic connectivity networks in cognitively normal elderly subjects. Brain 2014;137:3327–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehéricy S, Bardinet E, Tremblay L, et al. Motor control in basal ganglia circuits using fMRI and brain atlas approaches. Cereb Cortex 2006;16:149–161. [DOI] [PubMed] [Google Scholar]
- 28.Di Martino A, Scheres A, Margulies DS, et al. Functional connectivity of human striatum: a resting state fMRI study. Cereb Cortex 2008;18:2735–2747. [DOI] [PubMed] [Google Scholar]
- 29.Ishibashi K, Ishiwata K, Toyohara J, Murayama S, Ishii K. Regional analysis of striatal and cortical amyloid deposition in patients with Alzheimer's disease. Eur J Neurosci 2014;40:2701–2706. [DOI] [PubMed] [Google Scholar]
- 30.Margulies DS, Vincent JL, Kelly C, et al. Precuneus shares intrinsic functional architecture in humans and monkeys. Proc Natl Acad Sci USA 2009;106:20069–20074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleinschmidt A, Thilo KV, Büchel C, Gresty MA, Bronstein AM, Frackowiak RSJ. Neural correlates of visual-motion perception as object- or self-motion. Neuroimage 2002;16:873–882. [DOI] [PubMed] [Google Scholar]
- 32.Verghese J, Wang C, Lipton RB, Holtzer R. Motoric cognitive risk syndrome and the risk of dementia. J Gerontol A Biol Sci Med Sci 2013;68:412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verghese J, Annweiler C, Ayers E, et al. Motoric cognitive risk syndrome: multicountry prevalence and dementia risk. Neurol 2014;83:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider JA, Li J, Li Y, Wilson RS, Kordower JH, Bennett DA. Substantia nigra tangles are related to gait impairment in older persons. Ann Neurol 2006;59:166–173. [DOI] [PubMed] [Google Scholar]
- 35.Beach TG, Sue LI, Walker DG, et al. Striatal amyloid plaque density predicts Braak neurofibrillary stage and clinicopathological Alzheimer's disease: implications for amyloid imaging. J Alzheimers Dis 2012;28:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex 2014;1:103–116. [DOI] [PubMed] [Google Scholar]
- 37.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 38.Snijders A, van de Warrenburg B. Neurological gait disorders in elderly people: clinical approach and classification. Lancet 2007;6:63–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.