

Abstract
Proteins belonging to the AP-1 family of transcription factors are known to be involved in the regulation of neuronal viability. While strides have been made to elucidate the mechanisms of how individual members regulate cell death, much remains unknown. We find that the expression of one AP-1 member, c-Fos, is reduced in cerebellar granule neurons (CGNs) induced to die by low potassium (LK) treatment. Restoration and increase of this expression protect CGNs against LK-induced death, whereas knockdown induces death of otherwise healthy neurons. Furthermore, forced expression can protect cortical neurons against homocysteic acid (HCA)-induced toxicity. Taken together, this suggests that c-Fos is necessary for neuronal survival and that elevating c-Fos expression has a neuroprotective effect. Consistent with this idea is the finding that c-Fos expression is reduced selectively in the striatum in two separate mouse models of Huntington’s disease and forced expression protects against neuronal death resulting from mutant huntingtin (mut-Htt) expression. Interestingly, neuroprotection by c-Fos does not require its DNA-binding, transcriptional, or heteromerization domains. However, this protective activity can be inhibited by pharmacological inhibition of c-Abl, CK-I, and MEK-ERK signaling. Additionally, expression of point mutant forms of this protein has identified that mutation of a tyrosine residue, Tyr345, can convert c-Fos from neuroprotective to neurotoxic. We show that c-Fos interacts with histone deacetylase-3 (HDAC3), a protein that contributes to mut-Htt neurotoxicity and whose overexpression is sufficient to promote neuronal death. When co-expressed, c-Fos can protect against HDAC3 neurotoxicity. Finally, our study identifies a 21-amino acid region at the C-terminus of c-Fos that is sufficient to protect neurons against death induced by LK, HCA treatment, or mut-Htt expression when expressed via a plasmid transfection or as a cell-permeable peptide. This cell-permeable peptide, designated as Fos-CTF, could have potential as a therapeutic agent for neurodegenerative diseases.
Keywords: AP-1, Neuronal death, Neuronal survival, Neurodegeneration, Huntington’s disease
Neurodegenerative diseases represent a major medical, economic, and social problem. A common and unifying feature of neurodegenerative diseases is the abnormal loss of neurons for which there is no cure or treatment strategy. Identifying molecules that protect against or promote neuronal death is of utmost importance because these molecules or their downstream effectors can be targeted for the development of therapies against neurodegenerative disorders. Among the molecules involved in the regulation of neuronal survival and death are members of the AP-1 family of transcription factors [1–4]. The members include the Fos proteins (c-Fos, FosB, Fra-1, and Fra-2) and the Jun proteins (c-Jun, JunB, and JunD). These proteins function by binding as homodimers or heterodimers to gene promoters containing AP-1 sites regulating both basal and inducible transcription of these genes [1–4]. Dimerization of these proteins involves a leucine zipper (LZ) domain, whereas binding to target sequences involves a DNA-binding domain located adjacent to the LZ domain. Members of the ATF protein family also heteromerize with AP-1 proteins. Whereas Jun proteins can function as homodimers, Fos and ATF proteins heterodimerize with Jun proteins to form transcriptionally active AP-1. In addition to regulating neuronal survival, AP-1 proteins are involved in a wide array of physiological processes, and the deregulation of their activity has been implicated or shown to be key players in many pathological conditions [1–4]. The large number and variety of biological functions attributed to AP-1 can be explained, in part, by the numerous combinations of Fos, Jun, and ATF components that can make up the active transcription factor. It deserves mention that some AP-1 proteins such as c-Fos and c-Jun have been found to interact with other non-AP-1 and ATF proteins, although the biological significance of these interactions is not clear [5–9].
Many laboratories have described that c-Jun plays a pivotal role in the promotion of neuronal death, an action involving both an increase in its expression as well as its phosphorylation by c-Jun N-terminal kinase (JNK) [10–12]. In cerebellar granule neurons (CGNs) induced to die by low potassium (LK) treatment, it has been reported that c-Jun may promote death by heterodimerization with ATF2, whose activity is also dependent on JNK phosphorylation [13]. Intriguingly, another recent study reported that a complete knockdown of c-Jun and ATF2 using siRNA fails to protect against LK-induced death, suggesting that neither c-Jun nor ATF2 is critical for neuronal death [14]. In addition to c-Jun and ATF2, ATF3 has been proposed to be necessary for LK-induced apoptosis [15]. Overexpression of ATF3 kills neurons in HK medium, while small hairpin RNA (shRNA)-mediated suppression of ATF3 expression protects in LK [15]. In contrast to these proteins, which promote neuronal death, emerging evidence suggests that c-Fos plays a protective role against neurodegeneration. Mice lacking c-Fos in the hippocampus have been reported to display increased neuronal death following kainic acid administration [16]. Another study found that c-Fos expression is reduced in the striatum of R6/2 mice, a transgenic model of Huntington’s disease (HD), and this decrease is attenuated by administration of a small molecule inhibitor of kyurenine 3-monooxygenase with neuroprotective effects [17]. Reduced c-Fos expression has also been described in R6/1 mice, another transgenic HD mouse model [18].
In this study, we have investigated more directly whether c-Fos promotes neuronal survival. We find that c-Fos expression is reduced in CGNs induced to die by LK treatment. We confirmed and extended previous observations [17, 18] to show that c-Fos expression is also reduced in the striatum in both chemical and genetic mouse models of HD. Recapitulating this reduction by shRNA-mediated downregulation, results in the death of otherwise healthy neurons indicated that elevated c-Fos expression is necessary for neuronal survival. Consistently, restoring elevated c-Fos levels by ectopic expression protects cultured neurons from death. Interestingly, increased expression of c-Jun is not sufficient to induce cell death unless accompanied by reduced c-Fos expression. In addition to c-Fos itself, we have also identified a 21-amino acid peptide derived from the C-terminal region of the c-Fos protein that is highly neuroprotective in tissue culture models.
Materials and Methods
Materials
Unless otherwise indicated, all reagents and materials were purchased from Sigma-Aldrich (St. Louis, MO). Life technologies (Grand Island, NY) was used to purchase tissue culture-related materials. Antibodies used in this paper were as follows: Flag (catalog #F1804, Sigma-Aldrich), α-Tubulin (catalog #T9026, Sigma-Aldrich), GFP (B-2 catalog #sc-9996; FL catalog #sc-8334, Santa Cruz Biotechnology (Dallas, Texas)), c-Fos (H-125 catalog #sc-7202, Santa Cruz Biotechnology; 9F6 catalog #2250, Cell Signaling Technology, Danvers, MA), c-Jun (L70B11 catalog #2315, Cell Signaling Technology; H-79 catalog #sc-1694, Santa Cruz Biotechnology), Huntington (D7F7 catalog #5656, Cell Signaling Technology), ERK (p42/44 MAPK catalog #9102, Cell Signaling Technology), and pERK (Phospho-p42/44 MAPK catalog #9101, Cell Signaling Technology). Primary antibodies were used at dilutions from 0.1 to 1.0 ng/μl for Western blot analysis and 1.0 to 5.0 ng/μl for immunocytochemistry in 5 % bovine serum albumin (BSA). Secondary antibodies (Thermo Fisher Scientific, Waltham, MA) were used at concentrations 0.2–0.5 ng/μl for Western blots. DyLight 594 or Alexa Fluor® 488-conjugated secondary antibodies for immunocytochemistry were purchased from Jackson Immunoresearch Laboratories (West Grove, PA) and used at a concentration 2.0 ng/μl. Fos-CTF-R9 peptide was purchased from United Biosystems Inc. (Herndon, VA).
Plasmids
The Flag-c-Fos expression vector, its mutants, and HDAC3-Flag were obtained from Addgene (Cambridge, MA) (39). GFP-tagged and untagged c-Fos expression vector and their mutants were kind gift from Mark Piechaczyk [19, 20]. Flag-c-Fos Y345D was constructed by site-directed mutagenesis. FosB and ΔFosB plasmids were a gift from Eric J. Nestler of Mount Sinai Medical Center, NY. GFP- and RFP-tagged Htt plasmids were a kind gift from Dr. Troy Littleton (Massachusetts Institute of Technology, Cambridge, MA). JAZ-Flag expression plasmid was cloned in our lab.
Culture and Transfection of CGNs
CGNs were cultured from 7 to 8-day-old Wistar rats and plated on dishes coated with poly-L-lysine in Basal Eagle’s medium with Earle’s salts (BME) supplemented with 10 % fetal bovine serum, 25 mM KCl, 2 mM glutamine, and 100 μg/ml gentamicin [21, 22]. Cytosine arabinofuranoside (10 μM) was added to prevent proliferation of nonneuronal cells 20 h after plating. Transient transfection using the calcium phosphate method was performed after 5 days in vitro [23–25]. Transfection efficiency in these cultures ranges from 0.1 to 1.0 %. Eight to 24 h after transfection, cultures were treated with BME supplemented with high potassium, 25 mM KCL (HK), or low potassium, 5 mM KCL (LK), for 24 h. Control experiments indicated that viability of transfected cultures was similar to untransfected cultures. Successfully transfected neurons were detected by EGFP fluorescence or by immunostaining using anti-Flag antibody [23–25]. Nuclear staining with 4,6-diamidino-2-phenylindole hydrochloride (DAPI) (0.1 μg/ml) was used to determine viability of neurons [23–25]. Condensed or fragmented nuclei were considered apoptotic. Pharmacological inhibitors when used were added at the time of HK/LK treatment. These were used at the following concentrations: PD98059 at 40 μM, U0126 at 10 μM, wortmannin (WORT.) at 100 nM, LY294002 at 20 μM, KN62 at 50 μM, Trichostatin A (TSA) at 1 μM, IC261 at 10 μM, 4,5,6,7-tetrabromobenzotriazole (TBB) at 10 μM and Gleevec (GLEE.) at 50 μM. All pharmacological inhibitors were purchased from EMD Millipore (Darmstadt, Germany). Viability or phosphorylation of target proteins was used to confirm effectiveness of the inhibitors (data not shown). Animals were used in strict adherence to NIH guidelines and protocols approved by the Southern Methodist University IACUC.
Cortical Neuron Culture and Transfection
Cortical cultures were prepared from Wistar rats at embryonic day 16 or 17 as described previously [24, 26, 27]. Cultures were kept in Neurobasal medium with 1 % B27 supplement, 0.25 % L-glutamine, 1 % penicillin/streptomycin, 0.1 % HEPES, and 1.1 % sodium pyruvate without serum to minimize glial proliferation. Cultures were transfected on day 5 in vitro 24 h after transfection cultures were treated with 1 mM homocysteic acid (HCA). Immunocytochemistry was performed after 18 h of HCA treatment. Viability of successfully transfected cells was quantified following immunocytochemistry and DAPI staining as described above for CGNs.
Site-Directed Mutagenesis
Site-directed mutagenesis was accomplished by using QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) following the manufacturer’s instructions. All the constructs were sequenced to check for mutations and expressed in human embryonic kidney (HEK293T) cells to check for protein expression. The following primers were used to create site-directed mutants:
c-Fos Y345D forward: 5′ CCTTTGTCTTCACCGACCCCGAGGCTGAC 3′
c-Fos Y345D reverse: 5′ GTCAGCCTCGGGGTCGGTGAAGACAAAGG 3′
Cell Lines and Transfection
HEK293T cells were purchased from the ATCC (catalog # CRL-11268). HT22 cells were a kind gift from Dr. Rajiv Ratan (Burke Medical Research Institute, NY). Both cell lines were grown in DMEM supplemented with 10 % FBS and transfected using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions.
RT-PCR
RNA was extracted from cultured neurons by using TRIzol (Life Technologies) according to the manufacturer’s instructions. Three to 5 μg of RNA was used for cDNA synthesis using the SuperScript First-Strand Synthesis System for RT-PCR (Life Technologies) according to the manufacturer’s instructions. PCR was performed with GoTaq Green Master Mix (Promega, Madison, WI). The primers used for PCR amplification are listed in supplementary data Table 1.
shRNA-Mediated Suppression
The pLKO.1-TRC control vector (Addgene) containing a nonhairpin 18-bp insert was used as transfection control. Five c-Fos shRNAs were used in this study: TRCN0000042679, TRCN0000231168, TRCN0000231166, TRCN0000042678, and TRCN0000042680 (Sigma-Aldrich) designated as sh1-5, respectively. Ability to suppress expression was performed using HEK293T cells. Briefly, HEK293T cells were transfected with pLKO.1 or with c-Fos shRNA plasmids along with c-Fos Flag plasmid in the ratio 7:1. Lysates were harvested 24 h later and subjected to Western blot analysis as described above. To examine the effect of c-Fos knockdown on neuronal viability, CGNs were co-transfected on day 5 in vitro with either pLKO.1 control plasmid or one of the c-Fos shRNA-expressing plasmids along with CMV-EGFP in a ratio of 6.5:1. Twenty-four hours later, the neurons were treated with HK or LK containing medium for another 24 h, followed by immunocytochemistry. Viability of the GFP-expressing cells was determined using DAPI staining, as described above.
Western Blot Analysis
Cell lysate was prepared by using 1× cell lysis buffer (Cell Signaling Technology). Fifty to hundred μg of protein from each sample were mixed with 6× SDS sample buffer [300 mM Tris-HCl (pH 6.8 at 25 °C), 12 % SDS, 60 % glycerol, 5 % β-mercaptoethanol, 0.01 % bromophenol blue]. After boiling at 95 °C for 5 min, samples were subjected to SDS-PAGE and transferred electrophoretically onto polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA). The membranes were blocked in 5 % nonfat dry milk in TBST. Membranes were incubated with primary antibody overnight at 4 °C, followed by secondary antibody for 1 h at room temperature. Immunoreactivity was examined by enhanced chemiluminescence (GE Healthcare Bio-Sciences, Pittsburgh, PA) according to the manufacturer’s instruction.
Immunoprecipitation
Cell lysates were harvested with 300 μl of 1× cell lysis buffer. Lysates were centrifuged at 10,000g for 10 min at 4 °C. Thirty microliters of the protein lysate were mixed with 6× SDS, boiled at 95 °C for 5 min, and subjected to Western blot analysis to check for protein overexpression (in the case of transfected cell lines) and to check for equal protein usage for immunoprecipitation. Thirty microliters of Protein A/G Plus-Agarose beads (Santa Cruz Biotechnology) were preincubated with 1 μg of pull-down antibody in 300 μl of 1× cell lysis buffer for 1 h at 4 °C with constant rocking. Beads bound with pull-down antibody were precipitated by centrifugation at 3300g for 2 min, and the supernatant was discarded. Two hundred fifty microliters of protein lysate was precleared with 30 μl of Protein A/G Plus-Agarose beads to reduce non-specific binding. After centrifuging at 3300g for 2 min, supernatant from preclear was added to the beads bound with pull-down antibody and incubated overnight at 4 °C with constant rocking. Immunoprecipitate was collected by centrifugation at 3300g for 2 min and washed thrice with 500 μl of 1× cell lysis buffer. Thirty microliters of 3× SDS were added to the immunoprecipitate, boiled for 5 min at 95 °C, and then subjected to Western blot analysis [25, 27].
Live/Dead Assay
Live/Dead assay was done using LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells (catalog #L-3224, Life technologies) as per the manufacturer’s instructions.
3-Nitropropionic Acid Treatment
Ten-week-old C57BL/6 male mice were purchased from Charles River Laboratories, Inc. (Wilmington, MA). Administration of 3-nitropropionic acid (3-NP) was performed as previously described [28, 29]. Briefly, 3-NP was dissolved in water and the solution brought to pH 7.4 with sodium hydroxide. Freshly prepared 3-NP was administered in ten intraperitoneal injections (50–55 mg/kg twice a day for 5 days). Saline injections were used for control animals. Mice were euthanized on the first, third, and fifth day following injection, and their brains were dissected. Striatum was extracted from other brain parts (OBP) and messenger RNA (mRNA) prepared from the tissue for analysis of gene expression.
Analysis of the R6/2 Transgenic Mouse Model
Female mice hemizygous for B6CBA-Tg(HDexon1)62Gpb/1J (via ovarian transplant) were bred to B6CBAF1/J males; both obtained from Jackson Laboratories (Bar Harbor, ME). These animals have glutamine expansion of 160±5. Genotyping was performed by PCR using tail-tip DNA collected from offspring on day 10. At 13 weeks of age, wild-type and transgenic littermate (R6/2) pairs were euthanized by carbon dioxide inhalation, their brains were dissected. Striatum was extracted from other brain parts (OBP) and mRNA prepared from the tissue for analysis of gene expression [27].
Statistical analysis
All the graphs in this study were generated using GraphPad Prism 5 software. Unless otherwise mentioned in the figure legends, statistical analysis was done using unpaired two-tailed t test (Student’s t test), and the results are shown as mean standard deviation. The p values <0.05 were deemed statistically significant. All viability experiments were done in duplicate and repeated at least three times. One hundred fifty to two hundred transfected cells were counted for each experiment.
Results
c-Jun Is Not Sufficient to Induce Cell Death
Several laboratories have reported increased expression of c-Jun in CGNs following LK treatment, as well as in other in vitro and in vivo paradigms of neurodegeneration [10–12]. Indeed, c-Jun induction is often considered a marker of neuronal death. As observed in other paradigms of neuronal death, such as NGF-deprived sympathetic neurons [11], LK-induced death of CGNs is blocked by the inhibition of c-Jun activity [10]. LK-induced death of CGNs can also be completely blocked by addition of IGF-1, a neurotrophic factor for many neuronal types [22, 30]. Surprisingly, the expression of c-Jun in IGF-1-treated CGN cultures is even higher than that seen in LK-treated cultures both at the mRNA and protein level (Fig. 1a, b). This observation demonstrates that increased expression of c-Jun by itself is not detrimental to neuronal survival and raises the possibility that modulation of expression or activity of another AP-1 protein, perhaps in conjunction with c-Jun stimulation, is necessary for neuronal death to ensue.
Fig. 1.
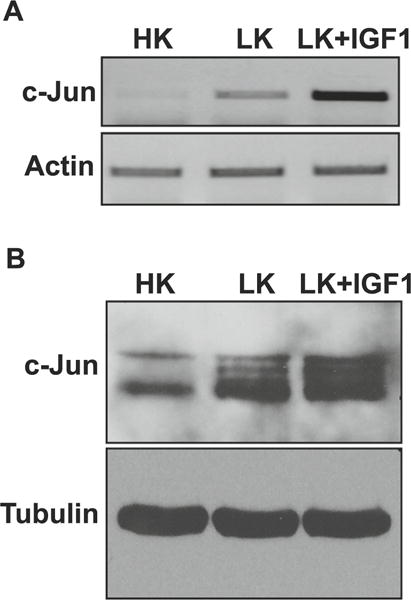
Effect of IGF-1 on c-Jun expression in CGNs. a RNA isolated from CGNs treated with HK, LK, or LK IGF-1 for 6 h was subjected to RT-PCR analysis for c-Jun expression. Actin serves as a loading control. b Protein extracted from CGNs treated with HK, LK, or LK IGF-1 for 6 h was subjected to Western blot analysis for c-Jun expression. Tubulin serves as a loading control
Expression of c-Fos and Other Proteins Is Modulated by LK Treatment
As a step toward identifying AP-1 proteins that regulate neuronal survival and death, we examined the mRNA expression of all members of the Fos and Jun subfamilies, as well as several members of the ATF family. In contrast to c-Jun, the expression of JunB and JunD was unchanged in neurons primed to die (Fig. 2a). Within the Fos family of proteins, the expression of Fra-1 was upregulated in LK-treated CGNs, whereas expression of c-Fos and FosB was substantially reduced by LK treatment (Fig. 2b). Among the ATF proteins, ATF3 was upregulated, whereas ATF6 and ATF6b showed reduced expression in LK (Fig. 2c). Because the expression of c-Fos mRNA was most highly altered, we focused on it. As observed at the mRNA level, c-Fos protein is also downregulated in CGNs within 3 h of LK treatment (Fig. 2d). The result obtained by Western blot analysis was confirmed by immunocytochemical analysis of LK-treated CGNs (Fig. 2e).
Fig. 2.
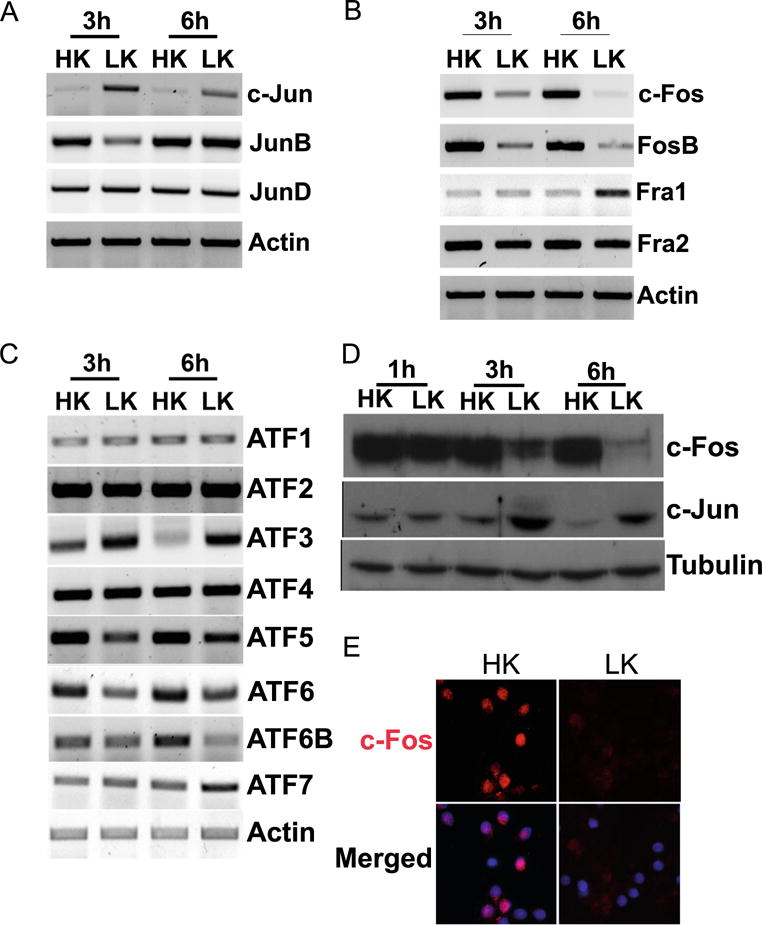
Expression profile of AP-1 members in postmitotic neurons. RNA isolated from CGNs treated with HK or LK for 3 and 6 h was subjected to RT-PCR analysis for Jun family (a), Fos family (b), and ATF family (c) expression. Actin serves as a loading control. d Whole-cell lysates prepared from CGNs treated with HK or LK for 1, 3, and 6 h were subjected to Western blot analysis using c-Fos and c-Jun antibody. Tubulin serves as a loading control RT-PCR, and Western blot were done at least three times from different samples. e CGNs were treated with HK or LK for 6 h. Immunocytochemistry with a c-Fos antibody was performed to visualize c-Fos levels. DAPI was used to visualize nuclei
c-Fos Is Necessary for Neuronal Survival Since elevated
c-Fos expression correlates with neuronal survival, we tested whether its expression was necessary for the viability of neurons by knocking down c-Fos expression in otherwise healthy neurons using separate shRNA constructs. To conduct this experiment, we first tested the ability of five separate shRNAs, designated sh1–sh5, to knockdown ectopically expressed rat c-Fos in HEK293T cells. Whereas sh2 and sh5 had an insignificant effect on ectopic c-Fos expression, sh3 and sh4 knocked down c-Fos robustly (Fig. 3a). A smaller knockdown was observed with sh1, treatment with which resulted in the production of a truncated c-Fos protein (Fig. 3a). Transfection with sh1, sh3, or sh4 reduced the survival of otherwise healthy neurons (Fig. 3b). In contrast, sh2 and sh5, which were ineffective at knocking down c-Fos expression, had no such effect. The finding that suppression of c-Fos is sufficient to induce cell death indicates that c-Fos expression is required for neuronal survival normally.
Fig. 3.
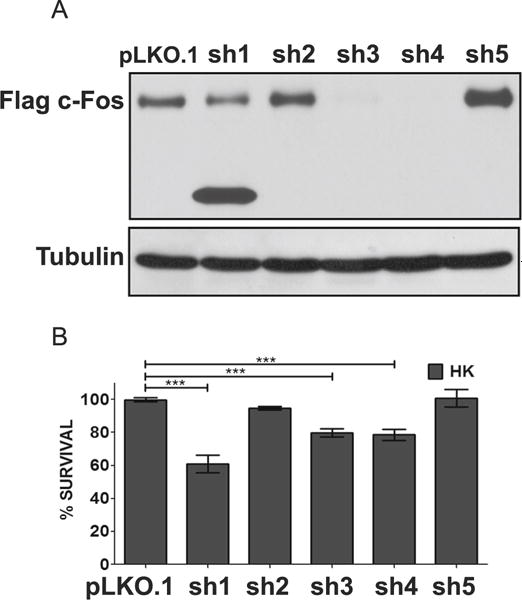
Suppression of c-Fos expression promotes neuronal death. a Efficiency of shRNA was tested with overexpression suppression using Flag-c-Fos plasmid co-expressed with either a control (pLKO.1) or an experimental shRNA (sh1-5) in the ratio 1:7. Protein lysates were prepared 24 h after transfection, and Western blot analysis was performed using anti-Flag antibody (N=3). b CGN cultures were cotransfected with plasmids expressing GFP and either pLKO.1 or one of the five c-Fos shRNA (sh1-5) in a 1:6.5 ratio. Twenty-four hours after transfection, cells were switched to serum-free HK medium. Immunocytochemistry was performed 24 h after the treatment, and cell viability of transfected cells was assessed and normalized to pLKO.1-transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.0001
If reduced, c-Fos expression is causally related to neuronal death, then raising c-Fos levels by forced expression should prevent the death of neurons exposed to apoptotic stimuli. As shown in Fig. 4a and as previously described [13], ectopic expression of c-Fos-Flag completely protected against LK-induced neuronal death. EGFP-c-Fos and untagged-c-Fos (cotransfected with EGFP to identify transfected neurons) were also expressed ectopically to rule out contributions of the Flag tag in neuroprotection. Neuroprotection was observed with both these c-Fos constructs (Fig. 4b, c). To examine if neuroprotection by c-Fos was restricted to CGNs or was a more general effect, we extended our analyses to cortical neurons. Treatment of embryonic cortical neurons with homocysteic acid (HCA) results in death as a result of oxidative stress [31, 32]. As observed in LK-treated CGNs, forced expression of c-Fos protects cortical neurons against HCA toxicity (Fig. 4d).
Fig. 4.
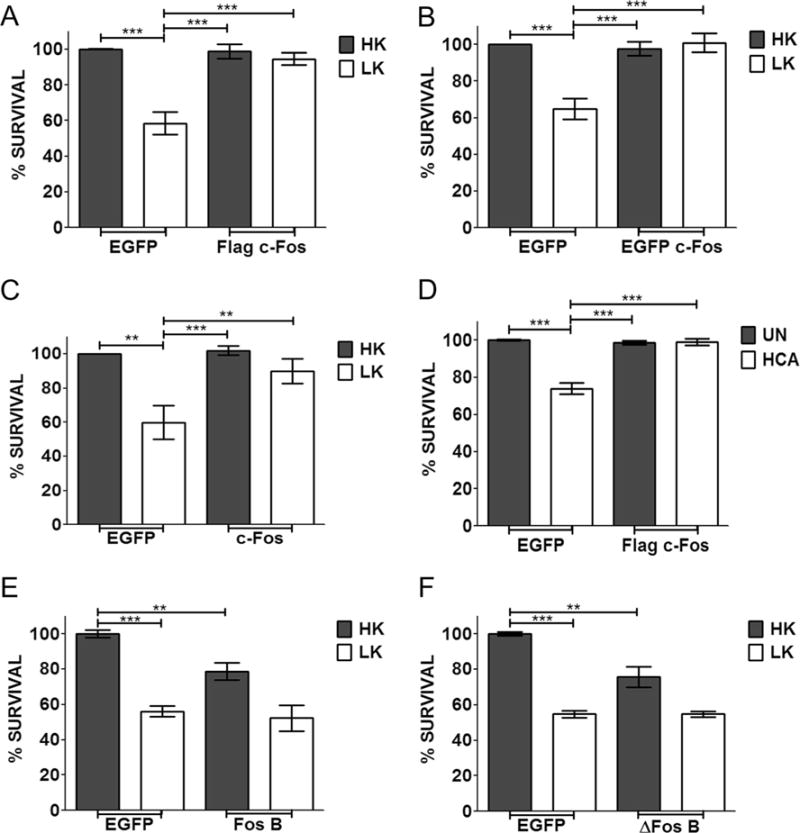
Effect of c-Fos and FosB overexpression on survival of postmitotic neurons. CGN cultures were transfected with EGFP or Flag-c-Fos (a), EGFP-c-Fos (b), untagged c-Fos (c), FosB (e), or ΔFosB (f) for 24 h and then treated with either HK or LK medium for 24 h. d Cortical cultures were transfected with Flag-c-Fos. After 24 h, they were either left untreated (UN) or treated with 1 mM HCA for 18 h. Immunocytochemistry was performed after treatment, and cell viability of transfected cells was assessed and normalized to EGFP transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.001 and ** represents p<0.005
Since FosB is also downregulated in LK-treated CGNs, we examined whether its overexpression was also neuroprotective. Despite its reduced expression under apoptotic conditions, elevation of FosB failed to protect against LK-induced death (Fig. 4e). On the contrary, ectopic expression of FosB induced apoptosis in otherwise healthy neurons and enhanced cell death in LK-treated CGN cultures. FosB is also expressed in a shorter form called ΔFosB. As observed with the longer form, expression of ΔFosB promoted neuronal death (Fig. 4f).
Inhibition of MEK-ERK, c-Abl, and Casein Kinase-I (CK-I) Signaling Reduces Neuroprotection by c-Fos
To gain insight into how c-Fos protects neurons, we examined if inhibition of signaling pathways and molecules known to promote neuronal survival impacts its ability to protect neurons. Inhibition of PI-3 kinase-Akt signaling using wortmannin and LY294002 had no effect on neuroprotection by c-Fos (Fig. 5a). Similarly, inhibition of calcium-calmodulin kinase (CaMK) with KN62, casein kinase-2 with TBB, or HDAC inhibitor TSA had no effect (Fig. 5a). Control experiments confirmed that the inhibitors were inhibiting their targets at the concentrations used in our analyses (data not shown). In contrast, treatment with two separate MEK inhibitors (U0126 and PD98059) and c-Abl inhibitor (Gleevec) caused a reduction in c-Fos-mediated neuroprotection (Fig. 5a). Additionally, neuroprotection by c-Fos was abrogated by IC261, an inhibitor of CK-I. It has previously been reported using nonneuronal systems that MEK-ERK pathway is involved in the stability of c-Fos [33, 34]. As observed by these other groups, we found that c-Fos mRNA and protein levels are reduced by treatment with MEK inhibitors raising the possibility that reduced stability might account for the effect of these inhibitors on c-Fos neuroprotection (Fig. 5b–d).
Fig. 5.
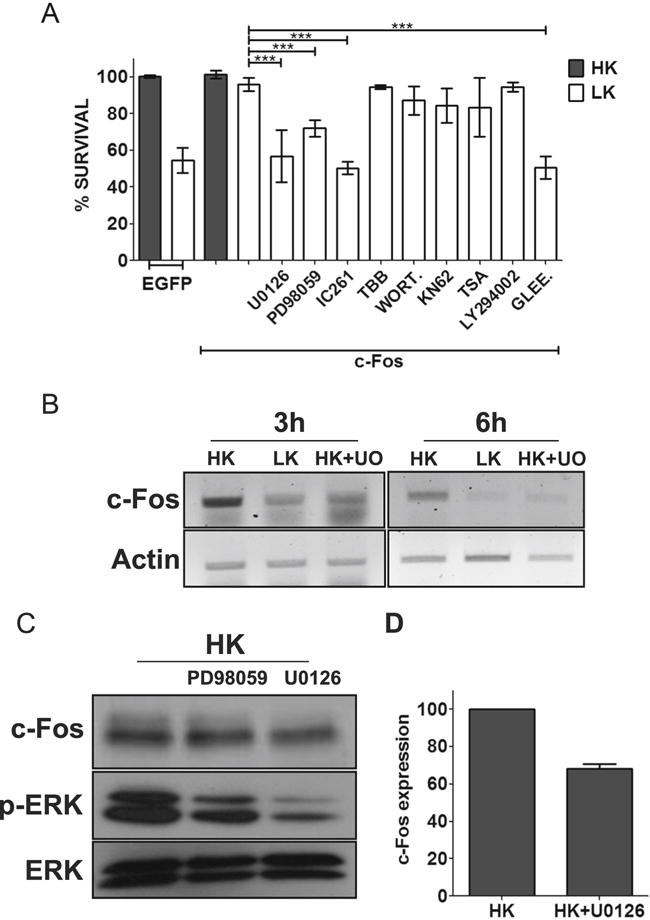
c-Fos protection is dependent on MEK-ERK pathway, casein kinase 1 (CK-I), and c-Abl. a CGN cultures, after 24 h of transfection with EGFP or Flag-c-Fos, were either treated with HK medium, LK medium, or LK medium supplemented with 10 μM U0126, 40 μM PD98059, 10 μM IC261, 10 μM TBB, 100 nM wortmannin (WORT.), 50 μM KN62, 1 μM trichostatin A, 10 μM LY294002, or 50 μm Gleevec (GLEE). Immunocytochemistry was performed 24 h after treatment, and cell viability of transfected cells was assessed and normalized to EGFP-transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.0001. Inhibitors against PI3 kinase-AKT, CaMK, HDACs, and CK-2 did not have any effect on c-Fos-mediated survival. However, c-Fos-induced protection was abolished by inhibitors against MEK-ERK signaling, CK-I, and c-Abl. b RNA isolated from CGNs treated with HK, LK, or LK+U0126 for 3 and 6 h was subjected to RT-PCR analysis for c-Fos. Actin serves as loading control. c Whole-cell lysates prepared from CGNs treated with HK, HK+ PD98059, or HK+U0126 for 3 h were subjected to Western blot analysis using anti c-Fos and anti p-ERK antibody. ERK serves as a loading control. d c-Fos protein levels were quantified and normalized to ERK levels using densitometric analysis of Western blots from three different experiments
Phosphorylation Sites Necessary for Mitogenic Stimulation of c-Fos Transcriptional Activity Are Not Necessary for Neuroprotection in Apoptotic Conditions
Following mitogen stimulation, the transcriptional activity and stability of c-Fos are enhanced by ERK phosphorylation at multiple residues within the C-terminal region [33, 35]. Phosphorylation at two residues, S362 and S374, is particularly important because this permits docking of ERK at a four-amino acid sequence (343FTYP346) called the DEF (docking site for ERK) [19, 34]. Docking of ERK at the DEF primes phosphorylation at other sites within the C-terminus transcriptional activation domain (TAD) resulting in an enhancement of c-Fos transcriptional activity [19, 34]. We find that c-Fos (S362/374A), a mutant in which the two Ser residues are replaced with Ala residues, is unable to protect neurons (Fig. 6b). Although this suggests that phosphorylation of residues is important for neuroprotection, c-Fos (S362/374A) is expressed at substantially lower levels than wild-type c-Fos, likely due to reduced protein stability (Fig. 6d). This is consistent with the reduced c-Fos expression we and others observe in the presence of ERK inhibitors [34, 35] (Fig. 5b, c). Arguing against a critical role for the two ERK phosphorylation sites in neuroprotection, our finding c-Fos (1–359), which lacks both sites, is completely protective. We also tested another mutant containing four substitutions, c-Fos (S362/364D; T325/331A). Disrupting phosphorylation at T325 and T331 greatly reduces the transcriptional activity of c-Fos in response to mitogen stimulation [35]. However, mutating these residues has no effect in the ability of c-Fos to be neuroprotective.
Fig. 6.
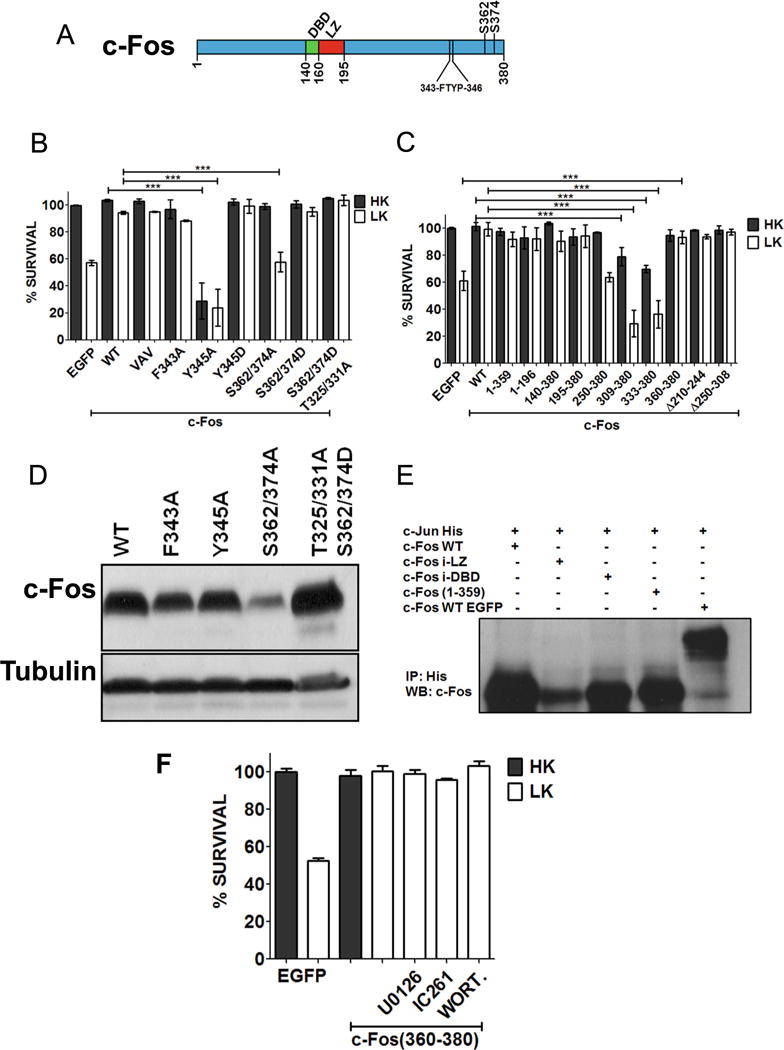
Domains and residues responsible for c-Fos neuroprotection. a Schematic diagram of c-Fos protein showing DNA binding (DBD) and leucine zipper (LZ) domain. b CGN cultures were transfected with c-Fos or point mutation constructs along with EGFP in ratio 6.5:1. Twenty-four hours after transfection, the cultures were treated with either HK or LK media. Immunocytochemistry was performed 24 h later, and cell viability of transfected cells was assessed and normalized to the EGFP-transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.0001. c CGN cultures were transfected with c-Fos deletion mutants. Twenty-four hours after transfection, the cultures were treated with either HK or LK media. Immunocytochemistry was performed 24 h later, and cell viability of transfected cells was assessed and normalized to the EGFP-transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.0001. d HEK293T cells were transfected with Flag-c-Fos or various mutant forms of it for 24 h. The expression of the various constructs was evaluated by Western blot analysis using anti-Flag antibody. e Co-immunoprecipitation of c-Jun with various c-Fos mutants. HEK293T cells were transfected with c-Jun-His along with untagged c-Fos, c-Fos insufficient (i)-LZ, c-Fos insufficient (i)-DBD, c-Fos (1–359), or EGFP-c-Fos. Twenty-four hours later, lysates were collected and subjected to immunoprecipitation using anti-His-tag antibody and were followed by Western blot analysis. f CGN cultures were transfected with EGFP-c-Fos (360–380). Twenty-four hours later, cultures were treated with HK medium, LK medium, or LK medium supplemented 10 μM U0126, 10 μM IC261, or 100 nM wortmannin (WORT.). Immunocytochemistry was performed 24 h after the treatment, and cell viability of transfected cells was assessed and normalized to EGFP-transfected control
Mutation of Tyr345 Transforms c-Fos from a Neuroprotective Protein to a Highly Toxic One
Mutation of the DEF domain by substituting either F343 or Y345 within the FTYP sequence with Ala disrupts the transcriptional activity of c-Fos in response to mitogen treatment as well as its ability to transform cells [35]. As shown in Fig. 6b, a construct in which F343 is mutated to an Ala, c-Fos (F343A), protects neurons just like wild-type c-Fos. Since both F343 and Y345 are required for ERK docking [35], this result suggests that docking of ERK is not required for neuroprotection. Interestingly, mutation of Y345 to an Ala residue not only abolishes neuroprotection but also transforms c-Fos into a neurotoxic protein (Fig. 6b). Rather than affecting docking of ERK, it is possible that mutation of Y345A abolishes phosphorylation of this residue by a tyrosine kinase. Consistent with this conclusion, mutating Y345 to a phosphomimetic Glu residue has no effect to neuroprotection, although such a mutation could be expected to disrupt ERK docking (Fig. 6b).
Neuroprotection by c-Fos Does Not Require DNA-Binding, Heteromerization with Other AP-1 Proteins, or Transcriptional Activity
To examine the contribution of various regions and domains of c-Fos to its neuroprotective activity, we expressed several deletion and point mutant forms of c-Fos in HK and LK-treated CGNs. Expression of a c-Fos (1–196), a mutant lacking the entire C-terminus half of the protein, was capable of protecting CGNs against LK-induced death (Fig. 6c). Previous studies have demonstrated that VAV-c-Fos which has a mutation of three leucine residues (L179V, L186A, and L193V within the LZ domain) destroys the ability of c-Fos to heteromerize with other AP-1 proteins [36–38]. Surprisingly, this mutant was just as effective in protecting neurons as wild-type c-Fos (Fig. 6b). We verified that this mutant was incapable of heteromerizing with c-Jun (Fig. 6e). We also used a construct that completely lacked the LZ domain and hence incapable of heteromerization. This mutant construct, c-Fos (195–380), was fully protective confirming that heteromerization with other AP-1 proteins was not necessary for neuroprotection (Fig. 6c). Expression of another mutant construct composed only of region of c-Fos spanning residues 250–380 failed to protect CGNs in LK (Fig. 6c).
Expression of C-terminus fragments of c-Fos had dramatic effects on neuronal viability. A construct spanning residues 309–380 or just 333–380 of c-Fos induced death in otherwise healthy neurons (in HK) and enhanced LK-mediated toxicity (Fig. 6c). Surprisingly, however, c-Fos (360–380), containing only the last 21 residues of c-Fos, was fully neuroprotective (Fig. 6c).
The small size of c-Fos (360–380) suggested that the mechanism by which it protected neurons was distinct from that utilized by full-length c-Fos. In support of this possibility is the finding that inhibition of CK-I or MEK-ERK signaling had no effect on neuroprotection by c-Fos (360–380) although these do block neuroprotection of full-length c-Fos (Fig. 6f).
c-Fos Is Protective Against Mutant Huntingtin Toxicity
To examine whether our findings were relevant to neurodegenerative diseases, we examined the expression of c-Fos in the R6/2 mouse model of Huntington’s disease (HD). When examined at 13 weeks, a stage when neuropathological and behavioral deficits are obvious, c-Fos expression is not only discernibly reduced in the striatum but also modestly in nonstriatal brain parts (Fig. 7a). We also examined expression in the 3-nitropropionic (3-NP) model of HD, a widely used chemical model of HD [39, 40]. Following 3-NP administration, mice display selective striatal degeneration starting at 3 days with substantial degeneration occurring by 5 days [29, 41]. As observed in the genetic model, c-Fos expression is reduced in the striatum of 3-NP administered mice coincident to neurodegeneration (Fig. 7b). There was no major change in c-Fos expression in nonstriatal brain tissue of 3-NP-injected mice. In contrast to c-Fos, expression of c-Jun is increased after 3-NP administration, particularly in the striatum (Fig. 7b). Taken together, these results show that c-Fos expression is reduced in mouse models of HD raising the possibility that providing elevated levels of c-Fos would protect against HD-associated neurodegeneration. To test this idea, we examined the effect of c-Fos overexpression in a cell culture model of HD, in which neuronal death is induced by expression of mutant Htt (mut-Htt). As shown in Fig. 8a, co-expression of c-Fos completely inhibits toxicity by mut-Htt. As observed in LK-treated CGNs, complete protection against mut-Htt was also seen by expression of EGFP-c-Fos (360–380) (Fig. 8b).
Fig. 7.
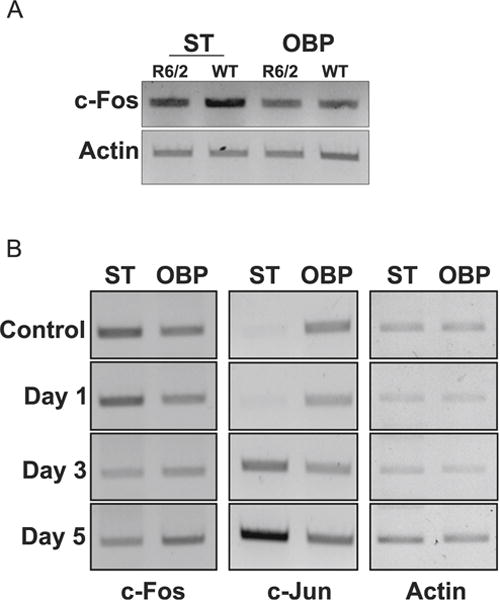
c-Fos is downregulated in mouse models of Huntington’s disease. a RT-PCR analysis of striatum (ST) and other brain parts (OBP) from 13-week-old control or R6/2 mice was performed for c-Fos. Actin serves as a loading control. b RT-PCR analysis of striatum (ST) and other brain parts (OBP) of 10-week-old mice injected with either saline (control) or 3-NP for 1, 3, or 5 days was performed for c-Fos and c-Jun. Actin serves as a loading control
Fig. 8.
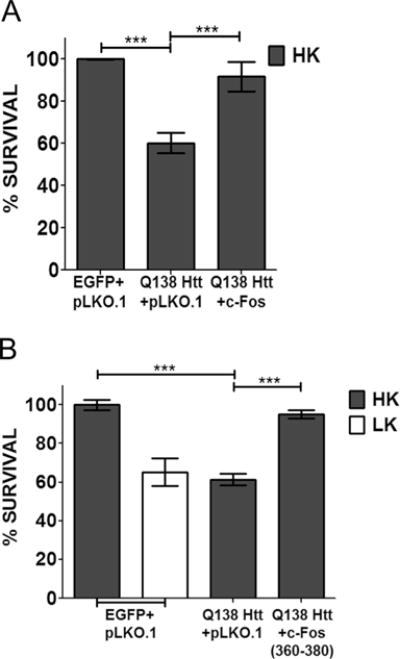
c-Fos can rescue neurons from mut-Htt-induced toxicity. CGN cultures were transfected with mut-Htt (Q138-Htt-GFP) along with pLKO.1 or Flag-c-Fos (a) or Q138-Htt-RFP along with pLKO.1 or EGFP-c-Fos (360–380) (b). Eight hours after transfection, cells were treated with either HK or LK medium. Immunocytochemistry was performed 24 h after treatment, and cell viability of transfected cells was assessed and normalized to an EGFP-transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.0001
c-Fos Is Protective Against HDAC3 Toxicity
Several studies have demonstrated that chemical inhibitors of histone deacetylases (HDACs) are protective in various invertebrate and mouse models of HD [42–47]. We previously reported that histone deacetylase-3 (HDAC3) is a key mediator of mut-Htt neurotoxicity and a likely target of the neuroprotective action of the HDAC inhibitors [26, 48]. We found that under normal conditions, the neurotoxic activity of HDAC3 is repressed through interaction with wild-type Htt [27, 48]. In the presence of mut-Htt, HDAC3 disassociates from wild-type Htt and induces neuronal death following its phosphorylation by GSK3β [26, 48]. Given that HDAC3 overexpression is neurotoxic by itself and in view of the essential role it plays in mut-Htt-induced neuronal death, we examined whether c-Fos could block the neurotoxic action of HDAC3. As shown in Fig. 9a, c-Fos does inhibit HDAC3 toxicity in CGNs maintained in HK but was less effective in LK. As observed with full-length c-Fos, expression of c-Fos (360–380) was able to protect CGNs from HDAC3-induced toxicity (Fig. 9b).
Fig. 9.
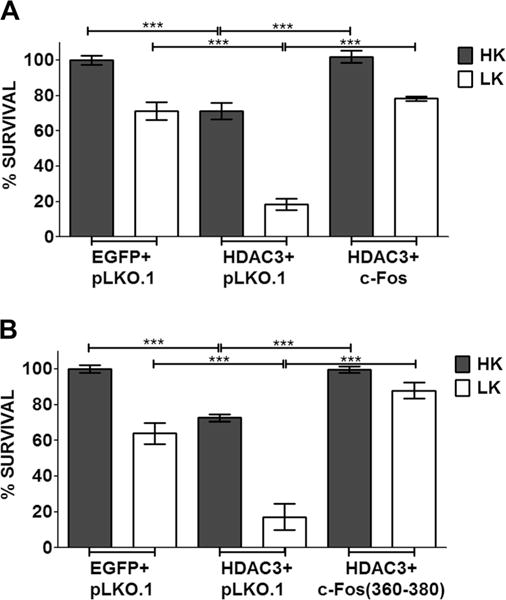
c-Fos rescues CGNs from HDAC3-induced apoptosis. CGN cultures were transfected with HDAC3-Flag along with pLKO.1, EGFP-c-Fos (a), or EGFP-c-Fos (360–380) (b). Eight hours after transfection, cells were treated with either HK or LK medium. Immunocytochemistry was performed 24 h after treatment, and cell viability of transfected cells was assessed and normalized to an EGFP-transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.0001
We previously described that in healthy neurons, wild-type Htt inhibits HDAC3 neurotoxicity by sequestering it through interaction [26]. It was possible that the protective effect of c-Fos also involved association with HDAC3. Indeed, co-immunoprecipitation studies performed in HEK293T cells in which both c-Fos and HDAC3 were co-expressed showed strong interaction between the two proteins (Fig. 10a). Although c-Fos is not expressed appreciably under normal conditions in HEK293Tcells, when overexpressed, it interacts with endogenous HDAC3 efficiently (Fig. 10b). Interestingly, c-Fos (360–380) also interacts with HDAC3 suggesting that, like c-Fos, its protective effect is also mediated through HDAC3 sequestration.
Fig. 10.
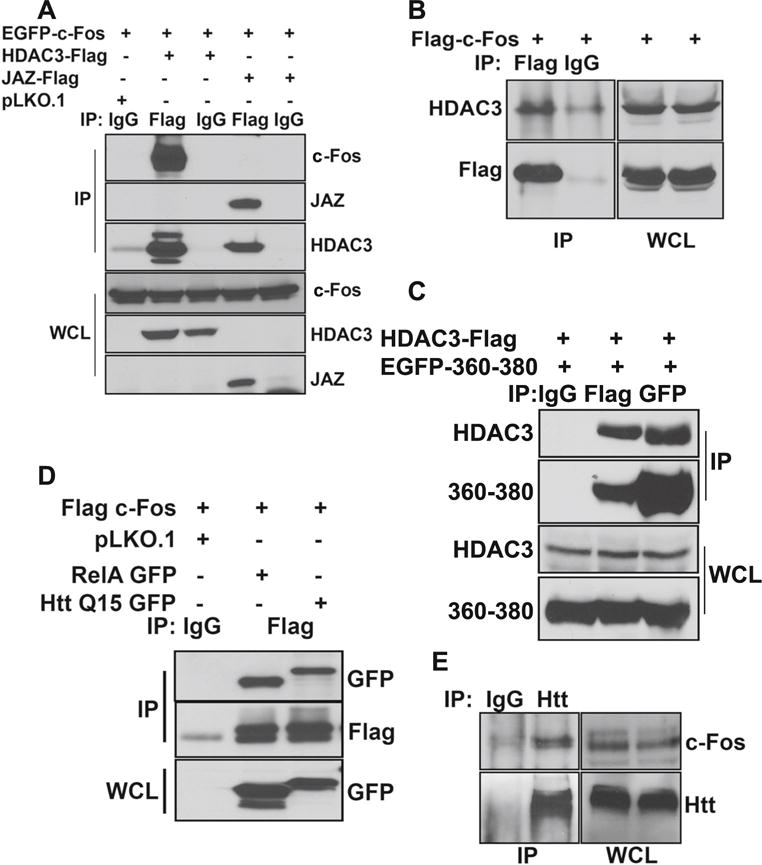
c-Fos interacts with HDAC3 and Htt. a HEK293T cells were transfected with EGFP-c-Fos along with pLKO.1, HDAC3-Flag, or JAZ-Flag. After 24 h, lysates were collected and were subjected to immunoprecipitation using either IgG or Flag antibody. Samples were then subjected to Western blot analysis. b HEK293T cells were transfected with Flag-c-Fos. Twenty-four hours later, lysates were collected and subjected to immunoprecipitation using either IgG or Flag antibody. Samples were then analyzed using Western blotting, and the presence of HDAC3 was detected. c HEK293T cells were transfected with HDAC3-Flag and EGFP-c-Fos (360–380). Twenty-four hours later, lysates were collected and subjected to immunoprecipitation using IgG, Flag, or GFP antibody. Samples were then analyzed using Western blot. d HEK293T cells were transfected with Flag-c-Fos along with pLKO.1, RelA-GFP, or Q15-Htt-GFP. After 24 h, lysates were collected and were subjected to immunoprecipitation using either IgG or Flag antibody. Samples were then subjected to Western blot analysis. e CGN cultures were treated with HK medium for 6 h. Lysates were collected, and immunoprecipitation was performed using IgG and Htt antibody. Samples were then subjected to Western blot analysis
Since HDAC3 interacts with wild-type Htt, and in view of our finding that c-Fos interacts with HDAC3, it was possible that c-Fos also associates with Htt as part of a c-Fos-HDAC3-Htt complex. As shown in Fig. 10d, co-immunoprecipitation analysis does demonstrate interaction between Htt and c-Fos when the two proteins are overexpressed in HEK293T cells. To verify that these two proteins also interact normally in neurons, we performed co-immunoprecipitation analysis using CGN lysates. Once again, interaction of endogenous Htt and c-Fos was observed in neurons (Fig. 10e).
Treatment with Fos-CTF Peptide Is Neuroprotective
As described above, an EGFP-tagged form of c-Fos (360–380) delivered into neurons by plasmid transfection had strong neuroprotective effects against LK-induced death of CGNs and against mut-Htt toxicity. To confirm and extend these findings, we synthesized a peptide spanning the region between residues 360–380 and added to the C-terminus of this 21-residue peptide 9 arginine residues (R9). Work by other laboratories, including our own, has shown that the R9 tag allows efficient delivery of peptides into cells [49–51]. As shown in Fig. 11a, treatment of cultured CGNs with c-Fos (360–380)-R9, henceforth designated as Fos C-terminal fragment (Fos-CTF), prevented LK-induced death. We have previously confirmed that R9 by itself has no effect on neuronal viability and cannot protect against LK-induced death of CGNs [49]. The increased expression of c-Jun in LK was blocked by Fos-CTF (Fig. 11b). HCA-induced toxicity of cortical neurons was also inhibited by Fos-CTF (Fig. 11c). We also tested whether Fos-CTF could inhibit against mut-Htt neurotoxicity. As shown in Fig. 11d, treatment with 10 μM of Fos-CTF had a robust protective effect against the neurotoxic effect of mut-Htt. Given its small size and its ability to inhibit mut-Htt toxicity, Fos-CTF could have value in the treatment of neurodegenerative disorders like HD.
Fig. 11.
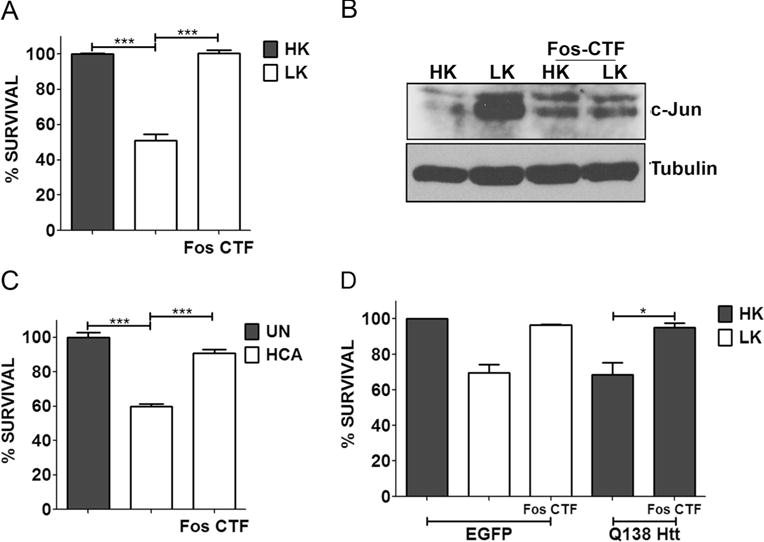
Fos-CTF is neuroprotective. a CGN cultures were treated with HK, LK, or LK+10 μM Fos-CTF for 24 h. c Cortical cultures were either left untreated (UN) or treated with 1 mM HCA with or without 10 μM Fos-CTF. Cell viability was assessed by performing Live-Dead assay 24 h and 18 h after treatment, respectively. b CGN cultures were treated with medium containing HK, LK, HK+10 μM Fos-CTF, or LK+10 μM Fos-CTF. Six hours after treatment, lysates were prepared and samples were subjected to Western blot analysis for c-Jun expression. Tubulin serves as a loading control. d CGNs were transfected with EGFP or Q138-Htt-GFP, 10 μM of Fos-CTF was added to the culture medium after transfection. Eight hours after transfection, cells were treated with HK, LK, HK+Fos-CTF, or LK+Fos-CTF medium. Immunocytochemistry was performed 24 h after the treatment and cell viability of transfected cells was assessed and normalized to EGFP transfected control. Results were obtained from at least three separate experiments done in duplicates. Statistical analysis using Student’s t test was performed where *** represents p<0.0001 and * represents p<0.05
Discussion
c-Fos expression is reduced in LK-treated CGNs and in two in vivo mouse models of HD. A knockdown of c-Fos expression in otherwise healthy neurons is sufficient to trigger death, whereas elevating c-Fos levels by ectopic expression protects CGNs from LK-induced death and cortical neurons from HCA toxicity. Furthermore, forced expression of c-Fos protects cultured neurons against death resulting from mut-Htt overexpression. Together, these findings show that c-Fos is necessary for neuronal survival, that reduction in its expression promotes neuronal death, and that overexpression of c-Fos can protect neurons against death resulting from varied stimuli. Our results confirm and extend the finding of Yuan et al. that c-Fos overexpression protects CGNs against LK-induced death [13]. The conclusion that c-Fos expression plays a protective role in neurons is also consistent with the finding that mice lacking c-Fos in the hippocampus display increased sensitivity to kainic acid-induced excitotoxicity [16]. Like us, other groups have found that c-Fos expression is reduced in the striatum of the R6/1 and R6/2 mouse models of HD [17, 18].
c-Fos is an AP-1 protein, which generally functions by heteromerizing with other AP-1 proteins, binding to DNA and activating transcription. Surprisingly, neuroprotection by c-Fos does not require heteromerization, DNA-binding, or residues and domains that play an important role in regulating its transcriptional activity at least in the context of mitogenic stimulation. This suggests a noncanonical and novel mechanism of action. Whereas pharmacological inhibition of PI3 kinase-Akt, casein kinase-II, CaMK, and HDAC signaling has no effect on neuroprotection by c-Fos, inhibition of MEK-ERK signaling reduces neuroprotection. This, however, is likely to be due to the previously described effect of MEK-ERK inhibition of c-Fos stability [35, 36] rather than a direct effect on its neuroprotective activity. Indeed, we find that pharmacological inhibition of MEK-ERK signaling or mutation of S362 and S374, residues known to be phosphorylated by ERK [36, 38], reduces the expression level of c-Fos in neurons. Strikingly, mutation of Y345 not only abolishes the neuroprotective activity of c-Fos but transforms it into a highly neurotoxic protein. Y345 lies within the DEF motif (343FTYP346) responsible for the docking of ERK to c-Fos, an event that is necessary for the stimulation of its transcriptional activity via ERK phosphorylation of other residues within the C-terminal region of c-Fos. However, the effect of Y345 on the neuroprotective activity of c-Fos is unlikely to be due to the disruption of ERK docking because mutation of F343, which is also essential for DEF function, has no effect on the ability of c-Fos to protect neurons. It is more likely that Y345 is the phosphorylation site of a tyrosine kinase and that its phosphorylation is essential for neuroprotection by c-Fos. In support of the idea that phosphorylation by a tyrosine kinase rather than ERK docking is the mechanism by which Y345 contributes to neuroprotection is the finding that mutation of this residue to a phosphomimetic one has no effect on neuroprotection by c-Fos, although this should reduce DEF function. In silico analysis suggests that Y345 could serve as a phosphorylation site for c-Abl or CK-I. Interestingly, pharmacological inhibitors of both c-Abl and CK-I abrogate the neuroprotective effect of c-Fos. More work is needed to confirm the requirement of c-Abl and CK-I to c-Fos neuroprotection and, if so, to determine whether any of these kinases act by phosphorylating Y345.
We report for the first time that c-Fos interacts with HDAC3. Knockdown experiments have shown that HDAC3 plays an essential role in promoting neuronal death in response to varied apoptotic stimuli, including mut-Htt expression [26, 48]. Furthermore, the overexpression of HDAC3 is sufficient to promote neuronal death. It has previously been suggested that the well-documented neuroprotective effects of HDAC inhibitors in diverse in vivo models of neurodegenerative diseases might be mediated through the inhibition of HDAC3 [52–54]. Indeed, selective inhibitors of HDAC3 prevent neurodegeneration and improve behavioral outcome in mouse models of HD [52–54]. We report that co-expression of c-Fos inhibits the neurotoxic effect of HDAC3. This raises the possibility that c-Fos protects neurons by inhibiting the neurotoxic activity of HDAC3 through direct interaction. Further experimentation is clearly necessary to uncover the relationship between c-Fos neuroprotection and HDAC3 neurotoxicity. We previously reported that wild-type Htt, but not mut-Htt, interacts efficiently with HDAC3 [26]. It is possible that c-Fos, HDAC3, and Htt are part of a neuroprotective protein complex and that, within this complex, HDAC3 has a beneficial role in neurons.
Interestingly, expression of just the C-terminal 21 amino acids of c-Fos and c-Fos (360–380) can protect neurons. In addition to delivering this fragment of c-Fos by plasmid transfection, we synthesized a cell-permeable peptide form of this region, Fos-CTF, which is also neuroprotective. The small size of this peptide renders it suitable for development as a therapeutic agent for neurodegenerative diseases. The mechanism by which Fos-CTF protect neurons remains to be determined, although it is likely to be distinct from the one utilized by c-Fos. One possibility is that the Fos-CTF acts by interacting with another protein that is essential for neuronal death and in doing so interferes with the neurotoxic action of that protein. Indeed, c-Fos (360–380) does interact with HDAC3. We previously described that neurotoxicity by HDAC3 involved interaction with HDAC1 [27]. It is possible that by binding HDAC3, c-Fos (360–380) prevents its ability to interact with HDAC1. Although disruption of HDAC1-HDAC3 interaction is a plausible mechanism, based on the presence of a PEST domain [20, 21] within this 21-amino acid region, it is also possible that neuroprotection involves sequestration of PEST-binding proteins reducing the degradation of certain neuroprotective proteins within the neurons.
In summary, we demonstrate that c-Fos is a protein that is necessary for neuronal survival and that can protect neurons when delivered at elevated levels. Neuroprotection by c-Fos occurs by a novel mechanism that does not require its heteromerization domain, its DNA-binding domain, or its transcriptional activity. Although further investigation is needed, we present evidence that c-Fos could act by inhibiting the neurotoxic activity of HDAC3 through direct interaction with it. Our study has also identified a 21-amino acid fragment of c-Fos with strong neuroprotective activity and which, based on its small size and effectiveness, may have clinical value.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01 NS040408 to SRD. We thank Eric J. Nestler of the Mount Sinai Medical Center, NY for the FosB and ΔFosB plasmids, Troy Littleton of MIT for the wild-type and mutant Htt plasmids, and Rajiv R. Ratan of the Burke Institute of Medical Research, NY, for the HT22 cell line, Jason A. Pfister, Jade M. Franklin and Chad C. Smith for reading the manuscript and providing comments.
Footnotes
Electronic supplementary material: The online version of this article (doi:10.1007/s12035-014-9058-1) contains supplementary material, which is available to authorized users.
Conflict of Interest The authors declare that they have no conflict of interest.
Contributor Information
Varun Rawat, Department of Biological Sciences, Southern Methodist University, Dedman Life Sciences Building, 6501 Airline Road, Dallas, TX 75275, USA; Department of Molecular and Cell Biology, University of Texas at Dallas, Richardson, TX 75080, USA.
Warren Goux, Department of Chemistry, University of Texas at Dallas, Richardson, TX 75080, USA.
Marc Piechaczyk, Institut de Génétique Moléculaire de Montpellier, Montpellier, France.
Santosh R. D’Mello, Email: sdmello@smu.edu, Department of Biological Sciences, Southern Methodist University, Dedman Life Sciences Building, 6501 Airline Road, Dallas, TX 75275, USA; Department of Molecular and Cell Biology, University of Texas at Dallas, Richardson, TX 75080, USA.
References
- 1.Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001;20:2413–2423. doi: 10.1038/sj.onc.1204380. [DOI] [PubMed] [Google Scholar]
- 2.Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- 3.Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- 4.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 5.D’Alonzo RC, Selvamurugan N, Karsenty G, Partridge NC. Physical interaction of the activator protein-1 factors c-Fos and c-Jun with Cbfa1 for collagenase-3 promoter activation. J Biol Chem. 2002;277:816–822. doi: 10.1074/jbc.M107082200. [DOI] [PubMed] [Google Scholar]
- 6.Gao Z, Ye J. Inhibition of transcriptional activity of c-JUN by SIRT1. Biochem Biophys Res Commun. 2008;376:793–796. doi: 10.1016/j.bbrc.2008.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivorra C, Kubicek M, Gonzalez JM, Sanz-Gonzalez SM, Alvarez-Barrientos A, O’Connor JE, Burke B, Andres V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006;20:307–320. doi: 10.1101/gad.349506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang CL, Chen JL, Hsu YP, Ou JT, Chang YS. Epstein-Barr virus BZLF1 gene is activated by transforming growth factor-beta through cooperativity of Smads and c-Jun/c-Fos proteins. J Biol Chem. 2002;277:23345–23357. doi: 10.1074/jbc.M107420200. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Zhao Y, Li H, Li Y, Cai X, Shen Y, Shi H, Li L, Liu Q, Zhang X, Ye L. The nuclear import of oncoprotein hepatitis B X-interacting protein depends on interacting with c-Fos and phosphorylation of both proteins in breast cancer cells. J Biol Chem. 2013;288:18961–18974. doi: 10.1074/jbc.M113.458638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 11.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-Jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenkel J. Activation of the c-Jun transcription factor following neurodegeneration in vivo. Neurosci Lett. 2004;361:36–39. doi: 10.1016/j.neulet.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Yuan Z, Gong S, Luo J, Zheng Z, Song B, Ma S, Guo J, Hu C, Thiel G, Vinson C, Hu CD, Wang Y, Li M. Opposing roles for ATF2 and c-Fos in c-Jun-mediated neuronal apoptosis. Mol Cell Biol. 2009;29:2431–2442. doi: 10.1128/MCB.01344-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjorkblom B, Vainio JC, Hongisto V, Herdegen T, Courtney MJ, Coffey ET. All JNKs can kill, but nuclear localization is critical for neuronal death. J Biol Chem. 2008;283:19704–19713. doi: 10.1074/jbc.M707744200. [DOI] [PubMed] [Google Scholar]
- 15.Chen HM, Wang L, D’Mello SR. Inhibition of ATF-3 expression by B-Raf mediates the neuroprotective action of GW5074. J Neurochem. 2008;105:1300–1312. doi: 10.1111/j.1471-4159.2008.05226.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Zhang D, McQuade JS, Behbehani M, Tsien JZ, Xu M. C-Fos regulates neuronal excitability and survival. Nat Genet. 2002;30:416–420. doi: 10.1038/ng859. [DOI] [PubMed] [Google Scholar]
- 17.Wacker JL, Huang SY, Steele AD, Aron R, Lotz GP, Nguyen Q, Giorgini F, Roberson ED, Lindquist S, Masliah E, Muchowski PJ. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J Neurosci. 2009;29:9104–9114. doi: 10.1523/JNEUROSCI.2250-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anglada-Huguet M, Giralt A, Perez-Navarro E, Alberch J, Xifro X. Activation of Elk-1 participates as a neuroprotective compensatory mechanism in models of Huntington’s disease. J Neurochem. 2012;121:639–648. doi: 10.1111/j.1471-4159.2012.07711.x. [DOI] [PubMed] [Google Scholar]
- 19.Ferrara P, Andermarcher E, Bossis G, Acquaviva C, Brockly F, Jariel-Encontre I, Piechaczyk M. The structural determinants responsible for c-Fos protein proteasomal degradation differ according to the conditions of expression. Oncogene. 2003;22:1461–1474. doi: 10.1038/sj.onc.1206266. [DOI] [PubMed] [Google Scholar]
- 20.Bossis G, Ferrara P, Acquaviva C, Jariel-Encontre I, Piechaczyk M. c-Fos proto-oncoprotein is degraded by the proteasome independently of its own ubiquitinylation in vivo. Mol Cell Biol. 2003;23:7425–7436. doi: 10.1128/MCB.23.20.7425-7436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yalcin A, Koulich E, Mohamed S, Liu L, D’Mello SR. Apoptosis in cerebellar granule neurons is associated with reduced interaction between CREB-binding protein and NF-kappaB. J Neurochem. 2003;84:397–408. doi: 10.1046/j.1471-4159.2003.01540.x. [DOI] [PubMed] [Google Scholar]
- 24.Dastidar SG, Landrieu PM, D’Mello SR. FoxG1 promotes the survival of postmitotic neurons. J Neurosci. 2011;31:402–413. doi: 10.1523/JNEUROSCI.2897-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardai FH, Verma P, Smith C, Rawat V, Wang L, D’Mello SR. Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J Neurosci. 2013;33:11833–11838. doi: 10.1523/JNEUROSCI.5831-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bardai FH, Price V, Zaayman M, Wang L, D’Mello SR. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem. 2012;287:35444–35453. doi: 10.1074/jbc.M112.394544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Ankati H, Akubathini SK, Balderamos M, Storey CA, Patel AV, Price V, Kretzschmar D, Biehl ER, D’Mello SR. Identification of novel 1,4-benzoxazine compounds that are protective in tissue culture and in vivo models of neurodegeneration. J Neurosci Res. 2010;88:1970–1984. doi: 10.1002/jnr.22352. [DOI] [PubMed] [Google Scholar]
- 29.Chin PC, Liu L, Morrison BE, Siddiq A, Ratan RR, Bottiglieri T, D’Mello SR. The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J Neurochem. 2004;90:595–608. doi: 10.1111/j.1471-4159.2004.02530.x. [DOI] [PubMed] [Google Scholar]
- 30.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 32.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen RH, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci U S A. 1993;90:10952–10956. doi: 10.1073/pnas.90.23.10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okazaki K, Sagata N. The Mos/MAP kinase pathway stabilizes c-Fos by phosphorylation and augments its transforming activity in NIH 3T3 cells. EMBO J. 1995;14:5048–5059. doi: 10.1002/j.1460-2075.1995.tb00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 36.Schuermann M, Neuberg M, Hunter JB, Jenuwein T, Ryseck RP, Bravo R, Muller R. The leucine repeat motif in Fos protein mediates complex formation with Jun/AP-1 and is required for transformation. Cell. 1989;56:507–516. doi: 10.1016/0092-8674(89)90253-5. [DOI] [PubMed] [Google Scholar]
- 37.Neuberg M, Adamkiewicz J, Hunter JB, Muller R. A Fos protein containing the Jun leucine zipper forms a homodimer which binds to the AP1 binding site. Nature. 1989;341:243–245. doi: 10.1038/341243a0. [DOI] [PubMed] [Google Scholar]
- 38.Cohen DR, Curran T. Analysis of dimerization and DNA binding functions in Fos and Jun by domain-swapping: involvement of residues outside the leucine zipper/basic region. Oncogene. 1990;5:929–939. [PubMed] [Google Scholar]
- 39.Borlongan CV, Koutouzis TK, Sanberg PR. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci Biobehav Rev. 1997;21:289–293. doi: 10.1016/s0149-7634(96)00027-9. [DOI] [PubMed] [Google Scholar]
- 40.Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem. 2005;95:1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Ankati H, Akubathini SK, Balderamos M, Storey CA, Patel AV, Price V, Kretzschmar D, Biehl ER, D’Mello SR. Identification of novel 1,4-benzoxazine compounds that are protective in tissue culture and in vivo models of neurodegeneration. J Neurosci Res. 2010;88:1970–1984. doi: 10.1002/jnr.22352. [DOI] [PubMed] [Google Scholar]
- 42.Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, Syed A, Purcell J, Chen Y, Sharma S, Sangrey GR, Darnell SB, Plasterer H, Sadri-Vakili G, Gottesfeld JM, Thompson LM, Rusche JR, Marsh JL, Thomas EA. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis. 2012;46:351–361. doi: 10.1016/j.nbd.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Hu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–43. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 45.Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci. 2011;31:1746–1751. doi: 10.1523/JNEUROSCI.5704-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao K, Ippolito G, Wang L, Price V, Kim MH, Cornwell G, Fulenchek S, Breen GA, Goux WJ, D’Mello SR. Neuron-selective toxicity of tau peptide in a cell culture model of neurodegenerative tauopathy: essential role for aggregation in neurotoxicity. J Neurosci Res. 2010;88:3399–3413. doi: 10.1002/jnr.22485. [DOI] [PubMed] [Google Scholar]
- 50.Zaidi N, Burster T, Sommandas V, Herrmann T, Boehm BO, Driessen C, Voelter W, Kalbacher H. A novel cell penetrating aspartic protease inhibitor blocks processing and presentation of tetanus toxoid more efficiently than pepstatin A. Biochem Biophys Res Commun. 2007;364:243–249. doi: 10.1016/j.bbrc.2007.09.114. [DOI] [PubMed] [Google Scholar]
- 51.Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 52.D’Mello SR. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009;22:513–524. doi: 10.1358/dnp.2009.9.1428871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 54.Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR. Putting the ‘HAT’ back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18:573–584. doi: 10.1517/13543780902810345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.