

See Corona and Landreth (doi:10.1093/awv300) for a scientific commentary on this article.
The ABCA1 transporter regulates cholesterol efflux to the lipid carriers, ApoE and ApoA1. Fitz et al. compare the phenotype of Apoe/Apoa1 double knockout mice to that of Abca1-/- animals. Surprisingly, double deletion of Apoe/Apoa1 decreases amyloid pathology and increases amyloid-β clearance, but worsens cognitive deficits and reduces dendritic complexity.
Keywords: ABCA1, APOE, APOA1, Alzheimer’s disease, behaviour

See Corona and Landreth (doi:10.1093/awv300) for a scientific commentary on this article.
The ABCA1 transporter regulates cholesterol efflux to the lipid carriers, ApoE and ApoA1. Fitz et al. compare the phenotype of Apoe/Apoa1 double knockout mice to that of Abca1-/- animals. Surprisingly, double deletion of Apoe/Apoa1 decreases amyloid pathology and increases amyloid-β clearance, but worsens cognitive deficits and reduces dendritic complexity.
Abstract
See Corona and Landreth (doi:10.1093/awv300) for a scientific commentary on this article.
ATP binding cassette transporter A1 (encoded by ABCA1) regulates cholesterol efflux from cells to apolipoproteins A-I and E (ApoA-I and APOE; encoded by APOA1 and APOE, respectively) and the generation of high density lipoproteins. In Abca1 knockout mice (Abca1ko), high density lipoproteins and ApoA-I are virtually lacking, and total APOE and APOE-containing lipoproteins in brain substantially decreased. As the ε4 allele of APOE is the major genetic risk factor for late-onset Alzheimer’s disease, ABCA1 role as a modifier of APOE lipidation is of significance for this disease. Reportedly, Abca1 deficiency in mice expressing human APP accelerates amyloid deposition and behaviour deficits. We used APP/PS1dE9 mice crossed to Apoe and Apoa1 knockout mice to generate Apoe/Apoa1 double-knockout mice. We hypothesized that Apoe/Apoa1 double-knockout mice would mimic the phenotype of APP/Abca1ko mice in regards to amyloid plaques and cognitive deficits. Amyloid pathology, peripheral lipoprotein metabolism, cognitive deficits and dendritic morphology of Apoe/Apoa1 double-knockout mice were compared to APP/Abca1ko, APP/PS1dE9, and single Apoa1 and Apoe knockouts. Contrary to our prediction, the results demonstrate that double deletion of Apoe and Apoa1 ameliorated the amyloid pathology, including amyloid plaques and soluble amyloid. In double knockout mice we show that 125I-amyloid-β microinjected into the central nervous system cleared at a rate twice faster compared to Abca1 knockout mice. We tested the effect of Apoe, Apoa1 or Abca1 deficiency on spreading of exogenous amyloid-β seeds injected into the brain of young pre-depositing APP mice. The results show that lack of Abca1 augments dissemination of exogenous amyloid significantly more than the lack of Apoe. In the periphery, Apoe/Apoa1 double-knockout mice exhibited substantial atherosclerosis and very high levels of low density lipoproteins compared to APP/PS1dE9 and APP/Abca1ko. Plasma level of amyloid-β42 measured at several time points for each mouse was significantly higher in Apoe/Apoa1 double-knockout then in APP/Abca1ko mice. This result demonstrates that mice with the lowest level of plasma lipoproteins, APP/Abca1ko, have the lowest level of peripheral amyloid-β. Unexpectedly, and independent of amyloid pathology, the deletion of both apolipoproteins worsened behaviour deficits of double knockout mice and their performance was undistinguishable from those of Abca1 knockout mice. Finally we observed that the dendritic complexity in the CA1 region of hippocampus but not in CA2 is significantly impaired by Apoe/Apoa1 double deletion as well as by lack of ABCA1. In conclusion: (i) plasma lipoproteins may affect amyloid-β clearance from the brain by the ‘peripheral sink’ mechanism; and (ii) deficiency of brain APOE-containing lipoproteins is of significance for dendritic complexity and cognition.
Introduction
The inheritance of ε4 allele of apolipoprotein E (APOE) is the major genetic risk factor for late-onset Alzheimer’s disease (Saunders et al., 1993); however the mechanisms underlying this association are elusive (Kim et al., 2009; Kanekiyo et al., 2014). Compared to APOE3 and APOE2 carriers, patients with Alzheimer’s disease with the APOE4 alelle are characterized by earlier onset of the disease and higher level of amyloid plaques (Huang and Mahley, 2014; Yu et al., 2014).
ATP binding cassette transporter A1 (ABCA1) regulates cholesterol and phospholipids efflux from cells to lipid-poor apolipoprotein A-I (ApoA-I, encoded by APOA1), as well as other apolipoprotein acceptors such as apolipoprotein E (APOE), thus mediating the generation of high density lipoproteins (reviewed in Koldamova et al., 2014). Prominent characteristics of patients with non-functional ABCA1 and Abca1 knockout mice (Abca1ko) is their virtual absence of circulating high density lipoproteins and ApoA-I in plasma and brain, and relatively low level of low density lipoproteins in the blood (Brooks-Wilson et al., 1999; Koldamova et al., 2014). In addition, mice lacking ABCA1 exhibit a substantial reduction of APOE-containing lipoproteins accompanied by a significant decrease of total level of APOE protein in the brain (Wahrle et al., 2004). Considering the prominent role that APOE plays in Alzheimer’s disease risk, the fact that ABCA1 functionality can affect APOE lipidation or its total level might be of significance for the disease. In mouse models of Alzheimer’s disease, Abca1 deficiency increased amyloid deposition in parallel with reduced APOE levels (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005; Lefterov et al., 2009). In contrast, Abca1 overexpression in PDAPP mice decreases amyloid burden (Wahrle et al., 2008). Recently, we demonstrated that Abca1 haplo-deficiency deferentially affects the phenotype of mice expressing human APOE3 or APOE4 isoforms suggesting that APOE4 isoform is more vulnerable to additional genetic effects (Fitz et al., 2012).
The mechanism by which Abca1 deficiency affects amyloid deposition when APOE is significantly reduced and there is virtually no ApoA-I is not understood and difficult to reconcile with the data from single Apoe and Apoa1 knockout mice. So far all published studies demonstrated that the deletion of Apoe diminished amyloid pathology in different APP mouse models (Bales et al., 1997; Kim et al., 2011). The expression of Apoa1 in APP transgenic mice reportedly affects cerebral amyloid angiopathy but has no effect on parenchymal amyloid plaques (Fagan et al., 2004; Lefterov et al., 2010; Lewis et al., 2010). Interestingly, when the deletion of Apoe was accompanied by the absence of another brain apolipoprotein, apolipoprotein J (clusterin, encoded by Clu), the mice had earlier onset and increased amyloid deposition (DeMattos et al., 2004).
In this study we used APP/PS1dE9 transgenic mice crossed to Apoe and Apoa1 knockout mice to generate Apoe/Apoa1 double-knockout mice (hereafter referred to as APP/DKO). We hypothesized that APP/DKO will replicate Alzheimer’s disease-like phenotype of APP/Abca1ko mice. We compared amyloid pathology, cognitive decline, and peripheral lipoprotein metabolism of APP/DKO to the phenotype of APP/Abca1ko mice and single knockout mice. Unexpectedly, and contrary to our prediction, the results demonstrated that the double deletion of Apoe and Apoa1 ameliorated the amyloid pathology and increased amyloid-β clearance. Furthermore, regardless of significantly decreased amyloid pathology, APP/DKO mice had substantial behaviour deficits and impaired dendrite morphology. Our data also suggest that the abnormal and very high level of plasma very-low-density lipoprotein/low-density lipoprotein (VLDL/LDL) in APP/DKO mice may affect amyloid-β clearance.
Materials and methods
Mice
APP/PS1ΔE9 [B6.Cg-Tg(APPswe, PSEN1ΔE9)85Dbo/Mmjax] transgenic mice referred to as APP/WT and mice with targeted disruption of mouse Apoa1 (B6.129P2-Apoa- Itm1Unc/J, referred to as Ako) or Apoe (B6.129P2-Apoetm1Unc/J, referred to as Eko), all strains on C57BL/6 J background, were purchased from Jackson Laboratory. Abca1 null mice on a C57BL/6 × DBA/1 mixed background (DBA/1-Abca1tm1Jdm/J, Abca1ko) were purchased from Jackson Laboratory and crossbred for 10 generations to pure C57BL/6 background in our laboratory. APP/WT mice were crossbred to either Ako, Eko, or Abca1ko mice to generate APP/Ako, APP/Eko or APP/Abca1ko mice, respectively. Additionally, APP/WT, Ako, and Eko mice were crossed to generate APP mice deficient in Apoa1 and Apoe (referred to as APP/DKO) and non-APP expressing littermates (DKO). APP/WT mice with wild-type mouse Apoa1, Apoe, and Abca1 were used as controls. For the brain homogenate injection study we used the brains of APP23 transgenic (Koldamova et al., 2005) or wild-type mice. All mice for each line were littermates and housed on a 12-h light/dark cycle with ad libitum food and water. For all experiments, male and female mice were used. All animal procedures were performed in accordance with PHS policies and University of Pittsburgh’s Institutional Animal Care and Use Committee. All reagents were purchased through Fisher Scientific unless noted.
Brain homogenate injections
Mice were anaesthetized with isoflurane, placed on a heating pad, surgical site shaven and disinfected. Bilateral stereotaxic injections of 2 µl of either APP23 or wild-type brain extract were infused into the hippocampus and overlying cortex (AP −2.5 mm, L ±2.0 mm, DV −1.0/−1.8 mm; 0.5 µl/min). Tris-buffered saline (TBS) brain extracts were obtained from a 23-month-old APP23 (Koldamova et al., 2005) and 24-month-old wild-type mouse. Following removal of the infusion needle the incision was sutured and mice monitored until fully recovered. Mice were infused at 3 months of age and amyloid pathology assessed 4 months later as described below.
Behavioural testing
Behavioural testing was performed during the light phase of the light/dark cycle. Contextual fear conditioning (Stoelting Co.) was performed as described previously (Fitz et al., 2014). Briefly, mice were placed in a conditioning chamber for 2 min before the onset of a tone (conditioned stimulus, duration of 30 s, 85 dB sound at 2800 Hz). In the last 2 s of the conditioned stimulus, mice were given a 2 s, 0.7 mA footshock through the floor, and this cycle was repeated. Mice remained in the chamber for 30 s before being returned to their housing cages. Twenty-four hours later contextual fear conditioning was assessed by measuring freezing behaviour for 5 min in the original chamber. Cued fear learning was assessed 24 h after contextual testing by placing mice in a novel context for 2 min, after which they were exposed to the conditioned stimulus for 3 min, and freezing behaviour measured. Freezing behaviour, defined as the absence of movement except that needed for breathing, was recorded and scored using AnyMaze software (Stoelting Co.).
Multiple blood draws
Multiple blood draws were performed from the lateral saphenous vein as described elsewhere (Hem et al., 1998). Mice were immobilized, hind leg held in the extended position, fur over the lateral saphenous vein shaved and the area cleaned with 70% alcohol wipes. A 25-gauge needle was used for saphenous venepuncture, gentle pressure was applied around the puncture sight and the equivalent of 0.5% of the animal’s bodyweight collected and centrifuged for 5 min before plasma collection. Plasma amyloid-β levels were assessed by sandwich enzyme-linked immunosorbent assay (ELISA) as described below.
Tissue processing
Mice were anaesthetized with Avertin (250 mg/kg of body weight, intraperitoneally) and blood collected through cardiac puncture followed by transcardial perfusion with 25 ml of cold 0.1 M phosphate-buffered saline (PBS), pH 7.4 (Fitz et al., 2014). Brains were rapidly removed and divided into hemispheres. The first hemisphere was quickly dissected into cortex, hippocampus, subcortical structures, and cerebellum and snap frozen on dry ice. The other hemisphere was drop fixed in 4% phosphate-buffered paraformaldehyde at 4°C for 48 h before storage in 30% sucrose. After minor arterial branching, adventitial and adipose tissues were removed, and the aorta was dissected from the heart to iliac arteries. The aortas were fixed in 10% neutral buffered formalin and stored at 4°C.
Histology and immunohistochemistry
All histological procedures for assessing amyloid pathology were as reported previously (Fitz et al., 2014). HistoPrep™-embedded hemibrains were cut in the coronal plane at 30 -μm sections and stored in a glycol-based cryoprotectant at −20°C until staining. Following staining all sections were cover slipped with gelvatol mounting media.
For X-34 staining, a series of five sections were selected 700 -μm apart, starting from a randomly chosen section ∼150 μm caudal to the first appearance of the CA3 and dentate gyrus. Mounted sections were washed in PBS for 10 min and stained with 1,4-bis(3-carboxy-4-hydroxyphenylethenyl)-benzene (X-34) (100 μm) for 10 min. Following the staining, slides were rinsed with PBS and destained with 0.2% NaOH in 80% ethanol for 2 min. Finally, sections were washed with PBS.
For 6E10 staining, adjacent sections to X-34 stained sections were immunostained with 6E10 antibody (SIG-39340; Covance) (Fitz et al., 2014). Briefly, free-floating sections were blocked for endogenous peroxidases, avidin-biotin quenched, and antigen retrieval was performed with 70% formic acid. Sections were then incubated in 6E10 biotin-labelled antibody (1:1000) at room temperature for 2 h before being developed with Vectastain ABC Elite kit and a DAB substrate (Vector Laboratories). Microscopic examination was performed using a Nikon Eclipse 80i microscope (Nikon Inc.; ×4 magnification). For quantitative analysis, staining in the cortex and hippocampus was defined as the percentage area covered by X-34 or 6E10 positivity using MetaMorph 7.0 software (Molecular Devices).
Neurite quantification
Neurite quantification was performed as previously described (Tapias et al., 2013; Tapias and Greenamyre, 2014). For immunofluorescence labelling, five sections per animal starting at the beginning of the dentate gyrus and every 300 μm were rinsed three times in PBS for 10 min and permeabilized with 1% Triton™ X-100 in PBS for 5 h at 4°C. The tissue was washed in PBS and blocked with 10% serum and 0.3% Triton™ X-100 in PBS for 30 min at room temperature. The sections were incubated for 72 h at 4°C with MAP2 primary antibody (1:2000; #MAB378, Millipore) and washed with PBS. Sections were incubated with Cy3-conjugated secondary antibody (1:500; Jackson-ImmunoResearch) for 2 h at room temperature followed by a final PBS wash and counterstain with H33342 nuclear reagent (1:3000). The FilamentTracer module of Imaris (Bitplane) was used to determine hippocampal neuronal patterning. The MAP2 labelling was used to quantify total neurite length, the number of segments, and the number of branches in hippocampal CA1 and CA2 regions. For neurite analysis, four to five confocal images were acquired for the CA1 region whereas three to four were captured for the CA2 region. Confocal fluorescence micrographs were obtained using ×60 magnification at very high resolution (100 µs). For unbiased examination, the size and the length of the neurites were the only parameters that required manual introduction. Neurite length was normalized to the number of H33342 stained nuclei.
Western blotting, ELISA and dot blotting
Frozen cortices and hippocampi were homogenized in TBS homogenization buffer (250 mM sucrose, 20 mM Tris base, 1 mM EDTA, and 1 mM EGTA, 1 ml per 100 mg of tissue) and protease inhibitors cocktail (Roche).
Amyloid-β ELISA was performed essentially as before (Fitz et al., 2012). Briefly, for extraction of soluble amyloid-β, samples of the initial brain homogenate were centrifuged at 100 000g for 1 h, supernatant (TBS extract) was used to determine soluble amyloid-β, non-plaque-associated amyloid-β. Insoluble amyloid-β and plaque-associated amyloid-β was extracted from the remaining pellet by mixing with 70% formic acid. ELISA for amyloid-β was performed using 6E10 as the capture antibody and anti-Aβ40 (G2-10 mAb) and anti-Aβ42 (G2-13 mAb) monoclonal antibodies conjugated to horseradish peroxidase (The Genetics Company) as detection antibodies. The final values of amyloid-β were calculated based on amyloid-β40 and amyloid-β42 peptide standards (American Peptide) and normalized amounts of amyloid-β were expressed as pmol/mg of total protein.
For western blot, proteins extracted with TBS or radioimmunoprecipitation assay (RIPA) buffer were resolved on sodium dodecyl sulphate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. For ABCA1 and APP detection, RIPA extracted proteins were resolved on 10% Tris-glycine gels and probed with anti-ABCA1 (Abcam) and 6E10 antibodies, respectively. For soluble amyloid-β and APP carboxy-terminal fragments, result of α-secretase cleavage (CTF-α), RIPA extracts were resolved on 4–12% BisTris gels probed with 6E10 antibody. β-Actin was used as a loading control for all western blots and detected by monoclonal antibody (Santa Cruz Biotechnology).
The level of soluble amyloid-β oligomers was measured by dot blot assay using TBS extract as before (Fitz et al., 2013). One microgram of protein was spotted on nitrocellulose membrane and probed with A11 antibody (1:2000), specific for oligomeric forms of amyloid-β and generously provided by Dr Charles Glabe (University of California, Irvine). The immunoreactive signals were visualized using enhanced chemiluminescence detection kit (GE Healthcare) and quantified densitometrically. To normalize for total protein, the blots were stripped and stained with Coomassie blue reagent. We performed two dot blots with A11 antibody and used the mean value for quantification. As an additional control the exact same amounts of sample protein were spotted on separate dot blots and probed with 6E10 antibody followed by Coomassie blue staining to normalize for total protein.
Amyloid-β clearance across the blood–brain barrier
14C-Inulin and 125I-amyloid-β40 were purchased from GE Healthcare. Amyloid-β42 obtained from Yale University was iodinated with 125I (Perkin Elmer) using standard protocol provided with the Pierce Iodination Beads (ThermoFisher Scientific), which allows for maintaining biological activity of proteins. The resulting components underwent centrifuge filter purification and resolved by HPLC and purity analysed by mass spectrometry.
Surgery and radiotracer application was performed as described before with slight modifications (Cirrito et al., 2005; Deane et al., 2008). Briefly, guide cannulas (22-gauge) with 5 mm projection from the pedestal (Plastics One) were stereotaxically implanted into the caudate-putamen of anaesthetized mice (AP +0.9 mm, L −1.9 mm, DV −2.9 mm). Animals were allowed to recover 4–6 h after cannula implantation, to allow the blood–brain barrier to partially repair and exclude large molecules. 14C-inulin and 125I-amyloid-β40 and 125I-amyloid-β42 were co-injected using a micropump, and animals sacrificed 30 min later and brains removed.
The percentage of radioactivity remaining in the brain after microinjection was determined as previously described (Deane et al., 2008). Briefly, % recovery in brain = 100 × (Nb/Ni), where Nb is the radioactivity remaining in the brain at the end of the experiment and Ni is the radioactivity microinjected into brain interstitial fluid, i.e. the disintegrations per minute for 14C-inulin and the counts per minute for trichloroacetic acid-precipitable 125I-radioactivity (intact amyloid-β). The percentage of amyloid-β (Aβ) cleared through the blood–brain barrier was calculated as [(1 − Nb(Aβ)/Ni(Aβ)) − (1 − Nb(inulin)/ Ni(inulin))] × 100, using a standard time of 30 min.
Fast protein liquid chromatography and cholesterol assay
Pooled serum samples (100 µl total) from five mice of each genotype were loaded on two Superose 6 columns (GE Healthcare) in an AKTA Explorer system (Amersham Pharmacia Biotech Inc.) and eluted with Dulbecco’s phosphate-buffered saline (2.7 mM KCl, 1.5 mM KH2PO4, 140 mM NaCl, 6.5 mM Na2HPO4, 1 mM EDTA, pH 7.2) at 0.2 ml/min. Total cholesterol in each fraction (0.4 ml) was determined using Amplex® Red Cholesterol Assay Kit (Molecular Probes) in duplicate. Fluorescence was read on a Synergy 2 microplate reader (Biotek).
In addition VLDL/LDL were separated from high density lipoproteins in serum samples using the high density lipoproteins and LDL/VLDL Cholesterol assay kit (AB65390, Abcam).
Statistical analyses
All results are reported as means ± standard error of the mean (SEM). All statistical analyses were performed in GraphPad Prism, version 5.0 (La Jolla, CA) and differences considered significant where P < 0.05.
Results
Effect of Apoe and Apoa1 double deficiency on amyloid-β level in APP/DKO mice
Previously, we and others demonstrated that the deletion of Abca1 increased amyloid plaques in APP transgenic mice (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005). The phenotype was accompanied by virtual absence of ApoA-I and a significant decrease of APOE protein in the brain. To examine how the deficiency of these two apolipoproteins affects amyloid deposition we generated Apoe/Apoa1 double-knockout mice and crossed them to APP/WT transgenic mice to obtain APP/DKO. The amyloid phenotype of APP/DKO was compared to APP mice lacking Abca1 (APP/Abca1ko) and to single Apoe (APP/Eko) and Apoa1 (APP/Ako) knockout mice. First, we evaluated the level of amyloid load using X-34 staining (Fig. 1A–C). Surprisingly, the assessment of compact amyloid plaques visualized by X-34 in cortex (Fig. 1B) and hippocampus (Fig. 1C) revealed that APP/DKO mice have significantly less amyloid load than APP/Abca1ko mice. Compact amyloid load of APP/DKO did not differ significantly from single APP/Eko and was significantly lower than APP/Ako mice. The levels of diffuse amyloid plaques visualized by 6E10 antibody confirmed this result (Fig. 1D–F). The effect of genotype on the level of X-34 and 6E10 plaques was not gender-dependent (data not shown). These experiments suggest that Apoe deficiency is the main factor that controls the effect of genotype on amyloid plaque load in APP/DKO mice and the deletion of Apoa1 does not contribute significantly.
Figure 1.

Lack of both APOE and ApoA-I in double knockout mice significantly decreases the level of amyloid-β plaques. Compact amyloid was visualized using X-34 (A–C) and diffuse amyloid plaques were stained with 6E10 anti-amyloid-β antibody (D–F). Analysis is by one-way ANOVA with Tukey’s post-test for multiple comparisons (shown on each graph). For both staining we used 6-7-month-old male and female mice: APP/WT (n = 10), APP/Abca1ko (n = 10), APP/DKO (n = 9), APP/Eko (n = 10), APP/Ako (n = 9). (A) Representative X-34 pictures for all genotypes. Quantification of X-34 staining in cortex (B) and in hippocampus (C). ANOVA for cortex and hippocampus is P < 0.0001. Post-test for hippocampus, APP/DKO versus APP/WT P < 0.05; APP/Eko versus APP/WT, P < 0.05. (D) Representative 6E10 staining for all genotypes. Quantification of 6E10 staining in cortex (E) and in hippocampus (F). ANOVA for cortex and hippocampus is P < 0.01.
Next we extracted soluble and insoluble amyloid-β from the brains of these mice and determined amyloid-β levels by ELISA. We found that the amount of soluble amyloid-β40 and amyloid-β42 were very low in APP/DKO and APP/Eko mice compared to the other three genotypes (Fig. 2A and B). To confirm the result, we also performed western blot to measure soluble amyloid-β from APP/WT, APP/DKO and APP/Abca1ko mice. As seen on the graph (Fig. 2C) and the representative picture (Fig. 2D), APP/DKO mice had significantly less amyloid-β compared to APP/Abca1ko and APP/WT mice. Western blots did not show significant difference in the levels of ABCA1, full length APP, and α-secretase cleavage (CTF-α) between the different genotypes (Fig. 2D). As expected, ApoA-I protein was missing in APP/DKO and APP/Ako mice. Similarly, APOE was not detected in APP/DKO and APP/Eko mice. As reported earlier, we detected a significant decrease of APOE protein in APP/Abca1ko mice and virtual absence of ApoA-I (Koldamova et al., 2005). To determine the level of APOE-containing high density lipoproteins particles in CSF we performed native gel electrophoresis. The results demonstrated substantial reduction of native APOE lipoproteins in the CSF of Abca1ko mice and their absence in double knockout mice (Fig. 2E). Finally, the results of insoluble amyloid-β40 and amyloid-β42 levels confirmed that double deletion of Apoe and Apoa1 reduced the level of deposited amyloid-β (Fig. 2F and G). Similar to the effect on amyloid plaques, the amount of insoluble amyloid-β in APP/DKO mice was significantly lower than in APP/Abca1ko and APP/Ako mice and indistinguishable from APP/Eko mice.
Figure 2.

Insoluble amyloid-β is increased in APP/Abca1ko and decreased in APP/DKO mice. Soluble and insoluble proteins were extracted from cortex and hippocampus of 6–7-month-old male and female mice as described in the methods and normalized to total protein (TP). (A) Soluble amyloid-β40 (Aβ40) and (B) amyloid-β42 (Aβ42) levels were determined by ELISA. For A and B, n = 8–13 mice per group. (C) Soluble amyloid-β in APP/WT, APP/Abca1ko and APP/DKO was also measured by western blotting. n = 10–12 per group. (D) The level of ABCA1, full length APP (APPfl), APOE, ApoA-I, CTF-α and soluble amyloid-β proteins was measured by western blotting. Pictures are representative of at least six mice per group. (E) Native gel electrophoresis of CSF from APP/WT, APP/Abca1ko and APP/DKO followed by western blotting for APOE. Loaded were: for APP/WT, 1 µl; for APP/Abca1ko and APP/DKO 10 µl per lane. Native markers are shown on the left. (F) Insoluble amyloid-β40 and (G) insoluble amyloid-β42 was measured by ELISA. For F and G, n = 8–13 per group. Analysis is by one-way ANOVA (For A, B, C, F and G, P < 0.0001) followed by Tukey’s post-test (shown on the graphs).
Soluble amyloid-β oligomers are increased in APP/Abca1ko and unchanged in APP/DKO mice
Previously we have shown that lack of one copy of Abca1 increases amyloid-β oligomers in APP23 mice (Lefterov et al., 2009). To examine whether the double deficiency of Apoe and Apoa1 affects amyloid-β oligomers, we used A11 conformation-specific antibody (Fig. 3) which recognizes soluble amyloid-β oligomers (Kayed et al., 2003). As shown in Fig. 3B, A11-positive oligomers detected in APP/DKO did not differ significantly from those in APP/WT mice. In contrast, A11-positive oligomers were significantly increased in APP/Abca1ko mice confirming our previous result (Lefterov et al., 2009). Although there is a trend towards an increase in APP/Abca1ko mice, no significant changes were detected in 6E10-positive aggregates.
Figure 3.

Soluble amyloid-β oligomers are increased in APP/Abca1ko and unchanged in APP/DKO mice. Amyloid-β oligomers were measured in the soluble fraction (cortex and hippocampus) by dot blot using conformation specific A-11 antibody. The intensity was normalized on total protein (TP) as measured by Coomassie blue staining. For comparison, dot blotting of the same samples probed with 6E10 antibody is shown. Analysis is by one-way ANOVA, P < 0.05, followed by Tukey’s post-test. n = 10 male and female mice per group. N.S. = not significant.
In conclusion, the data presented on Figs 1–3 demonstrate that the effect on amyloid phenotype is mainly determined by the deletion of Apoe and that the lack of Apoa1 does not significantly contribute to amyloid deposition in APP/DKO mice.
Abca1 deficiency but not lack of Apoe and Apoa1 delays amyloid-β40 and amyloid-β42 clearance from the brain
To examine how the lack of Apoe and Apoa1 affect amyloid-β efflux out of the brain we injected mice of all genotypes (not expressing human APP) with radioactively-labelled amyloid-β using a modification of previously published method (Deane et al., 2004; Cirrito et al., 2005). Mice that do not express human APP were used to avoid the interference of amyloid deposition on the clearance of exogenous amyloid-β. The results (Fig. 4A and B) demonstrate that Abca1 deficiency significantly decreased amyloid-β clearance through the blood–brain barrier and the effect is more pronounced on 125I-amyloid-β42 efflux. In contrast, the deficiency of Apoe in double knockout and Eko mice significantly increased amyloid-β efflux from CNS compared to APP/WT and APP/Abca1ko. The effect was not related to the disruption of blood–brain barrier as demonstrated by Evan’s blue experiments (Supplementary Fig. 1). Our conclusion is that the deficiency of Abca1 delays the export of amyloid-β from the brain and deficiency of Apoe increases amyloid-β clearance.
Figure 4.
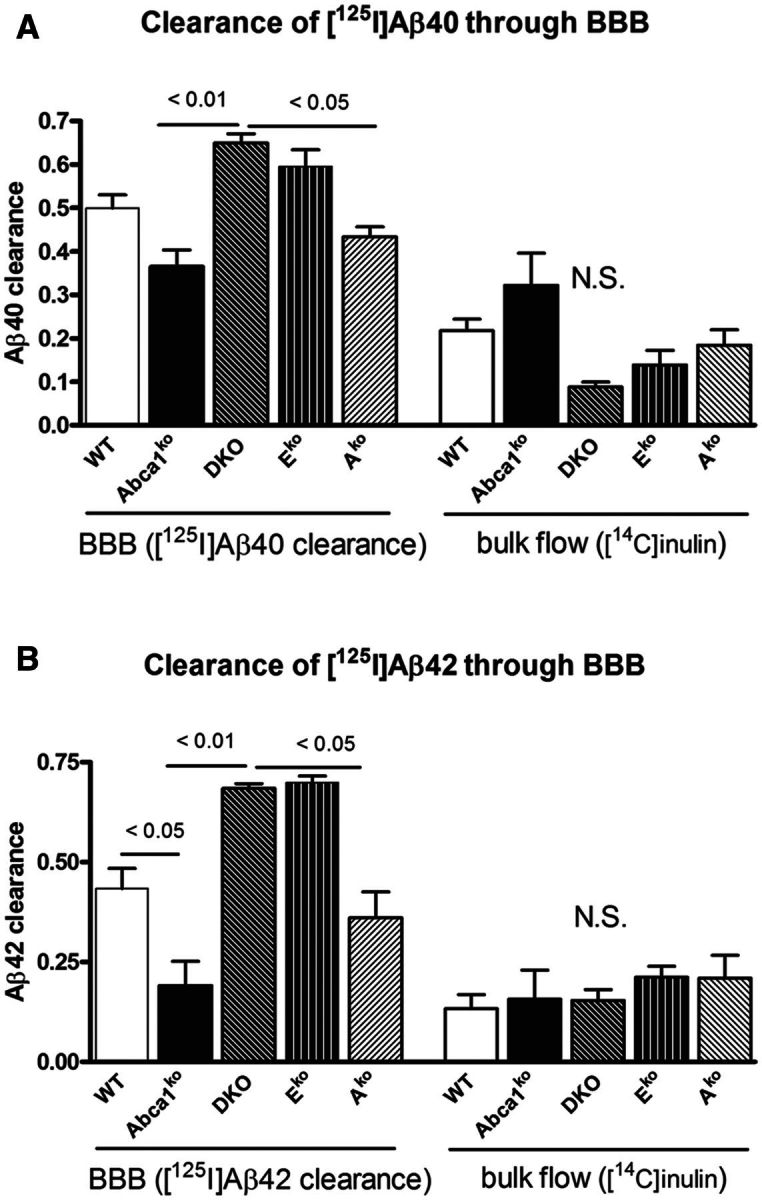
Abca1 deficiency but not the lack of Apoe and Apoa1 delays amyloid-β40 and amyloid-β42 clearance from the brain. (A and B) 5-6-month-old wild type (WT), Abca1ko, DKO, Eko and Ako mice were injected with 12 nmol 125I-amyloid-β40 (Aβ40, A) or 125I-amyloid-β42 (Aβ42, B) into the caudate-putamen. Clearance was assessed 30 min following this injection. 14C-inulin was co-injected in each experiment to assess bulk flow elimination from the interstitial fluid. n = 3–5 mice per group. One-way ANOVA followed by Tukey’s post-test was used to analyse the difference. N.S. = not significant; BBB = blood–brain barrier.
Effects of Abca1, Apoa1 and Apoe genotypes on the dissemination of exogenous amyloid-β seeds
Previous studies demonstrated that brain extract from old APP expressing mice injected into the brain of young mice effectively disseminates thus forming amyloid-β plaques in areas far from the injected side (Meyer-Luehmann et al., 2006). To test how the deficiency of Abca1 or Apoe affects the spreading of exogenous amyloid-β seeds, young pre-depositing APP mice of various genotypes were injected with amyloid-β seeds from 24-month-old APP23 mouse (labelled as ‘+’). The injected material was a soluble TBS extract from brain that usually contains amyloid-β monomers or oligomers, but not fibrils (Lefterov et al., 2009). The control mice of the same genotypes were injected with TBS extract from 24-month-old wild-type littermates (labelled as ‘−’). Three months later, amyloid deposition in these mice was examined using X-34 staining for compact amyloid. As seen in Fig. 5, all genotypes had significantly more amyloid when inoculated with APP23 extract than their counterparts injected with wild-type homogenate. However the different genotypes varied in the magnitude of their response to injected exogenous amyloid: whereas APP/Abca1ko augmented amyloid plaque level by >10-fold, the increase seen in APP/DKO mice was ∼2-fold. Similarly, APP/Eko mice displayed a similar response in spreading of exogenous amyloid-β seeds. The response of APP/WT and APP/Ako was intermediate. The appearance of amyloid plaques was consistent with the pattern of the amyloid deposits in APP/WT mouse model and differed significantly from that in APP23 mice. This suggests that injected amyloid-β served as a seed to increase the amyloid deposition typical of the host model and did not transmit the amyloid phenotype of the donor APP23.
Figure 5.
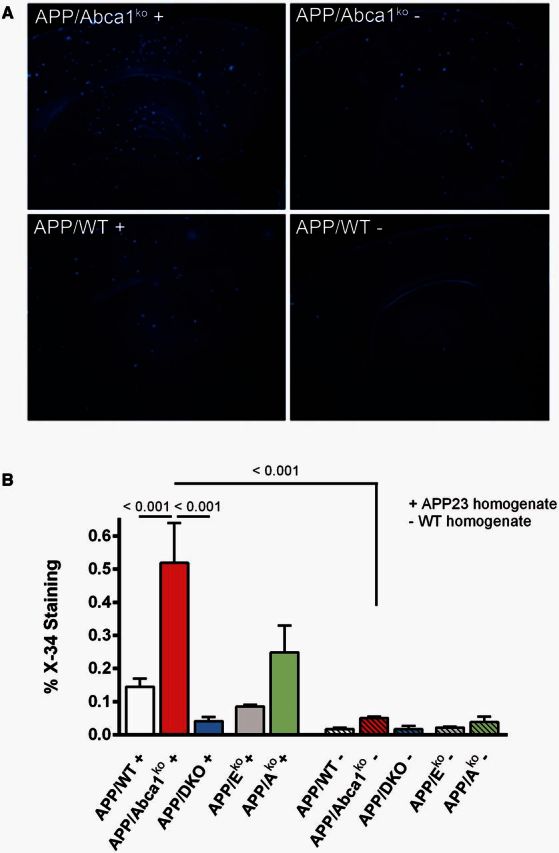
Effects of Abca1, Apoa1 and Apoe genotypes on the dissemination of exogenous amyloid-β seeds: Three-month-old pre-depositing APP mice of different genotypes were injected with amyloid-β seeds from 23-month-old APP23 mouse (labelled as ‘+’) and amyloid-β plaques evaluated 3 months later using X-34 staining. Control mice were injected with brain homogenate from 24-month-old wild-type mouse (labelled as ‘−’). Analysis is by one-way ANOVA (P < 0.001) with Tukey’s post-test (shown on the graph). n per group: APP/WT ( + ) = 8; APP/WT (−) = 6; APP/Abca1ko = 6 per each group; APP/DKO and APP/Eko = 4 mice/group and APP/Ako = 3 mice/group.
Effects of Abca1, Apoa1 and Apoe deletion on atherosclerosis, peripheral lipoproteins and amyloid-β level in plasma
The Apoe-knockout mouse is a well-described model for atherosclerosis (Plump et al., 1992). To determine how the deletion of both Apoe and Apoa1 affect atherosclerosis in APP transgenic mice, we examined the level of atherosclerotic plaques in the aorta and compared to single knockout mice. As expected, the level of atherosclerosis in APP/WT was negligible (0.34%) and significantly increased in APP/Eko mice (16.5%) (Supplementary Fig. 2A and B). Consistent with previous data for Abca1ko (Zhao et al., 2011) and Apoa1-knockout mice (Parolini et al., 2005), APP/Abca1ko and APP/Ako mice had very low levels of atherosclerotic plaques (0.28 and 0.23%, respectively). Surprisingly, the atherosclerosis in APP/DKO mice (12.4%) was statistically undistinguishable from the observed level in APP/Eko mice. We also noticed that wild-type littermates (Eko versus APP/Eko, P < 0.05 and DKO versus APP/DKO, P < 0.01) had less atherosclerotic plaques compared to their APP counterparts (Supplementary Fig. 2B). This suggests that amyloid-β deposition can contribute to atherosclerosis, however, only in genetically predisposed mice as the difference between APP/WT and wild-type mice was not apparent.
We next examined the plasma lipid profiles of APP/WT, APP/Abca1ko, APP/DKO, APP/Eko and APP/Ako mice at 12 months of age. As shown on Fig. 6A, the lipid profiles of APP/Eko and APP/DKO were consistent with their atherosclerotic phenotype and their VLDL/LDL levels were very high as shown previously for double knockout and ApoEko mice without APP transgene (Thorngate et al., 2003). In contrast, APP/Abca1ko lacked detectable high density lipoproteins. The high density lipoprotein fraction of APP/Ako mice was shifted to the left, towards lower density, consistent with high density lipoproteins containing mainly APOE. An alternative method to measure the level of LDL cholesterol was carried out and confirmed that VLDL/LDL levels in APP/Eko mice were 20 times higher and in APP/DKO mice 14 times higher than in APP/WT mice (Fig. 6B). Using the same method, VLDL/LDL fraction in in APP/Abca1ko mice were almost undetectable.
Figure 6.

Effects of Abca1, Apoa1 and Apoe deletion on peripheral lipoproteins and amyloid-β level in plasma. (A) Shown is fast protein liquid chromatography lipid profiles for APP/WT, APP/Abca1ko, APP/DKO, APP/Eko and APP/Ako mice. Filtered mouse serum was fractionated by size exclusion chromatography on Superose 6 columns in PBS, running at a 0.2 ml/min flow rate. Fractions were collected and assayed for cholesterol. Used were pooled samples of five mice per group. (B) VLDL/LDL and high density lipoprotein fractions were separated from serum of the same genotypes shown on A using kit from Abcam. Quantification of cholesterol level for VLDL/LDL fraction is shown. (C) Kinetic of amyloid-β42 level in plasma: blood was withdrawn from the lateral saphenous vein of APP/WT, APP/Abca1ko, APP/DKO mice at age of 4, 6 and 7 months and amyloid-β level determined by ELISA. n = 6–9 mice per group. Analysis for each time point is by one-way ANOVA (P < 0.05 for 4 and 7 months). *P < 0.05 by Tukey’s post-test when APP/Abca1ko was compared to APP/DKO is shown on the graph. (D) Ratio of amyloid-β42 in plasma and total level of VLDL/LDL fractions as a fold of APP/WT level. For amyloid-β42 was used the mean value from all time points for the respective genotype.
Amyloid-β was reported to bind plasma lipoproteins (LaDu et al., 2012). We reasoned that the very high level of VLDL/LDL in APP/Eko and APP/DKO mice can change the balance of amyloid-β on the two sides of the blood–brain barrier and increase its clearance from the brain. To examine how plasma amyloid-β level was affected by the genotype, blood was withdrawn from the lateral saphenous vein of APP/WT, APP/Abca1ko and APP/DKO mice at several time points for each mouse from 4 to 7 months of age and amyloid-β42 level determined. As seen in Fig. 6C, APP/DKO mice had the highest level of amyloid-β42 in plasma and APP/Abca1ko the lowest. Figure 6D shows a comparison of the ratio of plasma amyloid-β and VLDL/LDL fractions presented as a fold of APP/WT level. This result demonstrates that the mice with the lowest level of VLDL/LDL, namely APP/Abca1ko, have the lowest level of peripheral amyloid-β and vice versa for APP/DKO.
Lack of Abca1 and double deficiency of Apoa1 and Apoe equally increases behaviour deficits in APP mice and non-transgenic littermates
To examine the effects of Abca1 deletion and double deficiency of Apoa1/Apoe on memory, 7-month-old mice were tested in a contextual cued fear conditioning paradigm. Non-transgenic Abca1ko or double knockout mice were used as controls. As shown on Fig. 7A, during the contextual fear conditioning phase of testing, APP/Abca1ko mice demonstrated a significant reduction in freezing time when compared to APP/WT mice (one-way ANOVA, P < 0.01, post-test P < 0.05). Similarly, APP/DKO mice showed a significant reduction in freezing time when compared to APP/WT mice (post-test, P < 0.01). This demonstrates impaired fear memory in both APP/Abca1ko and APP/DKO mice compared to APP/WT mice. Cued phase of test, 48 h following the initial paradigm acquisition, showed no statistical difference in the performance of the mice (Fig. 7B). Importantly we observed exactly the same significant difference between non-transgenic control genotypes during the cued phase of the fear conditioning paradigm (Fig. 7C and D, respectively). This experiment suggests that double deficiency of Apoa1 and Apoe affects behaviour performance regardless of amyloid plaques or soluble amyloid-β level.
Figure 7.

Effects of Abca1, Apoa1 and Apoe deletion on contextual and cued fear conditioning memory. Seven-month-old mice of various genotypes were examined for changes in memory performance using a contextual cued fear conditioning paradigm. Twenty-four hours after the initial acquisition of the contextual fear conditioning (A) the contextual test demonstrates that deletion of Abca1 or ApoA1 and Apoe in APP expressing mice significantly worsens memory performance. Forty-eight hours after the initial acquisition the cued phase of test showed no statistical differences between genotype (B). Similar change in memory performance of non-APP expressing mice that had an Abca1 or ApoA1 and Apoe deletion was observed in the contextual (C) but not in cued (D) phase of training. Analysis by one-way ANOVA with Tukey’s post-test. n = 13–25 male and female mice per genotype. N.S. = not significant.
Deletion of Apoe and Apoa1 impacts dendrite architecture
To determine how the deletion of both Apoe and Apoa1 impacts dendrite morphology in APP transgenic mice, two different regions (CA1 and CA2) of the medial hippocampus were examined. Quantitative analysis revealed that CA1 region of hippocampus showed a significant decrease in dendrite length (P < 0.05), number of branch points (P < 0.05) and number of neurite segments (P < 0.05) in APP/Abca1ko mice when compared to APP/WT mice (Fig. 8A and B). Interestingly, deletion of both Apoe and Apoa1 resulted in a substantial negative impact on neurite morphometry. APP/DKO displayed a significant decrease in total neurite length (P < 0.01) and both number of branch points (P < 0.001) and segments (P < 0.001) relative to APP/WT mice. The data were normalized to the number of neurons and there was no significant change in the number of H3342-positive nuclei between all genotypes. Neurite architectural changes were restricted to the CA1 region of the hippocampus as we did not observe any gene effect in the CA2 region when comparing genotypes (Fig. 8C and D). Our data demonstrate that the deficiency of APOE and ApoA-I proteins potentiates neurite degeneration in the hippocampus, and this effect was CA1 region-specific.
Figure 8.


Deletion of Apoa1 and Apoe significantly affect dendritic morphology in CA1 but not in CA2 region of hippocampus. MAP2 and H3342 staining were used for dendritic tree reconstruction in the hippocampal CA, and CA2 regions of APP/WT, APP/Abca1ko and APP/DKO mice. Data were analysed from a total of four images (×60 confocal imaging) from each of the three sections for each mouse (n = 5 per group). The total dendritic length, branch points and segments were quantified using Imaris filament tracing macros and were normalized to the total H3342 positive nuclei of the CA1 (A and B) and CA2 (C and D) regions. Analysis by one-way ANOVA with Tukey’s post-test. Panel A shows representative images for CA1 region and B shows the quantification of neurites length, branches and segments. In C, representative images for CA2 region and D the quantification of neurites length, branches and segments in this region. Note that deletion of Abca1 as well as the deletion of Apoa1 and Apoe significantly impacts neurite length, segments and branch points only in the CA1 region of the hippocampus. N.S. = not significant.
Discussion
In this study we examined the effect of Apoe and Apoa1 double deletion on amyloid pathology, behaviour and neurite morphology in APP transgenic mice. The results were compared to the effect of Abca1, Apoe or Apoa1 single deletions in the same Alzheimer’s disease model. The conclusion is that in APP/DKO mice, the effect of Apoe deletion on amyloid-β deposition and clearance is dominant whereas the deficiency of Apoa1 does not induce changes of those parameters. In contrast, simultaneous deletion of Apoe and Apoa1 significantly aggravates memory impairment, has a negative impact on dendrite architecture, and increases aortic atherosclerosis.
As mentioned in the ‘Introduction’ section, APP mice lacking Abca1 display increased amyloid deposition accompanied by virtual absence of ApoA-I protein and significantly decreased level of APOE. This study was rationalized on the assumption that the deletion of both Apoe and Apoa1 in APP mice would affect amyloid phenotype in a similar manner as the lack of Abca1. Surprisingly, we discovered that APP/DKO mice differed significantly from APP/Abca1ko and in fact recapitulated the effect of Apoe deletion on amyloid deposition and clearance. Our data demonstrate that APP/DKO mice have significantly less X-34 (Fig. 1A–C) and 6E10-positive amyloid plaques (Fig. 1D–F) and insoluble amyloid-β (Fig. 2F and G) compared to APP/Abca1ko. Furthermore, APP/DKO similarly to APP/Eko mice, have very little soluble amyloid without any effect on APP processing (Fig. 2D) suggesting that amyloid-β clearance is accelerated by the lack of APOE. This conclusion is supported by experiments that measure clearance of radioactive amyloid-β through the blood–brain barrier shown on Fig. 4. Finally, as visible from Fig. 5, lack of Abca1 increased the dissemination of exogenous amyloid significantly more than Apoe and Apoa1 double deletion. Thus, APP/Abca1ko mice, which have virtually no ApoA-I and significantly decreased APOE protein, display substantially worsened amyloid pathology exemplified by increased level of amyloid plaques, soluble oligomers and decreased amyloid-β clearance than mice with global deletion of Apoe and Apoa1. The question that arises is why?
Several possible scenarios can explain why APP/Abca1ko showed worse amyloid phenotype and strongest reaction to exogenous amyloid: (i) lack of ABCA1 is accompanied by absence of APOE-containing high density lipoproteins in CNS (Wahrle et al., 2004) and this increases aggregation of the endogenous amyloid-β or exogenously injected amyloid-β seeds. Previously we have shown that high density lipoproteins particles decrease amyloid-β aggregation in vitro (Lefterov et al., 2010) therefore this can be a plausible explanation; (ii) clearance of amyloid by microglia, both endogenous and exogenous, is impaired in Abca1ko mice because of the lack of brain high density lipoproteins. Prior studies demonstrated the significance of APOE-containing lipoproteins for microglia-mediated amyloid-β clearance (Jiang et al., 2008; Terwel et al., 2011); and (iii) as the efflux of amyloid-β out of the brain in Abca1ko mice is significantly diminished it allows amyloid-β concentration in the brain to reach the critical level needed to form amyloid-β seeds and to accelerate aggregation. These scenarios, however, still do not clarify why the complete absence of APOE and ApoA-I proteins ameliorates the amyloid pathology in APP/DKO mice.
So far, in vivo data have consistently demonstrated that the absence of Apoe decreases compact amyloid plaques in the brain (Bales et al., 1997; Holtzman et al., 2000a, b; Fryer et al., 2003). In support of this, amyloid-β clearance experiments demonstrate that APOE delays amyloid-β efflux from the brain (Bell et al., 2007; Deane et al., 2008). More recent reports established that amyloid pathology is significantly decreased by eliminating only one Apoe allele in mice expressing human APOE4 or APOE3 isoforms (Kim et al., 2011; Bien-Ly et al., 2012). Therefore, the conclusion from studies with Apoe knockout and hemizygous mice is that the presence of APOE in the brain retains amyloid-β in CNS and delays its clearance. In regard to Apoa1, there are two studies demonstrating that Apoa1 deletion does not affect parenchymal amyloid plaques (Fagan et al., 2004; Lefterov et al., 2010). In addition, Apoa1 transgenic overexpression is not sufficient to affect the process of β-amyloidosis (Lewis et al., 2010). Interestingly, both Lefterov et al. (2010) and Lewis et al. (2010) demonstrated that ApoA-1 affects the level of vascular amyloid and cerebral amyloid angiopathy. In our present work, the APP/Ako mice did not show any significant difference in parenchymal load compared to APP/WT controls, and we did not examine cerebral amyloid angiopathy. At the same time, it seems difficult to reconcile the phenotype of APP/Abca1ko mice that have less APOE protein but significantly higher levels of amyloid when compared to APP/DKO and APP/Eko mice. Therefore in a search for an explanation for these seemingly unreconciled facts, we turned the attention to other phenotypic characteristics of Abca1ko and ApoEko mice.
A search of the literature and our data shown on Fig. 6 lead to the conclusion that a major difference between APP/Abca1ko and APP/Eko mice is their very distinct lipoprotein metabolism. ABCA1 regulates cholesterol efflux and high density lipoproteins metabolism, therefore the major consequence of its deletion is the near absence of high density lipoproteins and relatively low LDL (Fitz et al., 2012; Koldamova et al., 2014; Westerterp et al., 2014). In contrast, lack of APOE, a ligand essential for lipoprotein clearance, leads to severe hypercholesterolaemia and spontaneous atherosclerosis (Plump et al., 1992; Breslow, 1996). We have shown here that APP/DKO and APP/Eko mice have significantly more plasma cholesterol, mainly in the form of VLDL/LDL, compared to APP/WT mice. In contrast, APP/Abca1ko mice had the lowest level of plasma lipoproteins (high density lipoproteins and VLDL/LDL). One possibility which we have explored, was to measure how the level of plasma lipoproteins affects peripheral amyloid-β concentrations. As shown on Fig. 6C, the level of plasma amyloid-β42 in APP/DKO mice was significantly higher compared to APP/Abca1ko at all of the examined time points. This result suggests the probability that significantly higher plasma levels of VLDL/LDL in APP/DKO mice could stimulate amyloid-β efflux out of the brain by the ‘peripheral sink’ mechanism. Initially, the peripheral sink hypothesis for amyloid-β clearance was proposed by DeMattos et al. (2001) to explain the decreased amyloid load in the brain after peripheral administration of anti-amyloid-β antibody. Data from patients and experimental animals support this hypothesis. A cross-sectional study showed that an increased brain amyloid-β is accompanied by lower peripheral levels of amyloid-β in patients with mild cognitive impairment (Devanand et al., 2011) and another more recent report demonstrated that lower plasma amyloid-β levels are associated with an increased risk of Alzheimer’s disease (Chouraki et al., 2015). Studies in mice also support the peripheral sink hypothesis: for example increased levels of amyloid-β binding molecules in plasma such as gelsolin (Gregory et al., 2012) or circulating lipoprotein receptors (Sagare et al., 2007) can affect amyloid-β level in the brain. Further support are data from our group demonstrating that plasma high density lipoprotein levels in APOE3 and APOE4 mice with one copy of Abca1 is significantly decreased and negatively correlated to amyloid-β plaque load (Fitz et al., 2012). In the same study we showed that APOE3 and APOE4 mice with higher amyloid-β42 levels in plasma have fewer X-34-positive plaques, suggesting an increased efflux out of the brain (Fitz et al., 2012).
In regard to cognitive performance, we have previously shown that APP mice with one copy of Abca1 perform significantly worse in a Morris water maze paradigm than mice with intact Abca1 in correlation to the level of soluble amyloid-β oligomers (Lefterov et al., 2009). Herein, we confirmed the behaviour deficits caused by Abca1 deletion using another behaviour test—contextual fear conditioning. Surprisingly, APP/DKO mice displayed significant memory deficits in this test that are indistinguishable from APP/Abca1ko mice (Fig. 7). The effect was unrelated to amyloid deposition, as non-transgenic controls showed similar performance in this paradigm. The effect was also unrelated to the soluble amyloid-β oligomers as APP/DKO mice had a similar level of oligomers compared to APP/WT mice (Fig. 3A and B). Our conclusion is that lack of brain APOE-containing lipoproteins in the case of double knockout mice and their significant decrease in Abca1ko mice affects memory regardless of amyloid-β oligomers and amyloid-β plaques.
Consistent with the behaviour data, are the results obtained for dendritic architecture (Fig. 8). Both APP/DKO and APP/Abca1ko mice demonstrated significant impairment in neurite length and the number of branches and segments, compared to APP/WT mice. Previous studies demonstrated that APOE has an isoform-specific effect on dendrite morphology in mice and in patients (Schonheit et al., 2007; Dumanis et al., 2009; Nwabuisi-Heath et al., 2014). Deficiency of Apoe reduces the number of synapses (Diaz-Cintra et al., 2004), affects hippocampal reactive sprouting response following lesions in entorhinal cortex (Champagne et al., 2005), and decreases MAP2-positive staining of neurons in amygdala (Robertson et al., 2005). Apoe knockout mice challenged with kainic acid have a significant loss of synaptophysin-positive presynaptic terminals and MAP2-positive dendrites in the neocortex and hippocampus (Buttini et al., 1999). However, it remains elusive how APOE can cause these effects on neurons. One plausible explanation is that APOE (as the main carrier of cholesterol and phospholipids in the brain) delivers these lipids to neurons. APOE is secreted by glial cells and serves as an acceptor for cholesterol and phospholipids during the process of cholesterol efflux mediated by ABCA1 (Abad-Rodriguez et al., 2004). Regardless of evidence that adult neurons can synthesize cholesterol (Dietschy, 2009), they rely on exogenously supplied lipids especially during periods of increased demands such as repair and increased synaptic activity (Pfrieger and Barres, 1997). Therefore, the similarities in the dendritic morphology of APP/Abca1ko and APP/DKO mice could be explained by the fact that they both lack APOE-containing high density lipoproteins in CNS.
In conclusion, our study demonstrates that the absence of both APOE and ApoA-I proteins has a favourable effect on amyloid pathology probably as a result of the increased amyloid-β clearance. In contrast the double deletion of these apolipoproteins causes deficits in cognitive performance and dendritic morphology. Thus, these findings emphasize the importance of APOE and ApoA-I for cognition and presents novel opportunities for therapeutic intervention.
Supplementary Material
Acknowledgements
We are grateful to Dr Neale S. Mason for his technical assistance (Departments of Radiology and Psychiatry, University of Pittsburgh, Pittsburgh, Pa.).
Glossary
Abbreviations
- Abca1ko
Abca1 knockout
- APP
APP/PS1dE9 mouse model
- ApoA-I
apolipoprotein A-I
- ApoE
apolipoprotein E
- APP/DKO
APP mice with double deletion of Apoe and Apoa1
- Ako
Apoa1 knockout
- Eko
Apoe knockout
- VLDL/LDL
very-low-density lipoprotein/low-density lipoprotein
Funding
NIH: AG037481, AG037919, ES024233, ES021243, K01AG044490, Alzheimer’s Association: NIRG-12-242432, DOD: W81XWH-13-1-0384.
Supplementary material
Supplementary material is available at Brain online.
References
- Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, et al. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol 2004; 167: 953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 1997; 17: 263–4. [DOI] [PubMed] [Google Scholar]
- Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, et al. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 2007; 27: 909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Ab accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci 2012; 32: 4803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow JL. Mouse models of atherosclerosis. Science 1996; 272: 685–8. [DOI] [PubMed] [Google Scholar]
- Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nature Genet 1999; 22: 336–45. [DOI] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, et al. Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/- mice: isoform-specific effects on neurodegeneration. J Neurosci 1999; 19: 4867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne D, Rochford J, Poirier J. Effect of apolipoprotein E deficiency on reactive sprouting in the dentate gyrus of the hippocampus following entorhinal cortex lesion: role of the astroglial response. Exp Neurol 2005; 194: 31–42. [DOI] [PubMed] [Google Scholar]
- Chouraki V, Beiser A, Younkin L, Preis SR, Weinstein G, Hansson O, et al. Plasma amyloid-beta and risk of Alzheimer's disease in the Framingham Heart Study. Alzheimers Dement 2015; 11: 249–57.e1. doi: 10.1016/j.jalz.2014.07.001. Epub Sep 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 2005; 115: 3285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest 2008; 118: 4002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du YS, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004; 43: 333–44. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 2001; 98: 8850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, et al. ApoE and clusterin cooperatively suppress abeta levels and deposition. evidence that apoe regulates extracellular abeta metabolism in vivo. Neuron 2004; 41: 193–202. [DOI] [PubMed] [Google Scholar]
- Devanand DP, Schupf N, Stern Y, Parsey R, Pelton GH, Mehta P, et al. Plasma Abeta and PET PiB binding are inversely related in mild cognitive impairment. Neurology 2011; 77: 125–31. doi: 10.1212/WNL.0b013e318224afb7. Epub 2011 Jun 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Cintra S, Yong A, Aguilar A, Bi X, Lynch G, Ribak CE. Ultrastructural analysis of hippocampal pyramidal neurons from apolipoprotein E-deficient mice treated with a cathepsin inhibitor. J Neurocytol 2004; 33: 37–48. [DOI] [PubMed] [Google Scholar]
- Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem 2009; 390: 287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci 2009; 29: 15317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Christopher E, Taylor JW, Parsadanian M, Spinner M, Watson M, et al. ApoAI deficiency results in marked reductions in plasma cholesterol but no alterations in amyloid-{beta} pathology in a mouse model of Alzheimer's disease-like cerebral amyloidosis. Am J Pathol 2004; 165: 1413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Castranio EL, Carter AY, Kodali R, Lefterov I, Koldamova R. Improvement of memory deficits and amyloid-beta clearance in aged APP23 mice treated with a combination of anti-amyloid-beta antibody and LXR agonist. J Alzheimers Dis 2014; 41: 535–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican AA, Lefterov I, Koldamova R. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models". Science 2013; 340: 924-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I, et al. Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci 2012; 32: 13125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, et al. Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci 2003; 23: 7889–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory JL, Prada CM, Fine SJ, Garcia-Alloza M, Betensky RA, Arbel-Ornath M, et al. Reducing available soluble beta-amyloid prevents progression of cerebral amyloid angiopathy in transgenic mice. J Neuropathol Exp Neurol 2012; 71: 1009–17. doi: 10.97/NEN.0b013e3182729845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hem A, Smith AJ, Solberg P. Saphenous vein puncture for blood sampling of the mouse, rat, hamster, gerbil, guinea pig, ferret and mink. Lab Anim 1998; 32: 364–8. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Maia LF, Burgess BL, Blain JF, Naus KE, McIsaac SA, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem 2005; 280: 43243–56. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 2000a; 97: 2892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, et al. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Ann Neurol 2000b; 47: 739–47. [PubMed] [Google Scholar]
- Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis 2014; 72(Pt A): 3–12. doi: 0.1016/j.nbd.2014.08.025. Epub Aug 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008; 58: 681–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron 2014; 81: 740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300: 486–9. [DOI] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron 2009; 63: 287–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci 2011; 31: 18007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Fitz NF, Lefterov I. ATP-binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol Dis 2014; 72 Pt A: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 cnsiderably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem 2005; 280: 43224–35. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Munson GW, Jungbauer L, Getz GS, Reardon CA, Tai LM, et al. Preferential interactions between ApoE-containing lipoproteins and Abeta revealed by a detection method that combines size exclusion chromatography with non-reducing gel-shift. Biochim Biophys Acta 2012; 1821: 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterov I, Fitz NF, Cronican A, Lefterov P, Staufenbiel M, Koldamova R. Memory deficits in APP23/Abca1+/- mice correlate with the level of Abeta oligomers. ASN Neuro 2009; 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterov I, Fitz NF, Cronican AA, Fogg A, Lefterov P, Kodali R, et al. Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem 2010; 285: 36945–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL, Cao D, Lu H, Mans RA, Su YR, Jungbauer L, et al. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem 2010; 285: 36958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313: 1781–4. [DOI] [PubMed] [Google Scholar]
- Nwabuisi-Heath E, Rebeck GW, Ladu MJ, Yu C. ApoE4 delays dendritic spine formation during neuron development and accelerates loss of mature spines in vitro. ASN Neuro 2014; 6: e00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolini C, Chiesa G, Gong E, Caligari S, Cortese MM, Koga T, et al. Apolipoprotein A-I and the molecular variant apoA-I(Milano): evaluation of the antiatherogenic effects in knock-in mouse model. Atherosclerosis 2005; 183: 222–9. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science 1997; 277: 1684–7. [DOI] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992; 71: 343–53. [DOI] [PubMed] [Google Scholar]
- Robertson J, Curley J, Kaye J, Quinn J, Pfankuch T, Raber J. apoE isoforms and measures of anxiety in probable AD patients and Apoe-/- mice. Neurobiol Aging 2005; 26: 637–43. [DOI] [PubMed] [Google Scholar]
- Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med 2007; 13: 1029–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Schmader K, Breitner JC, Benson MD, Brown WT, Goldfarb L, et al. Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer's disease and in other amyloid-forming diseases. Lancet 1993; 342: 710–1. [DOI] [PubMed] [Google Scholar]
- Schonheit B, Glockner F, Ohm TG. Apolipoprotein E polymorphism and dendritic shape in hippocampal interneurons. Neurobiol Aging 2007; 28: 677–86. [DOI] [PubMed] [Google Scholar]
- Tapias V, Greenamyre JT. A rapid and sensitive automated image-based approach for in vitro and in vivo characterization of cell morphology and quantification of cell number and neurite architecture. Curr Protoc Cytom 2014; 68: 12.33.1–12.33.22. [DOI] [PubMed] [Google Scholar]
- Tapias V, Greenamyre JT, Watkins SC. Automated imaging system for fast quantitation of neurons, cell morphology and neurite morphometry in vivo and in vitro. Neurobiol Dis 2013; 54: 158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JA, Holtzman DM, et al. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J Neurosci 2011; 31: 7049–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorngate FE, Yancey PG, Kellner-Weibel G, Rudel LL, Rothblat GH, Williams DL. Testing the role of apoA-I, HDL, and cholesterol efflux in the atheroprotective action of low-level apoE expression. J Lpid Res 2003; 44: 2331–8. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, et al. Deletion of Abca1 increases A{beta} deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem 2005; 280: 43236–42. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest 2008; 118: 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem 2004; 279: 40987–93. [DOI] [PubMed] [Google Scholar]
- Westerterp M, Bochem AE, Yvan-Charvet L, Murphy AJ, Wang N, Tall AR. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ Res 2014; 114: 157–70. doi: 10.1161/CIRCRESAHA.114.300738. [DOI] [PubMed] [Google Scholar]
- Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu Rev Neurosci 2014; 37: 79–100. (doi): 10.1146/annurev-neuro-071013-14300. Epub 2014 Apr 21. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Pennings M, Vrins CL, Calpe-Berdiel L, Hoekstra M, Kruijt JK, et al. Hypocholesterolemia, foam cell accumulation, but no atherosclerosis in mice lacking ABC-transporter A1 and scavenger receptor BI. Atherosclerosis 2011; 218: 314–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.